
Galectin-1 in Cardiovascular Pathogenesis: Unraveling Dual Roles and Mechanistic Insights in Emerging Research

 ,
,  ,
,  ,
, 
 ,
, 
Abstract

1. Introduction
2. Galectin-1: A Multifaceted Player in Cardiovascular Health and Disease
2.1. Overview of Galectin-1 Structure and Function
2.2. Dual Role of Galectin-1 in Cardiovascular Diseases
2.2.1. Protective Effect
2.2.2. Pathological Implications
3. Insights from Recent Studies
3.1. Galectin-1 and Atherosclerosis
3.1.1. Anti-Inflammatory Actions
3.1.2. Endothelial Cell Function Modulation
3.2. Galectin-1 and Myocardial Infarction
3.2.1. Cardioprotective Mechanisms
3.2.2. Fibrosis and Remodeling
3.3. Galectin-1 and Heart Failure
3.3.1. Role in Cardiac Fibrosis
3.3.2. Impact on Immune Response
4. Clinical Implications and Therapeutic Potential
4.1. Galectin-1 as a Biomarker for Cardiovascular Risk Assessment
4.2. Targeting Galectin-1 for Therapeutic Intervention
4.2.1. Pharmacological Agents
4.2.2. Gene Therapy Approaches
5. Challenges and Future Directions
5.1. Limitations of Current Research
5.2. Unresolved Questions and Areas for Further Investigation
5.3. Potential Strategies to Overcome Challenges
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Gal-1 | Galectin-1 |
| CVD | Cardiovascular diseases |
| HF | Heart failure |
| CAD | Coronary artery disease |
| HRs | Hazard ratios |
| CIs | Confidence intervals |
| MI | Myocardial infarction |
| BMSCs | Bone marrow stromal cells |
| CRD | Carbohydrate recognition domain |
| MMP | Matrix metalloproteinases |
| TIMPs | Tissue inhibitors of metalloproteinases |
| ECM | Extracellular matrix |
| TNF-α | Tumor necrosis factor-alpha |
| IL-6 | Interleukin-6 |
| IL-1β | Interleukin-1 beta |
| IL-10 | Interleukin-10 |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| ICAM-1 | Intercellular adhesion molecule-1 |
| HFpEF | Heart failure with preserved ejection fraction |
| AAV | Adeno-associated viruses |
References
- Sotoudeheian, M.J.; Mirahmadi, S.M.; Pirhayati, M.; Azarbad, R.; Nematollahi, S.; Taghizadeh, M.; Pazoki-Toroudi, H. Understanding the Role of Galectin-1 in Heart Failure: A Comprehensive Narrative Review. Curr. Cardiol. Rev. 2024, 20, 9. [Google Scholar] [CrossRef] [PubMed]
- Sygitowicz, G.; Maciejak-Jastrzębska, A.; Sitkiewicz, D. The Diagnostic and Therapeutic Potential of Galectin-3 in Cardiovascular Diseases. Biomolecules 2021, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Seropian, I.M.; Cassaglia, P.; Miksztowicz, V.; González, G.E. Unraveling the role of galectin-3 in cardiac pathology and physiology. Front. Physiol. 2023, 14, 1304735. [Google Scholar] [CrossRef] [PubMed]
- Cassaglia, P.; Penas, F.; Betazza, C.; Estevez, F.F.; Miksztowicz, V.; Naya, N.M.; Llamosas, M.C.; Truant, S.N.; Wilensky, L.; Volberg, V.; et al. Genetic Deletion of Galectin-3 Alters the Temporal Evolution of Macrophage Infiltration and Healing Affecting the Cardiac Remodeling and Function after Myocardial Infarction in Mice. Am. J. Pathol. 2020, 190, 1789–1800. [Google Scholar] [CrossRef]
