


Association of Microsatellite Instability and Gene Expression Profile in Colorectal Carcinoma and Potential Implications for Therapy
 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Genome-Wide Gene Expression Assay
2.2. MSI Detection
2.3. Statistical Analysis
3. Results
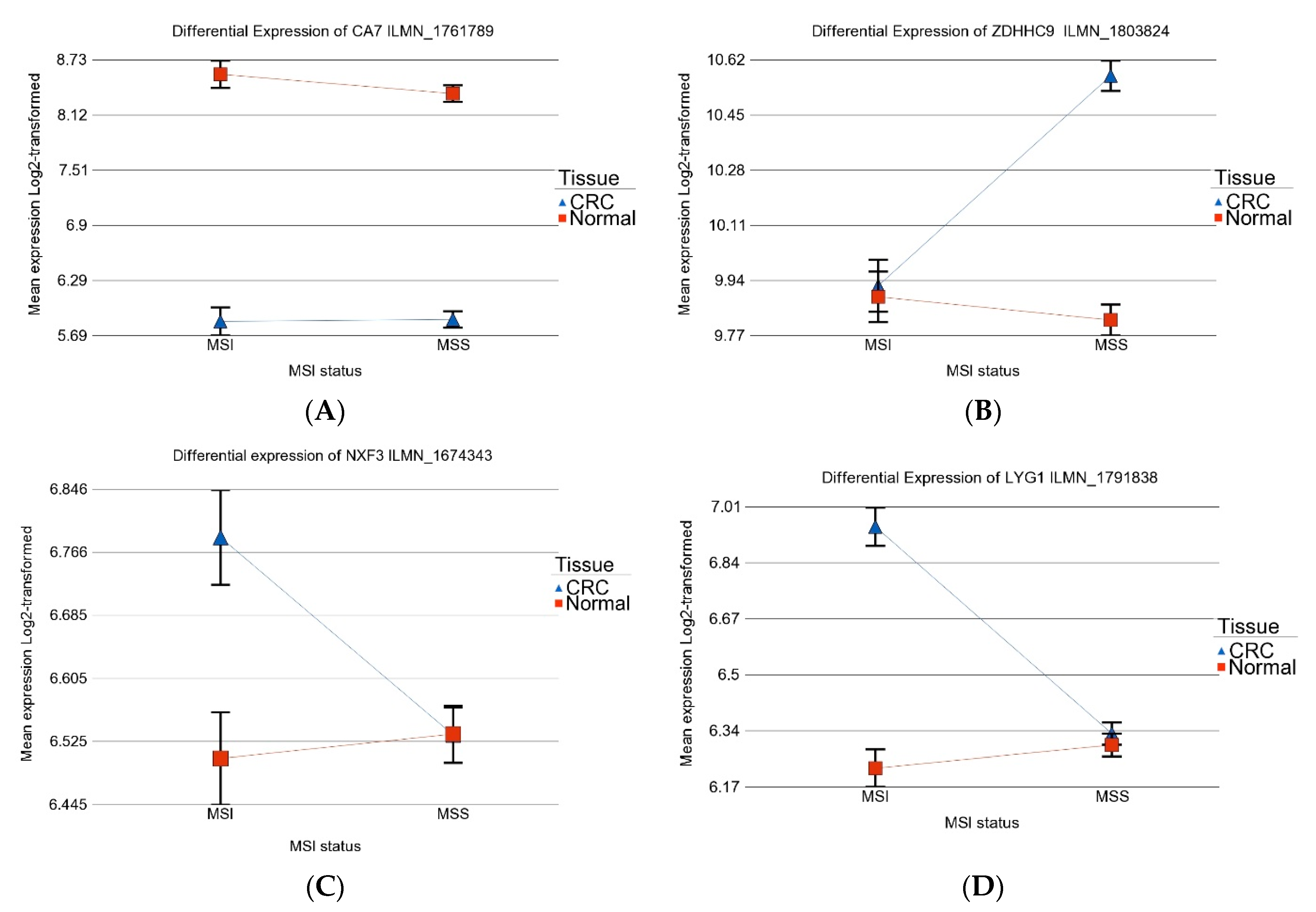
3.1. Gene-Level Analysis
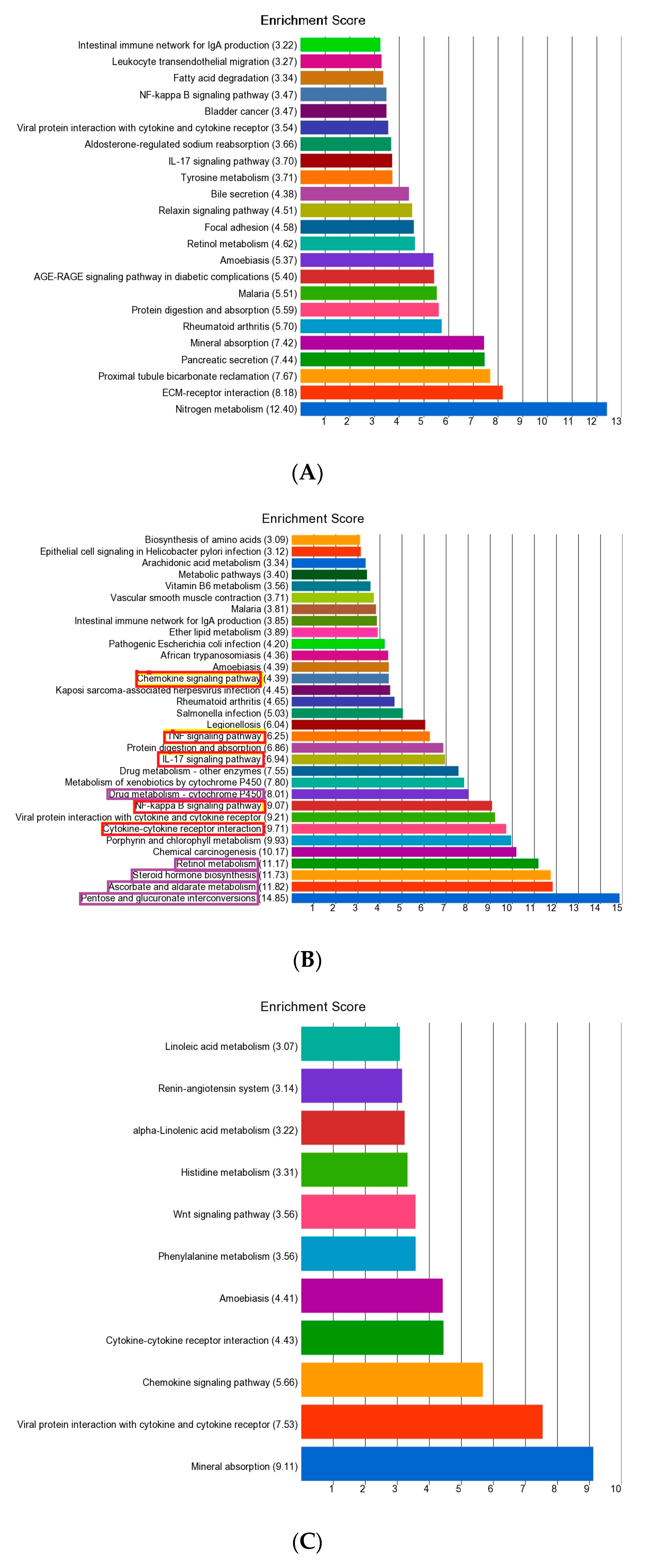
3.2. Enrichment Analysis of Differentially Expressed Genes
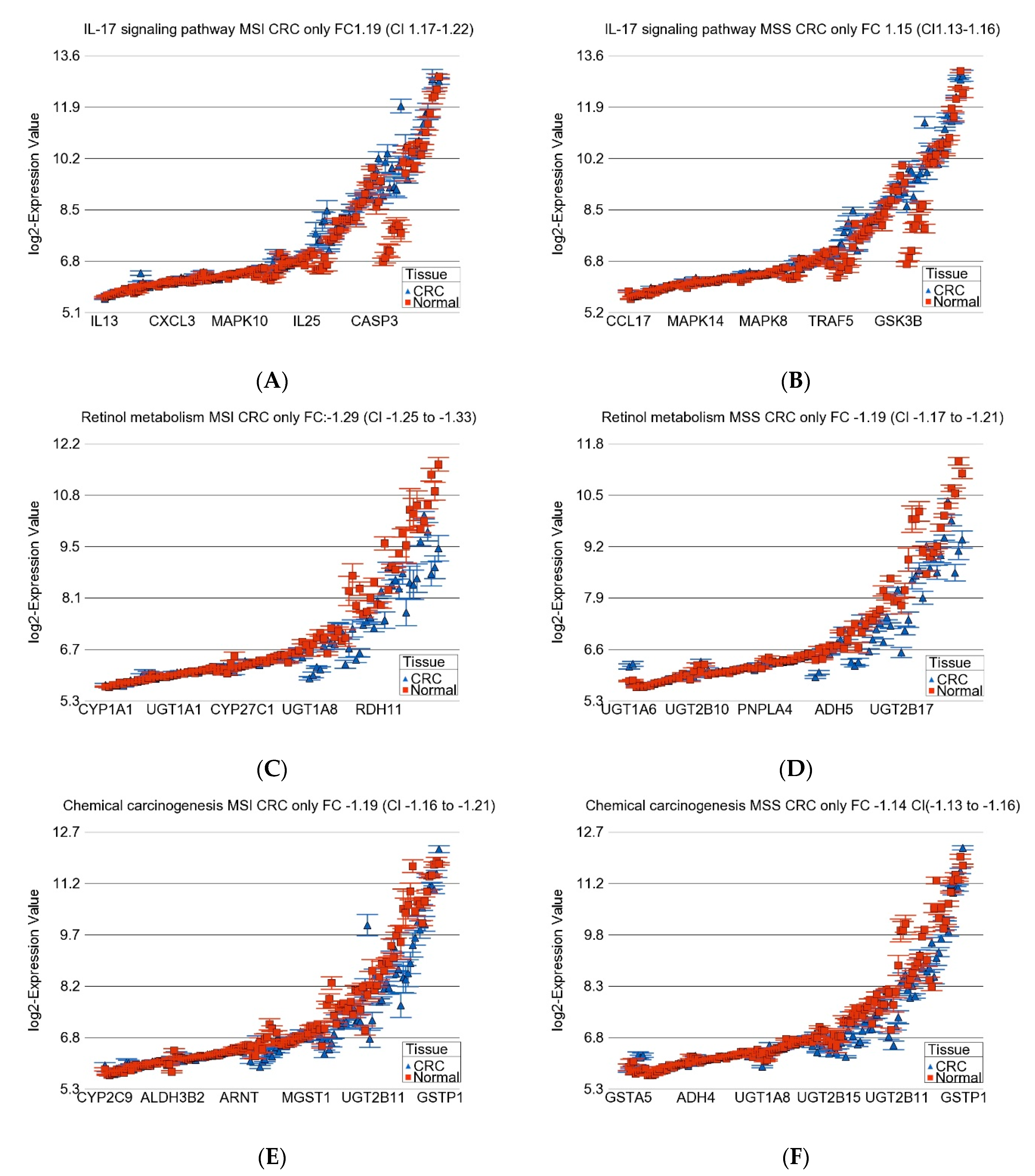
3.3. Gene-Set or Pathway Level Analysis
3.4. Differential Gene Expression: MSI vs. MSS Tumors
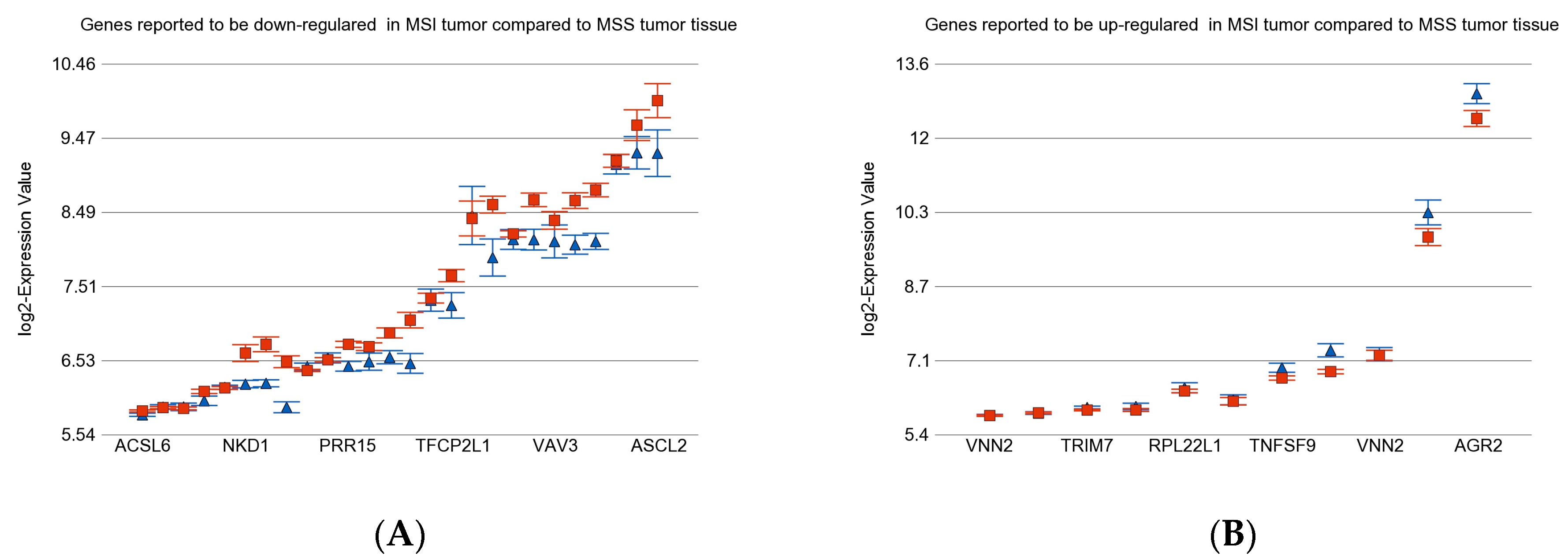
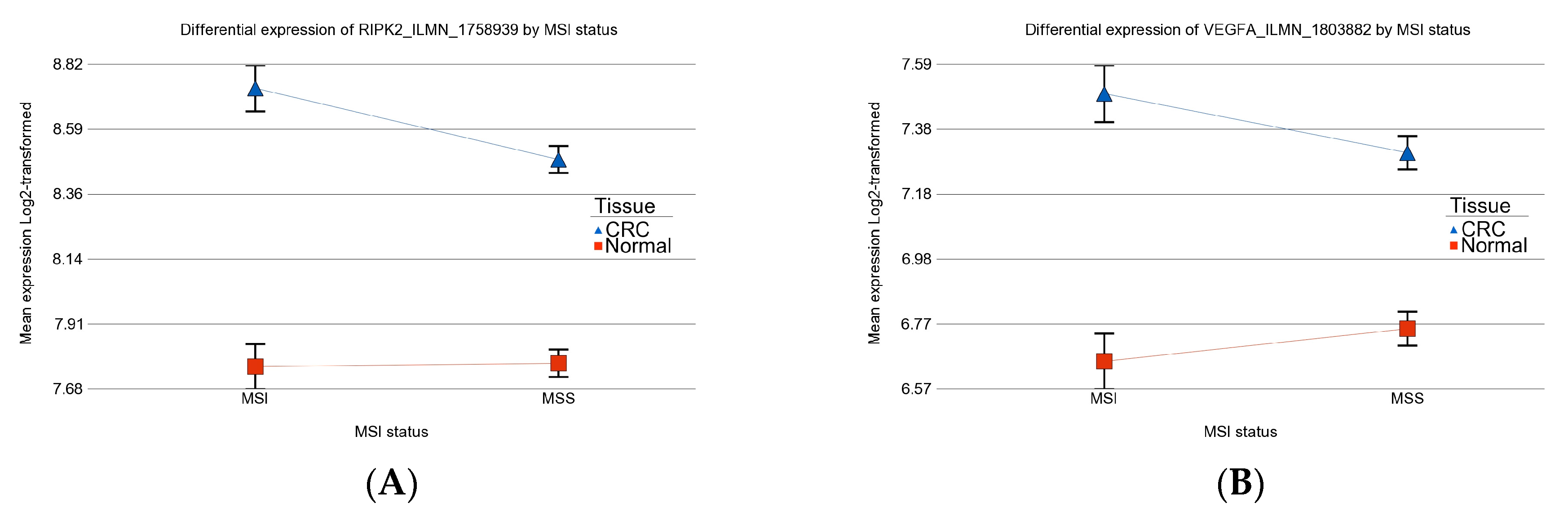
3.5. Previously Reported Genes in Our Data Set
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grady, W.M. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004, 23, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Nguyen, L.H.; Goel, A.; Chung, D.C. Pathways of Colorectal Carcinogenesis. Gastroenterology 2020, 158, 291–302. [Google Scholar] [CrossRef]
- Lin, A.; Zhang, J.; Luo, P. Crosstalk Between the MSI Status and Tumor Microenvironment in Colorectal Cancer. Front. Immunol. 2020, 11, 2039. [Google Scholar] [CrossRef]
- Kibriya, M.G.; Jasmine, F.; Pekow, J.; Munoz, A.; Weber, C.; Raza, M.; Kamal, M.; Ahsan, H.; Bissonnette, M. Pathways Related to Colon Inflammation Are Associated with Colorectal Carcinoma: A Transcriptome- and Methylome-Wide Study. Cancers 2023, 15, 2921. [Google Scholar] [CrossRef]
- Jasmine, F.; Haq, Z.; Kamal, M.; Raza, M.; da Silva, G.; Gorospe, K.; Paul, R.; Strzempek, P.; Ahsan, H.; Kibriya, M.G. Interaction between Microsatellite Instability (MSI) and Tumor DNA Methylation in the Pathogenesis of Colorectal Carcinoma. Cancers 2021, 13, 4956. [Google Scholar] [CrossRef]
