A Review of the Ocular Phenotype and Correlation with Genotype in Poretti–Boltshauser Syndrome


Abstract
1. Introduction
2. Materials and Methods
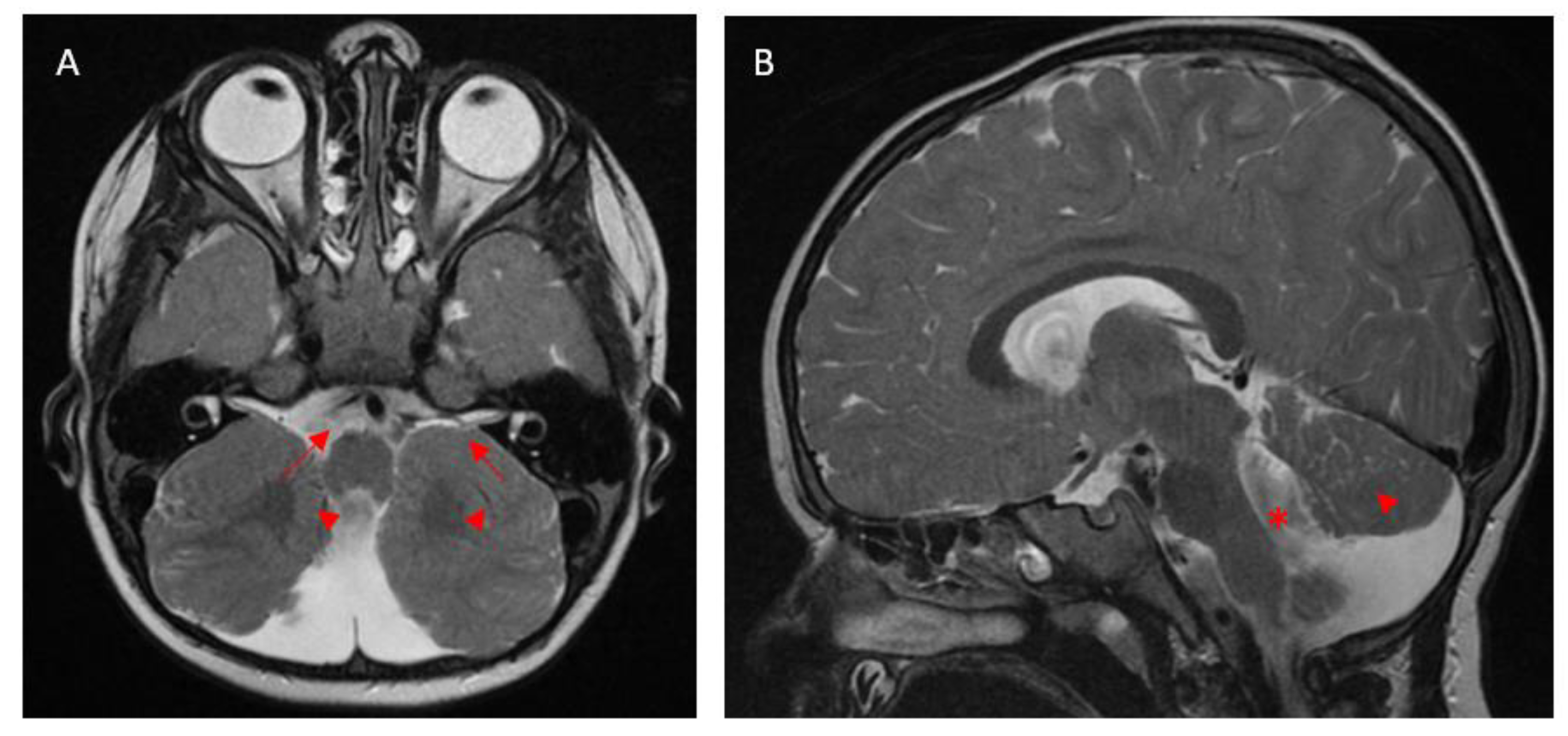
3. Results
3.1. Demographics and Clinical Presentation
3.2. Myopia and Its Associated Features
3.3. Strabismus and Ocular Motility Dysfunction
3.4. Retinal Dystrophy
3.5. Genotype–Phenotype Correlations in LAMA1
4. Discussion
4.1. Ocular Manifestations and Their Variability
4.2. Retinal Dystrophy and Electrodiagnostic Findings
4.3. Genotypic Influence and the Complexity of PBS
4.4. Clinical Mimics of PBS: Joubert and Knobloch Syndrome
4.5. Limitations
4.6. Clinical and Research Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PBS | Poretti–Boltshauser syndrome |
| EDT | Electrodiagnostic test |
| ERG | Electroretinogram |
| FEVR | Familial exudative vitreoretinopathy |
| KNO | Knobloch syndrome |
References
- Schiff, E.R.; Aychoua, N.; Nutan, S.; Davagnanam, I.; Moore, A.T.; Robson, A.G.; Patel, C.K.; Webster, A.R.; Arno, G. Variability of retinopathy consequent upon novel mutations in LAMA1. Ophthalmic Genet. 2022, 43, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, A.; Poretti, A.; Romani, M.; Ginevrino, M.; Mazza, T.; Aiello, C.; Zanni, G.; Baumgartner, B.; Borgatti, R.; Brockmann, K.; et al. Clinical, neuroradiological and molecular characterization of cerebellar dysplasia with cysts (Poretti–Boltshauser syndrome). Eur. J. Hum. Genet. 2016, 24, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Alahmadi, A.S.; Badawi, A.H.; Magliyah, M.S.; Albakri, A.; Schatz, P. Poretti-Boltshauser syndrome: A rare differential diagnosis to consider in pediatric high myopia with retinal degeneration. Ophthalmic Genet. 2021, 42, 96–98. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]
- Aldinger, K.A.; Mosca, S.J.; Tétreault, M.; Dempsey, J.C.; Ishak, G.E.; Hartley, T.; Phelps, I.G.; Lamont, R.E.; O’day, D.R.; Basel, D.; et al. Mutations in LAMA1 cause cerebellar dysplasia and cysts with and without retinal dystrophy. Am. J. Hum. Genet. 2014, 95, 227–234. [Google Scholar] [CrossRef]
- Marlow, E.; Chan, R.V.P.; Oltra, E.; Rusu, I.; Gupta, M.P. Retinal Avascularity and Neovascularization Associated with LAMA1 (laminin1) Mutation in Poretti-Boltshauser Syndrome. JAMA Ophthalmol. 2018, 136, 96–97. [Google Scholar] [CrossRef]
- Vilboux, T.; Malicdan, M.C.V.; Chang, Y.M.; Guo, J.; Zerfas, P.M.; Stephen, J.; Cullinane, A.R.; Bryant, J.; Fischer, R.; Brooks, B.P.; et al. Cystic cerebellar dysplasia and biallelicLAMA1mutations: A lamininopathy associated with tics, obsessive compulsive traits and myopia due to cell adhesion and migration defects. J. Med. Genet. 2016, 53, 318–329. [Google Scholar] [CrossRef]