- Mansour, A.A.; Krautter, F.; Zhi, Z.; Iqbal, A.J.; Recio, C. The interplay of galectins-1, -3, and -9 in the immune-inflammatory response underlying cardiovascular and metabolic disease. Cardiovasc. Diabetol. 2022, 21, 253. [Google Scholar] [CrossRef]
- Yu, X.; Qian, J.; Ding, L.; Yin, S.; Zhou, L.; Zheng, S. Galectin-1: A Traditionally Immunosuppressive Protein Displays Context-Dependent Capacities. Int. J. Mol. Sci. 2023, 24, 6501. [Google Scholar] [CrossRef]
- Li, R.; Shao, J.; Hu, C.; Xu, T.; Zhou, J.; Zhang, J.; Liu, Q.; Han, M.; Ning, N.; Fan, X.; et al. Metabolic risks remain a serious threat to cardiovascular disease: Findings from the Global Burden of Disease Study 2019. Intern. Emerg. Med. 2024, 19, 1299–1312. [Google Scholar] [CrossRef]
- GBD 2021 Risk Factors Collaborators. Global burden and strength of evidence for 88 risk factors in 204 countries and 811 subnational locations, 1990–2021: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2024, 403, 2162–2203. [Google Scholar] [CrossRef]
- Loh, K.W.Z.; Liang, M.C.; Soong, T.W.; Hu, Z. Regulation of cardiovascular calcium channel activity by post-translational modifications or interacting proteins. Pflug. Arch. 2020, 472, 653–667. [Google Scholar] [CrossRef]
- Drake, I.; Fryk, E.; Strindberg, L.; Lundqvist, A.; Rosengren, A.H.; Groop, L.; Ahlqvist, E.; Borén, J.; Orho-Melander, M.; Jansson, P.A. The role of circulating galectin-1 in type 2 diabetes and chronic kidney disease: Evidence from cross-sectional, longitudinal and Mendelian randomisation analyses. Diabetologia 2022, 65, 128–139. [Google Scholar] [CrossRef]
- Sanjurjo, L.; Schulkens, I.A.; Touarin, P.; Heusschen, R.; Aanhane, E.; Castricum, K.C.M.; De Gruijl, T.D.; Nilsson, U.J.; Leffler, H.; Griffioen, A.W.; et al. Chemokines modulate glycan binding and the immunoregulatory activity of galectins. Commun. Biol. 2021, 4, 1415. [Google Scholar] [CrossRef] [PubMed]
- Dickhout, A.; Tullemans, B.M.E.; Heemskerk, J.W.M.; Thijssen, V.; Kuijpers, M.J.E.; Koenen, R.R. Galectin-1 and platelet factor 4 (CXCL4) induce complementary platelet responses in vitro. PLoS ONE 2021, 16, e0244736. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, H.; Cruz-Adalia, A.; Del Hoyo, G.M.; Cibrián-Vera, D.; Bonay, P.; Pérez-Hernández, D.; Vázquez, J.; Navarro, P.; Gutierrez-Gallego, R.; Ramirez-Huesca, M.; et al. The leukocyte activation receptor CD69 controls T cell differentiation through its interaction with galectin-1. Mol. Cell. Biol. 2014, 34, 2479–2487. [Google Scholar] [CrossRef]
- Karmakar, S.; Stowell, S.R.; Cummings, R.D.; McEver, R.P. Galectin-1 signaling in leukocytes requires expression of complex-type N-glycans. Glycobiology 2008, 18, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, Y.Y.; Liu, W.J.; Fu, Q.; Zhao, J.; Liu, Y.B. DNA methylation of promoter region inhibits galectin-1 expression in BMSCs of aged mice. Am. J. Physiol. Cell Physiol. 2024, 326, C429–C441. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Zhang, Y.; Yu, J.; Tang, L. Galectin-1 deletion in mice causes bone loss via impaired osteogenic differentiation potential of BMSCs. FASEB J. 2022, 36, e22516. [Google Scholar] [CrossRef]