- Jasmine, F.; Rahaman, R.; Dodsworth, C.; Roy, S.; Paul, R.; Raza, M.; Paul-Brutus, R.; Kamal, M.; Ahsan, H.; Kibriya, M.G. A genome-wide study of cytogenetic changes in colorectal cancer using SNP microarrays: Opportunities for future personalized treatment. PLoS ONE 2012, 7, e31968. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, M.G.; Raza, M.; Jasmine, F.; Roy, S.; Paul-Brutus, R.; Rahaman, R.; Dodsworth, C.; Rakibuz-Zaman, M.; Kamal, M.; Ahsan, H. A genome-wide DNA methylation study in colorectal carcinoma. BMC Med. Genom. 2011, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, M.G.; Raza, M.; Kamal, M.; Haq, Z.; Paul, R.; Mareczko, A.; Pierce, B.L.; Ahsan, H.; Jasmine, F. Relative Telomere Length Change in Colorectal Carcinoma and Its Association with Tumor Characteristics, Gene Expression and Microsatellite Instability. Cancers 2022, 14, 2250. [Google Scholar] [CrossRef]
- Janavicius, R.; Matiukaite, D.; Jakubauskas, A.; Griskevicius, L. Microsatellite instability detection by high-resolution melting analysis. Clin. Chem. 2010, 56, 1750–1757. [Google Scholar] [CrossRef]
- Morifuji, M.; Hiyama, E.; Murakami, Y.; Imamura, Y.; Sueda, T.; Yokoyama, T. Fluorescent-based BAT-26 analysis for distinct screening of microsatellite instability in colorectal cancers. Int. J. Oncol. 2003, 22, 807–813. [Google Scholar] [CrossRef]
- Findeisen, P.; Kloor, M.; Merx, S.; Sutter, C.; Woerner, S.M.; Dostmann, N.; Benner, A.; Dondog, B.; Pawlita, M.; Dippold, W.; et al. T25 repeat in the 3′ untranslated region of the CASP2 gene: A sensitive and specific marker for microsatellite instability in colorectal cancer. Cancer Res. 2005, 65, 8072–8078. [Google Scholar] [CrossRef]
- Deschoolmeester, V.; Baay, M.; Wuyts, W.; Van Marck, E.; Van Damme, N.; Vermeulen, P.; Lukaszuk, K.; Lardon, F.; Vermorken, J.B. Detection of microsatellite instability in colorectal cancer using an alternative multiplex assay of quasi-monomorphic mononucleotide markers. J. Mol. Diagn. 2008, 10, 154–159. [Google Scholar] [CrossRef]