- Kamate, M.; Goudar, N.; Hattiholi, V. Antenatal presentation and supratentorial brain abnormalities in a child with Poretti-Boltshauser syndrome. Brain Dev. 2022, 44, 139–141. [Google Scholar] [CrossRef]
- Elmas, M.; Gogus, B.; Solak, M. Understanding What You Have Found: A Family with a Mutation in the LAMA1 Gene with Literature Review. Clin. Med. Insights Case Rep. 2020, 13, 1179547620948666. [Google Scholar] [CrossRef]
- Raftopoulos, K.M.M.; Chiang, F.S.N.; Gama, L.d.M.; Farage, L.; Medina, C.T.N. Phenotypic variability in two siblings with Poretti-Boltshauser syndrome. Glob. Med. Genet. 2024, 12, 100010. [Google Scholar] [CrossRef]
- Geerts, C.; Sznajer, Y.; D’Haenens, E.; Dumitriu, D.; Nassogne, M.-C. Phenotypic spectrum of patients with Poretti-Boltshauser syndrome: Patient report of antenatal ventriculomegaly and esophageal atresia. Eur. J. Med. Genet. 2023, 66, 104692. [Google Scholar] [CrossRef]
- Ikeda, K.; Tamagake, A.; Kubota, T.; Izumi, R.; Yamaguchi, T.; Yanagi, K.; Misu, T.; Aoki, Y.; Kaname, T.; Aoki, M. Case Report: An Adult Case of Poretti-Boltshauser Syndrome Diagnosed by Medical Checkup. Cerebellum 2024, 23, 2205–2207. [Google Scholar] [CrossRef] [PubMed]
- Faizi, N.; Casteels, I.; Termote, B.; Coucke, P.; De Baere, E.; De Bruyne, M.; Balikova, I. High myopia and vitreal veils in a patient with Poretti– Boltshauser syndrome due to a novel homozygous LAMA1 mutation. Ophthalmic Genet. 2022, 43, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; da Cruz, N.F.S.; Lopez-Font, F.; Staropoli, P.; Berrocal, A. Exudative Vitreoretinopathy with a Coats-Like Response in Poretti-Boltshauser Syndrome. J. Vitr. Dis. 2024, 8, 24741264241296465. [Google Scholar] [CrossRef] [PubMed]
- Wente, S.; Schröder, S.; Buckard, J.; Büttel, H.-M.; von Deimling, F.; Diener, W.; Häussler, M.; Hübschle, S.; Kinder, S.; Kurlemann, G.; et al. Nosological delineation of congenital ocular motor apraxia type Cogan: An observational study. Orphanet J. Rare Dis. 2016, 11, 104. [Google Scholar] [CrossRef]
- Masson, R.; Piretti, E.; Pellegrin, S.; Gusson, E.; Poretti, A.; Valente, E.M.; Cantalupo, G. Early-onset head titubation in a child with Poretti-Boltshauser syndrome. Neurology 2017, 88, 1478–1479. [Google Scholar] [CrossRef]
- Wang, P.; Jia, X.; Xiao, X.; Li, S.; Long, Y.; Liu, M.; Li, Y.; Li, J.; Xu, Y.; Zhang, Q. An Early Diagnostic Clue for COL18A1- and LAMA1-Associated Diseases: High Myopia with Alopecia Areata in the Cranial Midline. Front. Cell Dev. Biol. 2021, 9, 644947. [Google Scholar] [CrossRef]
- Banerjee, A.; Vyas, S.; Sankhyan, N. Cerebellar Cysts and Dysplasias: More Diagnoses to Consider. Pediatr. Neurol. 2019, 98, 91–92. [Google Scholar] [CrossRef]
- Edwards, M.M.; Mammadova-Bach, E.; Alpy, F.; Klein, A.; Hicks, W.L.; Roux, M.; Simon-Assmann, P.; Smith, R.S.; Orend, G.; Wu, J.; et al. Mutations in Lama1 disrupt retinal vascular development and inner limiting membrane formation. J. Biol. Chem. 2010, 285, 7697–7711. [Google Scholar] [CrossRef]