- Faust, K.; Freitag, N.; Barrientos, G.; Hartel, C.; Blois, S.M. Galectin-Levels Are Elevated in Infants Born Preterm Due to Amniotic Infection and Rapidly Decline in the Neonatal Period. Front. Immunol. 2020, 11, 599104. [Google Scholar] [CrossRef]
- Bartolomé, M.V.; López, L.M.; Gil-Loyzaga, P. Galectine-1 expression in cochleae of C57BL/6 mice during aging. Neuroreport 2001, 12, 3107–3110. [Google Scholar] [CrossRef]
- Jover, E.; Martín-Núñez, E.; Garaikoetxea, M.; Matilla, L.; Blanco-Colio, L.M.; Pérez-Sáez, J.M.; Navarro, A.; Fernández-Celis, A.; Gainza, A.; Álvarez, V.; et al. Sex-dependent expression of galectin-1, a cardioprotective β-galactoside-binding lectin, in human calcific aortic stenosis. FASEB J. 2024, 38, e23447. [Google Scholar] [CrossRef]
- Takeuchi, T.; Oyama, M.; Tamura, M.; Arata, Y.; Hatanaka, T. Reduced form of Galectin-1 Suppresses Osteoclastic Differentiation of Human Peripheral Blood Mononuclear Cells and Murine RAW264 Cells In Vitro. Biomolecules 2024, 14, 121. [Google Scholar] [CrossRef]
- Hu, Z.; Li, G.; Wang, J.W.; Chong, S.Y.; Yu, D.; Wang, X.; Soon, J.L.; Liang, M.C.; Wong, Y.P.; Huang, N.; et al. Regulation of Blood Pressure by Targeting Ca(V)1.2-Galectin-1 Protein Interaction. Circulation 2018, 138, 1431–1445. [Google Scholar] [CrossRef]
- Wang, J.; Thio, S.S.; Yang, S.S.; Yu, D.; Yu, C.Y.; Wong, Y.P.; Liao, P.; Li, S.; Soong, T.W. Splice variant specific modulation of CaV1.2 calcium channel by galectin-1 regulates arterial constriction. Circ. Res. 2011, 109, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.T.; Chen, I.M.; Hsu, F.F.; Chen, Y.H.; Tsai, M.S.; Hsu, Y.W.; Leu, H.B.; Huang, P.H.; Chen, J.W.; Liu, F.T.; et al. Gal-1 (Galectin-1) Upregulation Contributes to Abdominal Aortic Aneurysm Progression by Enhancing Vascular Inflammation. Arter. Thromb. Vasc. Biol. 2021, 41, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.H.; Huang, S.S.; Kuo, C.S.; Wang, S.C.; Tsai, Y.L.; Lu, Y.W.; Chang, C.C.; Huang, P.H.; Lin, S.J. Galectin-1 is associated with the severity of coronary artery disease and adverse cardiovascular events in patients undergoing coronary angiography. Sci. Rep. 2020, 10, 20683. [Google Scholar] [CrossRef] [PubMed]
- Chou, R.H.; Tsai, C.T.; Lu, Y.W.; Guo, J.Y.; Lu, C.T.; Tsai, Y.L.; Wu, C.H.; Lin, S.J.; Lien, R.Y.; Lu, S.F.; et al. Elevated serum galectin-1 concentrations are associated with increased risks of mortality and acute kidney injury in critically ill patients. PLoS ONE 2021, 16, e0257558. [Google Scholar] [CrossRef]
- Seropian, I.M.; González, G.E.; Maller, S.M.; Berrocal, D.H.; Abbate, A.; Rabinovich, G.A. Galectin-1 as an Emerging Mediator of Cardiovascular Inflammation: Mechanisms and Therapeutic Opportunities. Mediat. Inflamm. 2018, 2018, 8696543. [Google Scholar] [CrossRef]