- Downey, T. Analysis of a multifactor microarray study using Partek genomics solution. Methods Enzymol. 2006, 411, 256–270. [Google Scholar] [CrossRef]
- Pačínková, A.; Popovici, V. Cross-platform Data Analysis Reveals a Generic Gene Expression Signature for Microsatellite Instability in Colorectal Cancer. Biomed. Res. Int. 2019, 2019, 6763596. [Google Scholar] [CrossRef]
- Jaafar, R.F.; Ibrahim, Z.; Ataya, K.; Hassanieh, J.; Ard, N.; Faraj, W. Receptor-Interacting Serine/Threonine-Protein Kinase-2 as a Potential Prognostic Factor in Colorectal Cancer. Medicina 2021, 57, 709. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, C.P.; Leung, S.Y.; Chen, X.; Yuen, S.T.; Kim, Y.H.; Bair, E.; Pollack, J.R. A gene expression signature of genetic instability in colon cancer. Cancer Res. 2005, 65, 9200–9205. [Google Scholar] [CrossRef] [PubMed]
- Kruhøffer, M.; Jensen, J.L.; Laiho, P.; Dyrskjøt, L.; Salovaara, R.; Arango, D.; Birkenkamp-Demtroder, K.; Sørensen, F.B.; Christensen, L.L.; Buhl, L.; et al. Gene expression signatures for colorectal cancer microsatellite status and HNPCC. Br. J. Cancer 2005, 92, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Y.; Zhu, Z.; Yang, J.; Tan, Y.; Wang, Y.; Xu, X. Development of novel models for predicting mismatch repair protein deficiency and relevant disease-free survival in colorectal cancer patients. Int. J. Color. Dis. 2022, 37, 1449–1464. [Google Scholar] [CrossRef]
- Pira, G.; Uva, P.; Scanu, A.M.; Rocca, P.C.; Murgia, L.; Uleri, E.; Piu, C.; Porcu, A.; Carru, C.; Manca, A.; et al. Landscape of transcriptome variations uncovering known and novel driver events in colorectal carcinoma. Sci. Rep. 2020, 10, 432. [Google Scholar] [CrossRef]