- Powell, L.; Olinger, E.; Wedderburn, S.; Ramakumaran, V.S.; Kini, U.; Clayton-Smith, J.; Ramsden, S.C.; Rice, S.J.; Barroso-Gil, M.; Wilson, I.; et al. Identification of LAMA1 mutations ends diagnostic odyssey and has prognostic implications for patients with presumed Joubert syndrome. Brain Commun. 2021, 3, fcab163. [Google Scholar] [CrossRef]
- Kirk, E.P.; Barlow-Stewart, K.; Selvanathan, A.; Josephi-Taylor, S.; Worgan, L.; Rajagopalan, S.; Cowley, M.J.; Gayevskiy, V.; Bittles, A.; Burnett, L.; et al. Beyond the panel: Preconception screening in consanguineous couples using the TruSight One “clinical exome”. Anesth. Analg. 2019, 21, 608–612. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ocular Manifestations | Number of Patients |
|---|---|
| Myopia | 39 |
| Strabismus | 27 |
| Ocular motor apraxia | 26 |
| Retinal/chorioretinal atrophy | 18 |
| Nystagmus | 17 |
| Retinal dystrophy | 16 |
| Optic disc hypoplasia or atrophy | 6 |
| Cataract | 6 |
| Amblyopia | 2 |
| Iris hypoplasia | 1 |
| Number of Patients with Each Genotype | Variant 1 | Variant 2 | Myopia | Retinal Atrophy | Retinal Dystrophy | Ocular Motor Apraxia | Strabismus | Nystagmus |
|---|---|---|---|---|---|---|---|---|
| n = 3 | c.1492delC | c.1492delC | 100% | 100% | 100% | 66.7% | 33.3% | 33.3% |
| n = 6 | c.2935delA | c.2935delA | 50% | 0 | 33.3% | 83.3% | 33.3% | 0 |
| n = 4 | c.6701delC | c.768+1G>A, c.8557-1G>C | 100% | 100% | 25% | 50% | 100% | 50% |
| n = 2 | c.2816_2817delAT | c.555T>G | 100% | 50% | 100% | 50% | 50% | 50% |
| n = 2 | c.664C>T | c.2331C>G | 100% | 0 | 0 | 0 | 100% | 100% |
| n = 2 | c.3881G>A | deletion of exons 31–32 | 100% | 0 | 0 | 50% | 100% | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Published by MDPI on behalf of the Lithuanian University of Health Sciences. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moon, W.Y.; Shah, S.; ElMeshad, N.; De Silva, S.R. A Review of the Ocular Phenotype and Correlation with Genotype in Poretti–Boltshauser Syndrome. Medicina 2025, 61, 881. https://doi.org/10.3390/medicina61050881
Moon WY, Shah S, ElMeshad N, De Silva SR. A Review of the Ocular Phenotype and Correlation with Genotype in Poretti–Boltshauser Syndrome. Medicina. 2025; 61(5):881. https://doi.org/10.3390/medicina61050881
Chicago/Turabian StyleMoon, Won Young, Sanil Shah, Nervine ElMeshad, and Samantha R. De Silva. 2025. "A Review of the Ocular Phenotype and Correlation with Genotype in Poretti–Boltshauser Syndrome" Medicina 61, no. 5: 881. https://doi.org/10.3390/medicina61050881
APA StyleMoon, W. Y., Shah, S., ElMeshad, N., & De Silva, S. R. (2025). A Review of the Ocular Phenotype and Correlation with Genotype in Poretti–Boltshauser Syndrome. Medicina, 61(5), 881. https://doi.org/10.3390/medicina61050881