- Roldán-Montero, R.; Pérez-Sáez, J.M.; Cerro-Pardo, I.; Oller, J.; Martinez-Lopez, D.; Nuñez, E.; Maller, S.M.; Gutierrez-Muñoz, C.; Mendez-Barbero, N.; Escola-Gil, J.C.; et al. Galectin-1 prevents pathological vascular remodeling in atherosclerosis and abdominal aortic aneurysm. Sci. Adv. 2022, 8, eabm7322. [Google Scholar] [CrossRef]
- Law, H.L.; Wright, R.D.; Iqbal, A.J.; Norling, L.V.; Cooper, D. A Pro-resolving Role for Galectin-1 in Acute Inflammation. Front. Pharmacol. 2020, 11, 274. [Google Scholar] [CrossRef]
- Tsai, M.S.; Chiang, M.T.; Tsai, D.L.; Yang, C.W.; Hou, H.S.; Li, Y.R.; Chang, P.C.; Lin, H.H.; Chen, H.Y.; Hwang, I.S.; et al. Galectin-1 Restricts Vascular Smooth Muscle Cell Motility Via Modulating Adhesion Force and Focal Adhesion Dynamics. Sci. Rep. 2018, 8, 11497. [Google Scholar] [CrossRef]
- Ou, D.; Ni, D.; Li, R.; Jiang, X.; Chen, X.; Li, H. Galectin-1 alleviates myocardial ischemia-reperfusion injury by reducing the inflammation and apoptosis of cardiomyocytes. Exp. Ther. Med. 2022, 23, 143. [Google Scholar] [CrossRef]
- Seropian, I.M.; Cerliani, J.P.; Toldo, S.; Van Tassell, B.W.; Ilarregui, J.M.; González, G.E.; Matoso, M.; Salloum, F.N.; Melchior, R.; Gelpi, R.J.; et al. Galectin-1 controls cardiac inflammation and ventricular remodeling during acute myocardial infarction. Am. J. Pathol. 2013, 182, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Fan, W.; Lei, J.; Zhou, Y.; Xu, H.; Kapoor, I.; Zhu, G.; Wang, J. Galectin-1 attenuates cardiomyocyte hypertrophy through splice-variant specific modulation of Ca(V)1.2 calcium channel. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, V.; Miller, M.C.; Blanchet, X.; Duan, R.; Leberzammer, J.; Duchene, J.; Soehnlein, O.; Megens, R.T.; Ludwig, A.K.; Dregni, A.; et al. Chemokines and galectins form heterodimers to modulate inflammation. EMBO Rep. 2020, 21, e47852. [Google Scholar] [CrossRef]
- Perillo, N.L.; Pace, K.E.; Seilhamer, J.J.; Baum, L.G. Apoptosis of T cells mediated by galectin-1. Nature 1995, 378, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Chellan, B.; Narayani, J.; Appukuttan, P.S. Galectin-1, an endogenous lectin produced by arterial cells, binds lipoprotein(a) [Lp(a)] in situ: Relevance to atherogenesis. Exp. Mol. Pathol. 2007, 83, 399–404. [Google Scholar] [CrossRef]
- Al-Obaidi, N.; Mohan, S.; Liang, S.; Zhao, Z.; Nayak, B.K.; Li, B.; Sriramarao, P.; Habib, S.L. Galectin-1 is a new fibrosis protein in type 1 and type 2 diabetes. FASEB J. 2019, 33, 373–387. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Chou, R.H.; Lu, Y.W.; Chang, C.C.; Kuo, C.S.; Huang, P.H.; Chen, J.W.; Lin, S.J. Associations between galectin-1, left ventricular diastolic dysfunction, and heart failure with preserved ejection fraction. J. Cardiol. 2022, 79, 371–375. [Google Scholar] [CrossRef]
- Kuo, C.S.; Chou, R.H.; Lu, Y.W.; Tsai, Y.L.; Huang, P.H.; Lin, S.J. Increased circulating galectin-1 levels are associated with the progression of kidney function decline in patients undergoing coronary angiography. Sci. Rep. 2020, 10, 1435. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, T.A.; Teuber, J.P.; Kuwabara, Y.; Subramani, A.; Lin, S.J.; Kanisicak, O.; Vagnozzi, R.J.; Zhang, W.; Brody, M.J.; Molkentin, J.D. Palmitoylation-dependent regulation of cardiomyocyte Rac1 signaling activity and minor effects on cardiac hypertrophy. J. Biol. Chem. 2023, 299, 105426. [Google Scholar] [CrossRef]