- Scanu, A.M.; De Miglio, M.R. Therapeutic Landscapes in Colorectal Carcinoma. Medicina 2023, 59, 821. [Google Scholar] [CrossRef]
- Liu, C.; Liu, R.; Wang, B.; Lian, J.; Yao, Y.; Sun, H.; Zhang, C.; Fang, L.; Guan, X.; Shi, J.; et al. Blocking IL-17A enhances tumor response to anti-PD-1 immunotherapy in microsatellite stable colorectal cancer. J. Immunother. Cancer 2021, 9, e001895. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.X.; Istl, A.C.; Quan, D.; Skaro, A.; Tang, E.; Zheng, X. PD-1 and PD-L1 inhibitors in cold colorectal cancer: Challenges and strategies. Cancer Immunol. Immunother. 2023, 72, 3875–3893. [Google Scholar] [CrossRef]
- Wang, X.; Yang, L.; Huang, F.; Zhang, Q.; Liu, S.; Ma, L.; You, Z. Inflammatory cytokines IL-17 and TNF-α up-regulate PD-L1 expression in human prostate and colon cancer cells. Immunol. Lett. 2017, 184, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Bras-Gonçalves, R.A.; Rosty, C.; Laurent-Puig, P.; Soulié, P.; Dutrillaux, B.; Poupon, M.F. Sensitivity to CPT-11 of xenografted human colorectal cancers as a function of microsatellite instability and p53 status. Br. J. Cancer 2000, 82, 913–923. [Google Scholar] [CrossRef]
- Fallik, D.; Borrini, F.; Boige, V.; Viguier, J.; Jacob, S.; Miquel, C.; Sabourin, J.C.; Ducreux, M.; Praz, F. Microsatellite instability is a predictive factor of the tumor response to irinotecan in patients with advanced colorectal cancer. Cancer Res. 2003, 63, 5738–5744. [Google Scholar]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef]
- Jacob, S.; Aguado, M.; Fallik, D.; Praz, F. The role of the DNA mismatch repair system in the cytotoxicity of the topoisomerase inhibitors camptothecin and etoposide to human colorectal cancer cells. Cancer Res. 2001, 61, 6555–6562. [Google Scholar]