- Krautter, F.; Recio, C.; Hussain, M.T.; Lezama, D.R.; Maione, F.; Chimen, M.; Iqbal, A.J. Characterisation of endogenous Galectin-1 and -9 expression in monocyte and macrophage subsets under resting and inflammatory conditions. Biomed. Pharmacother. 2020, 130, 110595. [Google Scholar] [CrossRef]
- Sundblad, V.; Morosi, L.G.; Geffner, J.R.; Rabinovich, G.A. Galectin-1: A Jack-of-All-Trades in the Resolution of Acute and Chronic Inflammation. J. Immunol. 2017, 199, 3721–3730. [Google Scholar] [CrossRef] [PubMed]
- Anginot, A.; Espeli, M.; Chasson, L.; Mancini, S.J.; Schiff, C. Galectin 1 modulates plasma cell homeostasis and regulates the humoral immune response. J. Immunol. 2013, 190, 5526–5533. [Google Scholar] [CrossRef]
- Kotterman, M.; Whittlesey, K.; Brooks, G.; Croze, R.; Schmitt, C.; Szymanski, P.; Nye, J.; Quezada, M.; Beliakoff, G.; Johnson, L.; et al. P1516Novel cardiotropic AAV variant C102 vectors show superior gene delivery & reduced immunogenicity in non-human primates, transduction of human cardiomyocytes, & correction of Fabry disease phenotype. Eur. Heart J. 2019, 40, ehz748.0278. [Google Scholar] [CrossRef]
- Wei, J.; Wu, Y.; Sun, Y.; Chen, D. Galectin-1 Regulates RNA Expression and Alternative Splicing of Angiogenic Genes in HUVECs. Front. Biosci. 2023, 28, 74. [Google Scholar] [CrossRef]
- La, M.; Cao, T.V.; Cerchiaro, G.; Chilton, K.; Hirabayashi, J.; Kasai, K.; Oliani, S.M.; Chernajovsky, Y.; Perretti, M. A novel biological activity for galectin-1: Inhibition of leukocyte-endothelial cell interactions in experimental inflammation. Am. J. Pathol. 2003, 163, 1505–1515. [Google Scholar] [CrossRef]
- Al-Salam, S.; Hashmi, S. Galectin-1 in early acute myocardial infarction. PLoS ONE 2014, 9, e86994. [Google Scholar] [CrossRef]
- Hermenean, A.; Oatis, D.; Herman, H.; Ciceu, A.; D’Amico, G.; Trotta, M.C. Galectin 1-A Key Player between Tissue Repair and Fibrosis. Int. J. Mol. Sci. 2022, 23, 5548. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Bu, M.; Yu, P.; Li, Y.; Chong, Y. Regulation of wound healing and fibrosis by galectins. J. Mol. Med. 2022, 100, 861–874. [Google Scholar] [CrossRef]
- Kremastiotis, G.; Handa, I.; Jackson, C.; George, S.; Johnson, J. Disparate effects of MMP and TIMP modulation on coronary atherosclerosis and associated myocardial fibrosis. Sci. Rep. 2021, 11, 23081. [Google Scholar] [CrossRef]
- Wienke, J.; Mertens, J.S.; Garcia, S.; Lim, J.; Wijngaarde, C.A.; Yeo, J.G.; Meyer, A.; van den Hoogen, L.L.; Tekstra, J.; Hoogendijk, J.E.; et al. Biomarker profiles of endothelial activation and dysfunction in rare systemic autoimmune diseases: Implications for cardiovascular risk. Rheumatology 2021, 60, 785–801. [Google Scholar] [CrossRef]
- Yousefi, F.; Shabaninejad, Z.; Vakili, S.; Derakhshan, M.; Movahedpour, A.; Dabiri, H.; Ghasemi, Y.; Mahjoubin-Tehran, M.; Nikoozadeh, A.; Savardashtaki, A.; et al. TGF-β and WNT signaling pathways in cardiac fibrosis: Non-coding RNAs come into focus. Cell Commun. Signal. 2020, 18, 87. [Google Scholar] [CrossRef]
- Parichatikanond, W.; Luangmonkong, T.; Mangmool, S.; Kurose, H. Therapeutic Targets for the Treatment of Cardiac Fibrosis and Cancer: Focusing on TGF-β Signaling. Front. Cardiovasc. Med. 2020, 7, 34. [Google Scholar] [CrossRef]
- Li, Y.Y.; McTiernan, C.F.; Feldman, A.M. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc. Res. 2000, 46, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, A.; Prado, A.F.; Antonio, R.C.; Issa, J.P.; Gerlach, R.F. Matrix metalloproteinases are involved in cardiovascular diseases. Basic Clin. Pharmacol. Toxicol. 2014, 115, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Bosch, N.; Vilariño, N.; Alameda, F.; Mojal, S.; Arumí-Uria, M.; Carrato, C.; Aldecoa, I.; Ribalta, T.; Vidal, N.; Bellosillo, B.; et al. Gal-1 Expression Analysis in the GLIOCAT Multicenter Study: Role as a Prognostic Factor and an Immune-Suppressive Biomarker. Cells 2023, 12, 843. [Google Scholar] [CrossRef] [PubMed]
- Novák, J.; Takács, T.; Tilajka, Á.; László, L.; Oravecz, O.; Farkas, E.; Than, N.G.; Buday, L.; Balogh, A.; Vas, V. The sweet and the bitter sides of galectin-1 in immunity: Its role in immune cell functions, apoptosis, and immunotherapies for cancer with a focus on T cells. Semin. Immunopathol. 2025, 47, 24. [Google Scholar] [CrossRef]
- Kovalová, A.; Prouza, V.; Zavřel, M.; Hájek, M.; Dzijak, R.; Magdolenová, A.; Pohl, R.; Voburka, Z.; Parkan, K.; Vrabel, M. Selection of Galectin-Binding Ligands from Synthetic Glycopeptide Libraries. Chempluschem 2023, 89, e202300567. [Google Scholar] [CrossRef]
- Sethi, A.; Sanam, S.; Alvala, M. Non-carbohydrate strategies to inhibit lectin proteins with special emphasis on galectins. Eur. J. Med. Chem. 2021, 222, 113561. [Google Scholar] [CrossRef]
- Gu, Y.; Zhao, Y.; Zhang, Z.; Hao, J.; Zheng, Y.; Liu, Q.; Liu, Y.; Shi, L. An Antibody-like Polymeric Nanoparticle Removes Intratumoral Galectin-1 to Enhance Antitumor T-Cell Responses in Cancer Immunotherapy. ACS Appl. Mater. Interfaces 2021, 13, 22159–22168. [Google Scholar] [CrossRef]
- Rodríguez-Remírez, M.; Del Puerto-Nevado, L.; Fernández-Aceñero, M.J.; Cruz-Ramos, M.; García-García, L.; Solanes, S.; Molina-Roldán, E.; García-Foncillas, J.; Cebrián, A. Targeting Galectin-1 by Aflibercept Strongly Enhances Its Antitumor Effect in Neuroendocrine Carcinomas. Neuroendocrinology 2021, 111, 146–157. [Google Scholar] [CrossRef]
- Leung, Z.; Ko, F.C.F.; Tey, S.K.; Kwong, E.M.L.; Mao, X.; Liu, B.H.M.; Ma, A.P.Y.; Fung, Y.M.E.; Che, C.M.; Wong, D.K.H.; et al. Galectin-1 promotes hepatocellular carcinoma and the combined therapeutic effect of OTX008 galectin-1 inhibitor and sorafenib in tumor cells. J. Exp. Clin. Cancer Res. 2019, 38, 423. [Google Scholar] [CrossRef] [PubMed]