- Ak, S.; Tunca, B.; Yilmazlar, T.; Tezcan, G.; Cecener, G.; Egeli, U.; Ozturk, E.; Yerci, O.; Ertürk, E.; Zorluoglu, A. Microsatellite instability status affects gene expression profiles in early onset colorectal cancer patients. J. Surg. Res. 2013, 185, 626–637. [Google Scholar] [CrossRef]
- Jou, E.; Rodriguez-Rodriguez, N.; McKenzie, A.N.J. Emerging roles for IL-25 and IL-33 in colorectal cancer tumorigenesis. Front. Immunol. 2022, 13, 981479. [Google Scholar] [CrossRef]
- Cui, G.; Yuan, A.; Pang, Z.; Zheng, W.; Li, Z.; Goll, R. Contribution of IL-33 to the Pathogenesis of Colorectal Cancer. Front. Oncol. 2018, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Gowhari Shabgah, A.; Amir, A.; Gardanova, Z.R.; Olegovna Zekiy, A.; Thangavelu, L.; Ebrahimi Nik, M.; Ahmadi, M.; Gholizadeh Navashenaq, J. Interleukin-25: New perspective and state-of-the-art in cancer prognosis and treatment approaches. Cancer Med. 2021, 10, 5191–5202. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.; Vyas, M.; Al-Attar, A.; Durrant, S.; Scholefield, J.; Durrant, L. High levels of cleaved caspase-3 in colorectal tumour stroma predict good survival. Br. J. Cancer 2013, 108, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Przybylla, R.; Shang, Y.; Dai, M.; Krohn, M.; Krämer, O.H.; Mullins, C.S.; Linnebacher, M. Microsatellite Status and IκBα Expression Levels Predict Sensitivity to Pharmaceutical Curcumin in Colorectal Cancer Cells. Cancers 2022, 14, 1032. [Google Scholar] [CrossRef]
- Hegde, M.; Girisa, S.; BharathwajChetty, B.; Vishwa, R.; Kunnumakkara, A.B. Curcumin Formulations for Better Bioavailability: What We Learned from Clinical Trials Thus Far? ACS Omega 2023, 8, 10713–10746. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, X.; Chu, Y.; Li, J.; Wang, W.; Hu, X.; Zhou, F.; Zhang, H.; Zhou, L.; Kuai, R.; et al. Analysis of transcript-wide profile regulated by microsatellite instability of colorectal cancer. Ann. Transl. Med. 2022, 10, 169. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Category | MSI | MSS | Chi Square p-Value |
|---|---|---|---|---|
| Sex | Male | 4 | 24 | 0.14 |
| Female | 14 | 29 | ||
| Location | Distal | 11 | 47 | 0.023 |
| Proximal | 7 | 6 | ||
| Stage | Stage-1 | 3 | 9 | 0.68 |
| Stage-2 | 7 | 15 | ||
| Stage-3 | 8 | 29 | ||
| Tumor Grade | Low | 14 | 43 | 1 |
| High | 4 | 10 | ||
| LymphNode | Present | 8 | 29 | 0.63 |
| Absent | 10 | 24 | ||
| TIL | 0 | 12 | 27 | 0.37 |
| 1 | 6 | 26 | ||
| Signet ring | Absent | 14 | 37 | 0.72 |
| Present | 4 | 16 | ||
| LVinvasion | Absent | 12 | 24 | 0.19 |
| Present | 6 | 29 | ||
| PNinvasion | Absent | 15 | 40 | 0.72 |
| Present | 3 | 13 | ||
| KRAS | Mutant | 5 | 15 | 0.79 |
| Wild | 13 | 38 | ||
| BRAF | Mutant | 1 | 0 | 0.56 |
| Wild | 17 | 53 |
| MSI | MSS | ||||||
|---|---|---|---|---|---|---|---|
| Gene Sets | Interaction | Fold Change | (95% CI) | p | Fold Change | (95% CI) | p |
| p | |||||||
| IL-17 signaling pathway | 0.000957916 | 1.19 | (1.16–1.21) | 1.02 × 10−59 | 1.15 | (1.13–1.16) | 8.11 × 10−104 |
| Retinol metabolism | 0.000177561 | −1.27 | (−1.31–1.23) | 7.35 × 10−56 | −1.20 | (−1.21–1.17) | 1.19368 × 10−86 |
| Chemical carcinogenesis | 6.15 × 10−5 | −1.18 | (−1.21–1.15) | 1.11 × 10−48 | −1.15 | (−1.16–1.13) | 1.66064 × 10−90 |
| Drug metabolism—cytochrome P450 | 0.000231622 | −1.22 | (−1.25–1.18) | 5.92 × 10−43 | −1.17 | (−1.19–1.15) | 1.74586 × 10−76 |
| Intestinal immune network for IgA production | 2.37 × 10−8 | −1.15 | (−1.18–1.10) | 9.73 × 10−16 | −1.29 | (−1.31–1.26) | 7.7175 × 10−132 |
| Steroid hormone biosynthesis | 0.000179774 | −1.22 | (−1.25–1.18) | 1.01 × 10−41 | −1.14 | (−1.16–1.12) | 6.24018 × 10−53 |
| Pentose and glucuronate interconversions | 3.37 × 10−6 | −1.35 | (−1.40–1.28) | 2.24 × 10−40 | −1.23 | (−1.26–1.19) | 1.17089 × 10−54 |