- Paz, H.; Joo, E.J.; Chou, C.H.; Fei, F.; Mayo, K.H.; Abdel-Azim, H.; Ghazarian, H.; Groffen, J.; Heisterkamp, N. Treatment of B-cell precursor acute lymphoblastic leukemia with the Galectin-1 inhibitor PTX008. J. Exp. Clin. Cancer Res. 2018, 37, 67. [Google Scholar] [CrossRef] [PubMed]
- Orozco, C.A.; Martinez-Bosch, N.; Guerrero, P.E.; Vinaixa, J.; Dalotto-Moreno, T.; Iglesias, M.; Moreno, M.; Djurec, M.; Poirier, F.; Gabius, H.J.; et al. Targeting galectin-1 inhibits pancreatic cancer progression by modulating tumor-stroma crosstalk. Proc. Natl. Acad. Sci. USA 2018, 115, E3769–E3778. [Google Scholar] [CrossRef] [PubMed]
- Herman, K.D.; Holyer, I.; Humphries, D.C.; Roper, J.A.; Peterson, K.; Zetterberg, F.R.; Pedersen, A.; MacKinnon, A.C.; Slack, R.J. Pharmacological Characterization of GB1908, a Selective Galectin-1 Carbohydrate Binding Domain Inhibitor for the Treatment of Cancer. Pharmacology 2025, 3, 1–14. [Google Scholar] [CrossRef]
- Wang, H.C.; Gao, A.C.; Xia, R.; Wu, C.T.; Hsu, S.W.; Chen, C.H.; Shih, T.C. Inhibition of Galectin-1 and Androgen Receptor Axis Enhances Enzalutamide Treatment in Enzalutamide Resistant Prostate Cancer. Cancers 2025, 17, 351. [Google Scholar] [CrossRef]
- Carvalho, L.; Assis, R.A.; Montenegro, C.; da Rosa, M.M.; Pereira, M.C.; Pitta, M.; de Melo Rêgo, M.J.B. Galectin Plasmatic Levels Reveal a Cluster Associated with Disease Aggressiveness and Kidney Damage in Multiple Myeloma Patients. Int. J. Mol. Sci. 2024, 25, 13499. [Google Scholar] [CrossRef]
- Zhang, H.; Zhan, Q.; Huang, B.; Wang, Y.; Wang, X. AAV-mediated gene therapy: Advancing cardiovascular disease treatment. Front. Cardiovasc. Med. 2022, 9, 952755. [Google Scholar] [CrossRef]
- Switala, L.; Di, L.; Gao, H.; Asase, C.; Klos, M.; Rengasamy, P.; Fedyukina, D.; Maiseyeu, A. Engineered nanoparticles promote cardiac tropism of AAV vectors. J. Nanobiotechnol. 2024, 22, 223. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, J.H.; Jung, E.J.; Son, Y.S.; Park, H.J.; Kim, J.M.; Park, T.; Jeong, S.H.; Lee, J.; Kim, T.H.; et al. Therapeutic Targeting of the Galectin-1/miR-22-3p Axis Regulates Cell Cycle and EMT Depending on the Molecular Subtype of Breast Cancer. Cells 2025, 14, 310. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
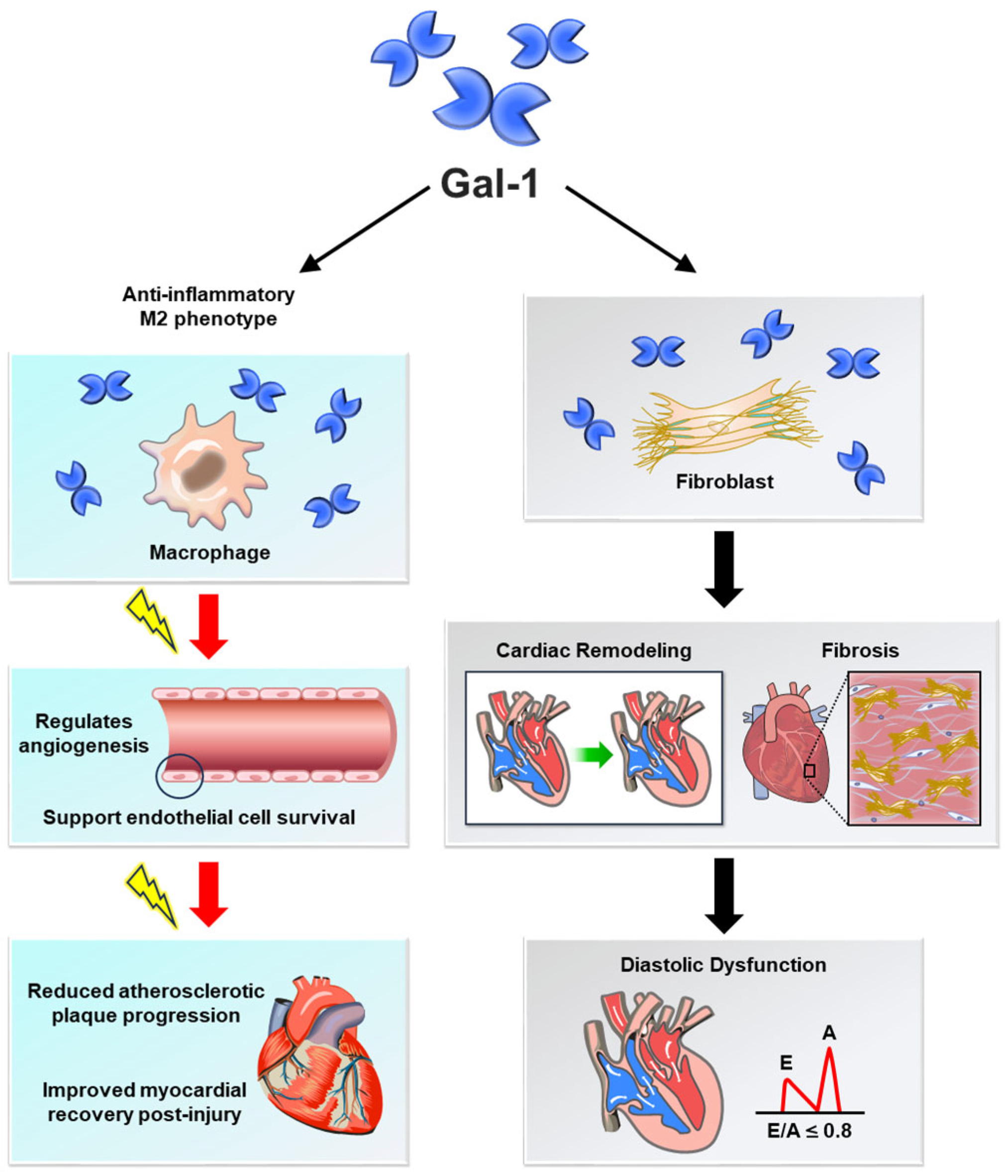
| Protective Effects of Gal-1 | |
|---|---|
| Function | Protective Effects |
| Vascular Homeostasis | Promotes endothelial cell survival and angiogenesis, mitigating ischemic damage and supporting vascular integrity [1,27]. |
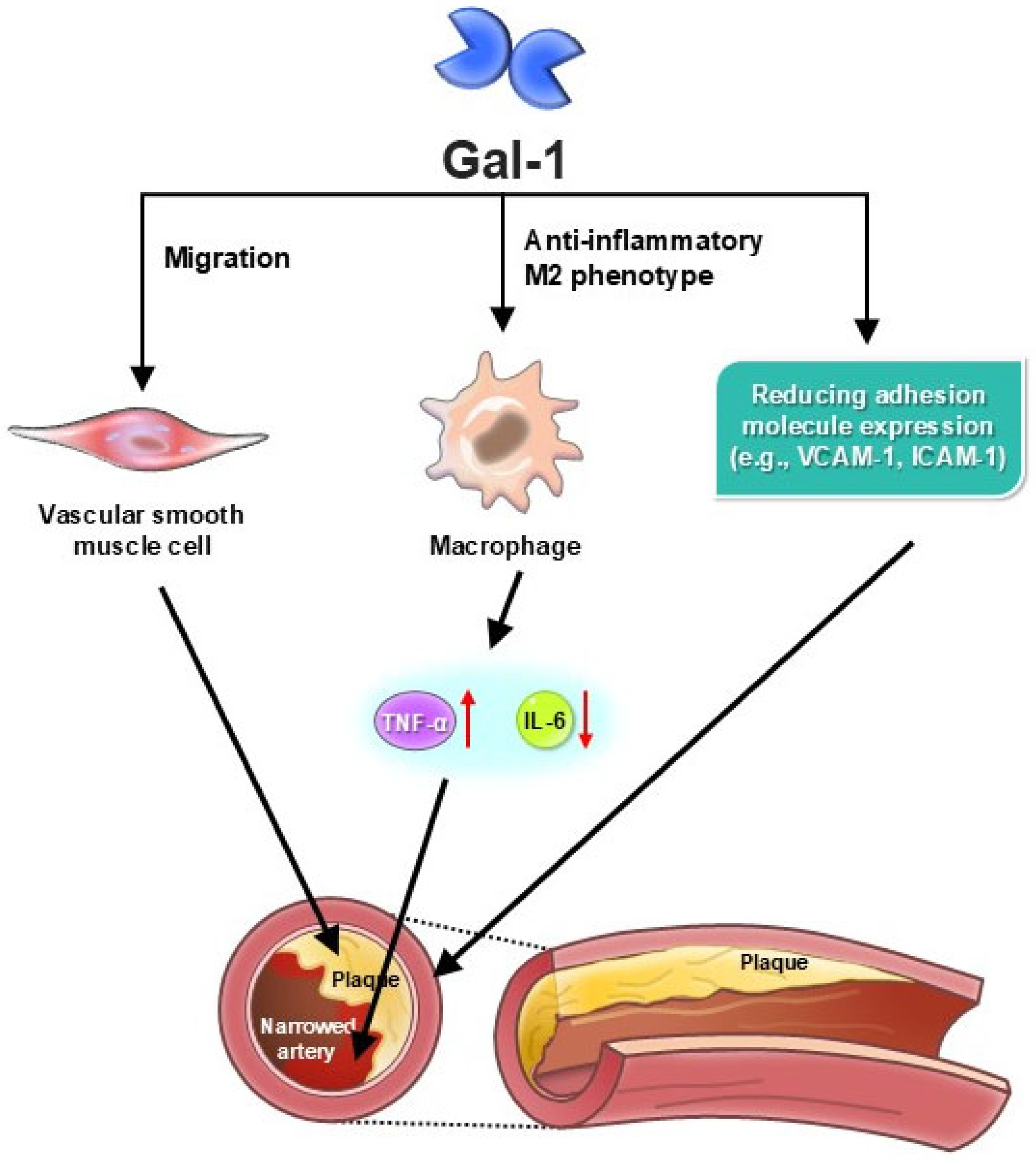
| Inflammation Regulation | Suppresses pro-inflammatory cytokines (e.g., TNF-α, IL-6) and encourages M2 macrophage polarization, reducing chronic vascular inflammation [5,28]. |
| Atherosclerosis Progression | Reduces expression of adhesion molecules (VCAM-1, ICAM-1), limiting monocyte infiltration and plaque formation [11,29]. |
| Myocardial Protection | Alleviates ischemia–reperfusion injury by reducing cardiomyocyte apoptosis and oxidative stress, preserving cardiac function [30,31]. |
| Heart Failure Modulation | Facilitates tissue repair and limits fibrotic remodeling, preserving myocardial function [19,32]. |