| Metabolism of xenobiotics by cytochrome P450 | 0.000177715 | −1.17 | (−1.20–1.14) | 3.48 × 10−32 | −1.14 | (−1.15–1.11) | 1.60731 × 10−56 |
| Vascular smooth muscle contraction | 2.30 × 10−6 | −1.07 | (−1.08–1.05) | 2.87 × 10−27 | −1.06 | (−1.06–1.05) | 3.17997 × 10−54 |
| Ether lipid metabolism | 0.591294 | −1.13 | (−1.15–1.10) | 6.57 × 10−23 | −1.11 | (−1.12–1.09) | 1.49968 × 10−47 |
| Ascorbate and aldarate metabolism | 8.54 × 10−7 | −1.29 | (−1.35–1.23) | 1.31 × 10−26 | −1.20 | (−1.22–1.16) | 1.9392 × 10−35 |
| Porphyrin and chlorophyll metabolism | 4.22 × 10−5 | −1.17 | (−1.20–1.13) | 3.23 × 10−22 | −1.12 | (−1.14–1.10) | 2.08756 × 10−33 |
| Salmonella infection | 3.63 × 10−7 | 1.11 | (1.08–1.12) | 8.10 × 10−25 | 1.06 | (1.04–1.07) | 1.21337 × 10−24 |
| Protein digestion and absorption | 0.0136228 | 1.08 | (1.05–1.09) | 1.34 × 10−15 | 1.08 | (1.06–1.09) | 9.45574 × 10−42 |
| Amoebiasis | 0.0187527 | 1.09 | (1.07–1.11) | 4.57 × 10−19 | 1.06 | (1.04–1.06) | 1.29366 × 10−21 |
| Drug metabolism—other enzymes | 2.02 × 10−8 | −1.11 | (−1.13–1.08) | 3.14 × 10−18 | −1.06 | (−1.07–1.04) | 3.68932 × 10−19 |
| Legionellosis | 8.09 × 10−8 | 1.11 | (1.07–1.13) | 9.76 × 10−17 | 1.06 | (1.04–1.07) | 1.91509 × 10−16 |
| Malaria | 0.000186005 | 1.18 | (1.13–1.21) | 1.12 × 10−18 | 1.08 | (1.05–1.10) | 8.28788 × 10−12 |
| Biosynthesis of amino acids | 3.22 × 10−12 | 1.06 | (1.03–1.07) | 2.82 × 10−10 | 1.05 | (1.04–1.06) | 1.95178 × 10−24 |
| TNF signaling pathway | 1.67 × 10−6 | 1.06 | (1.04–1.07) | 3.71 × 10−18 | 1.03 | (1.01–1.03) | 2.34661 × 10−9 |
| Rheumatoid arthritis | 4.08 × 10−17 | 1.15 | (1.12–1.17) | 2.39 × 10−26 | 1.02 | (1.00–1.03) | 0.0193784 |
| Epithelial cell signaling in Helicobacter pylori infection | 0.52238 | 1.05 | (1.03–1.07) | 1.18 × 10−8 | 1.04 | (1.03–1.05) | 1.09674 × 10−13 |
| Vitamin B6 metabolism | 0.00322795 | 1.14 | (1.05–1.21) | 0.000387 | 1.12 | (1.07–1.16) | 7.67061 × 10−8 |
| Arachidonic acid metabolism | 0.059206 | −1.02 | (−1.04–1.00) | 0.0502923 | −1.02 | (−1.03–1.01) | 0.000402162 |
| Viral protein interaction with cytokine and cytokine receptor | 2.43 × 10−9 | 1.02 | (−1.00–1.03) | 0.120965 | −1.06 | (−1.06–1.04) | 6.93335 × 10−18 |
| NF-kappa B signaling pathway | 9.57 × 10−7 | 1.02 | (1.00–1.03) | 0.0397081 | −1.03 | (−1.04–1.02) | 1.01891 × 10−11 |
| African trypanosomiasis | 0.0268193 | 1.04 | (1.00–1.07) | 0.0214121 | −1.01 | (−1.03–1.00) | 0.171386 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kibriya, M.G.; Jasmine, F.; Khamkevych, Y.; Raza, M.; Kamal, M.; Bissonnette, M.; Ahsan, H. Association of Microsatellite Instability and Gene Expression Profile in Colorectal Carcinoma and Potential Implications for Therapy. Medicina 2024, 60, 348. https://doi.org/10.3390/medicina60030348
Kibriya MG, Jasmine F, Khamkevych Y, Raza M, Kamal M, Bissonnette M, Ahsan H. Association of Microsatellite Instability and Gene Expression Profile in Colorectal Carcinoma and Potential Implications for Therapy. Medicina. 2024; 60(3):348. https://doi.org/10.3390/medicina60030348
Chicago/Turabian StyleKibriya, Muhammad G., Farzana Jasmine, Yuliia Khamkevych, Maruf Raza, Mohammed Kamal, Marc Bissonnette, and Habibul Ahsan. 2024. "Association of Microsatellite Instability and Gene Expression Profile in Colorectal Carcinoma and Potential Implications for Therapy" Medicina 60, no. 3: 348. https://doi.org/10.3390/medicina60030348
APA StyleKibriya, M. G., Jasmine, F., Khamkevych, Y., Raza, M., Kamal, M., Bissonnette, M., & Ahsan, H. (2024). Association of Microsatellite Instability and Gene Expression Profile in Colorectal Carcinoma and Potential Implications for Therapy. Medicina, 60(3), 348. https://doi.org/10.3390/medicina60030348