| Calcium Channel Regulation | Modulates Ca(V)1.2 activity, reducing arterial constriction and preventing hypertension [21,22]. |
| Pathological Effects of Gal-1 | |
| Function | Protective Effects |
| Vascular Homeostasis | Disrupts endothelial function by increasing vascular permeability and driving inflammation, contributing to atherosclerosis and aneurysm progression [23,24]. |
| Inflammation Regulation | Enhances leukocyte recruitment and activation, perpetuating vascular inflammation and worsening cardiovascular disease [33,34]. |
| Atherosclerosis Progression | Promotes foam cell formation, lipid accumulation, and vascular smooth muscle cell migration, accelerating plaque instability [24,35]. |
| Myocardial Protection | Contributes to maladaptive post-myocardial infarction remodeling and fibrosis, impairing cardiac performance [36,37]. |
| Heart Failure Modulation | Induces fibrosis and excessive extracellular matrix deposition, leading to ventricular stiffening and diastolic dysfunction [38,39]. |
| Calcium Channel Regulation | Disrupts calcium signaling in cardiomyocytes, contributing to contractile dysfunction and arrhythmias [9,32]. |
| Disease Entity | Sample Size | Assay Type | Median/Mean Gal-1 Level (ng/mL) | Clinical Endpoints | Hazard Ratio (HR) (95% CI) |
|---|---|---|---|---|---|
| Critically Ill Patients | 350 | ELISA | Median: 39–70 (tertiles) | 90-day mortality | HR: 3.21 (1.90–5.42) [25] |
| Acute Kidney Injury (AKI) | 350 | ELISA | Median: 39–70 (tertiles) | AKI within 48 h | HR: 2.88 (1.20–6.88) [25] |
| Coronary Artery Disease (CAD) | 200 | ELISA | Median: 56.3 (CAD) vs. 32.1 (controls) | Fibrosis, vascular stiffening | p < 0.01 [24] |
| Heart Failure (HF) | 180 | ELISA | Mean: 68.5 (HF) vs. 40.2 (non-HF) | Left ventricular diastolic dysfunction, hospitalization | HR: 2.45 (1.62–3.71) [37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, P.-Y.; Cheng, C.-Y.; Chen, C.-C.; Chen, H.-Y.; Liu, J.-C.; Hao, W.-R.; Cheng, T.-H.; Chen, J.-J. Galectin-1 in Cardiovascular Pathogenesis: Unraveling Dual Roles and Mechanistic Insights in Emerging Research. Medicina 2025, 61, 1020. https://doi.org/10.3390/medicina61061020
Chen P-Y, Cheng C-Y, Chen C-C, Chen H-Y, Liu J-C, Hao W-R, Cheng T-H, Chen J-J. Galectin-1 in Cardiovascular Pathogenesis: Unraveling Dual Roles and Mechanistic Insights in Emerging Research. Medicina. 2025; 61(6):1020. https://doi.org/10.3390/medicina61061020
Chicago/Turabian StyleChen, Po-Yuan, Chun-Yao Cheng, Chun-Chao Chen, Huan-Yuan Chen, Ju-Chi Liu, Wen-Rui Hao, Tzu-Hurng Cheng, and Jin-Jer Chen. 2025. "Galectin-1 in Cardiovascular Pathogenesis: Unraveling Dual Roles and Mechanistic Insights in Emerging Research" Medicina 61, no. 6: 1020. https://doi.org/10.3390/medicina61061020
APA StyleChen, P.-Y., Cheng, C.-Y., Chen, C.-C., Chen, H.-Y., Liu, J.-C., Hao, W.-R., Cheng, T.-H., & Chen, J.-J. (2025). Galectin-1 in Cardiovascular Pathogenesis: Unraveling Dual Roles and Mechanistic Insights in Emerging Research. Medicina, 61(6), 1020. https://doi.org/10.3390/medicina61061020