Rare c.302C>T TTR Variant Associated with Transthyretin Amyloidosis

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Evaluation
2.2. Genetic Evaluation
2.2.1. DNA Extraction
2.2.2. Next Generation Sequencing
2.2.3. Sanger Sequencing
2.2.4. Real-Time PCR Analysis
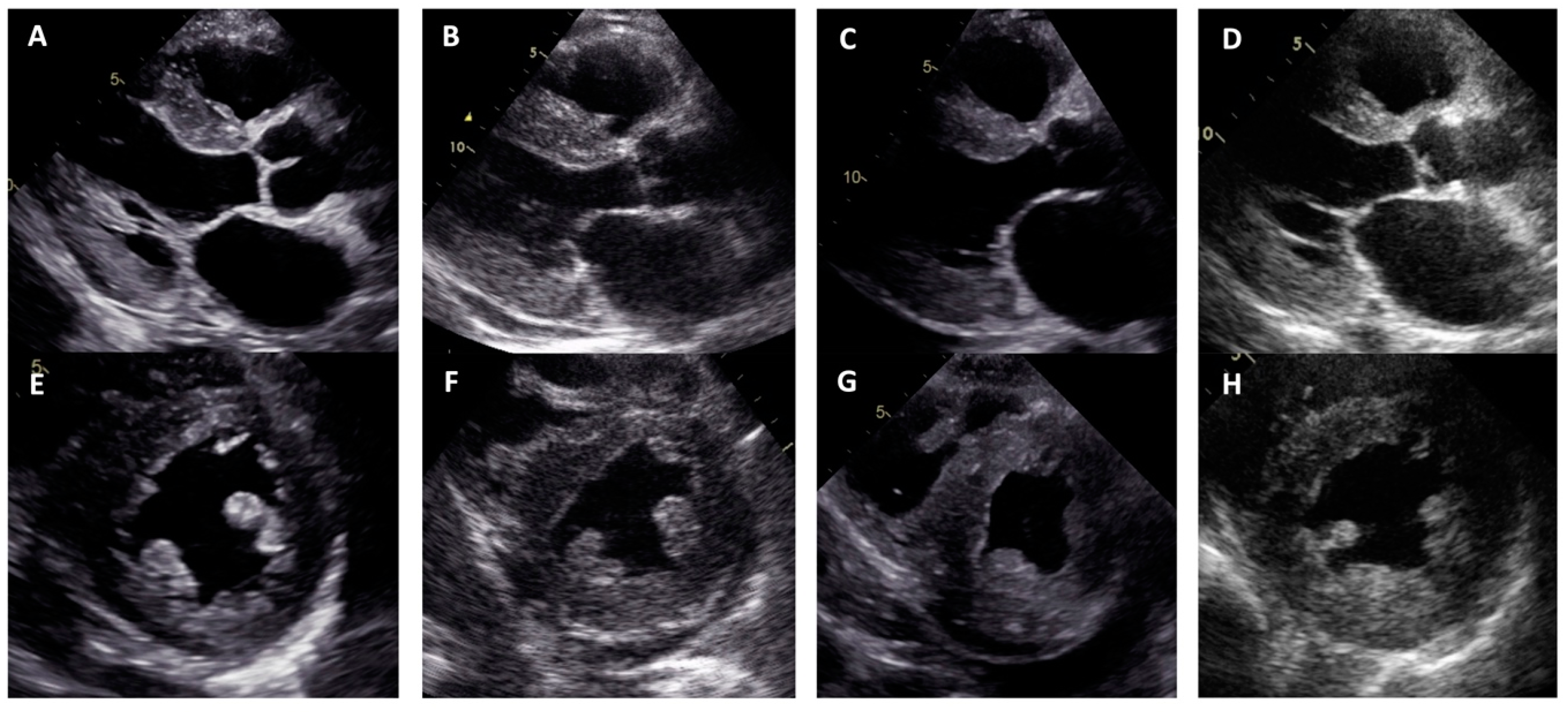
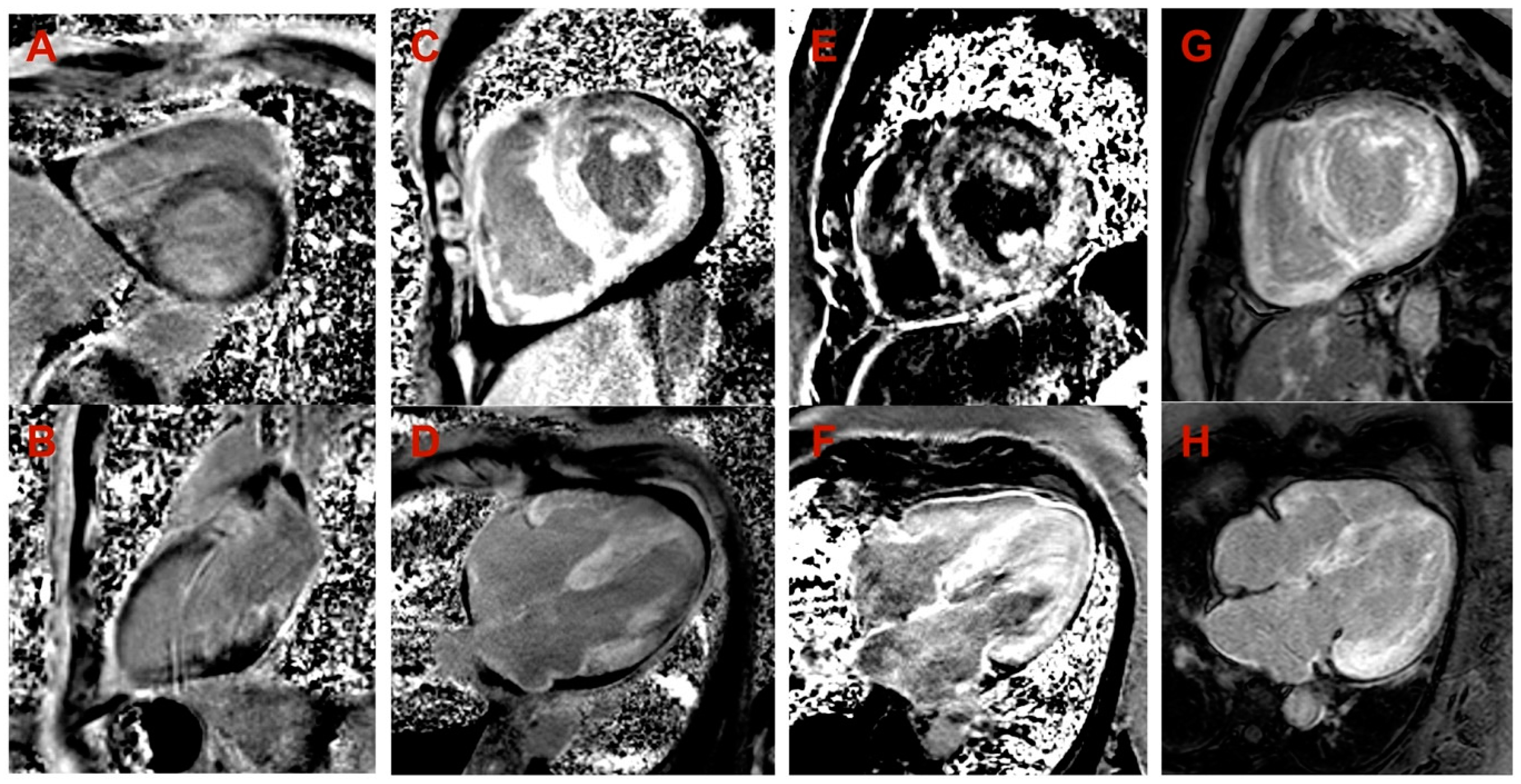
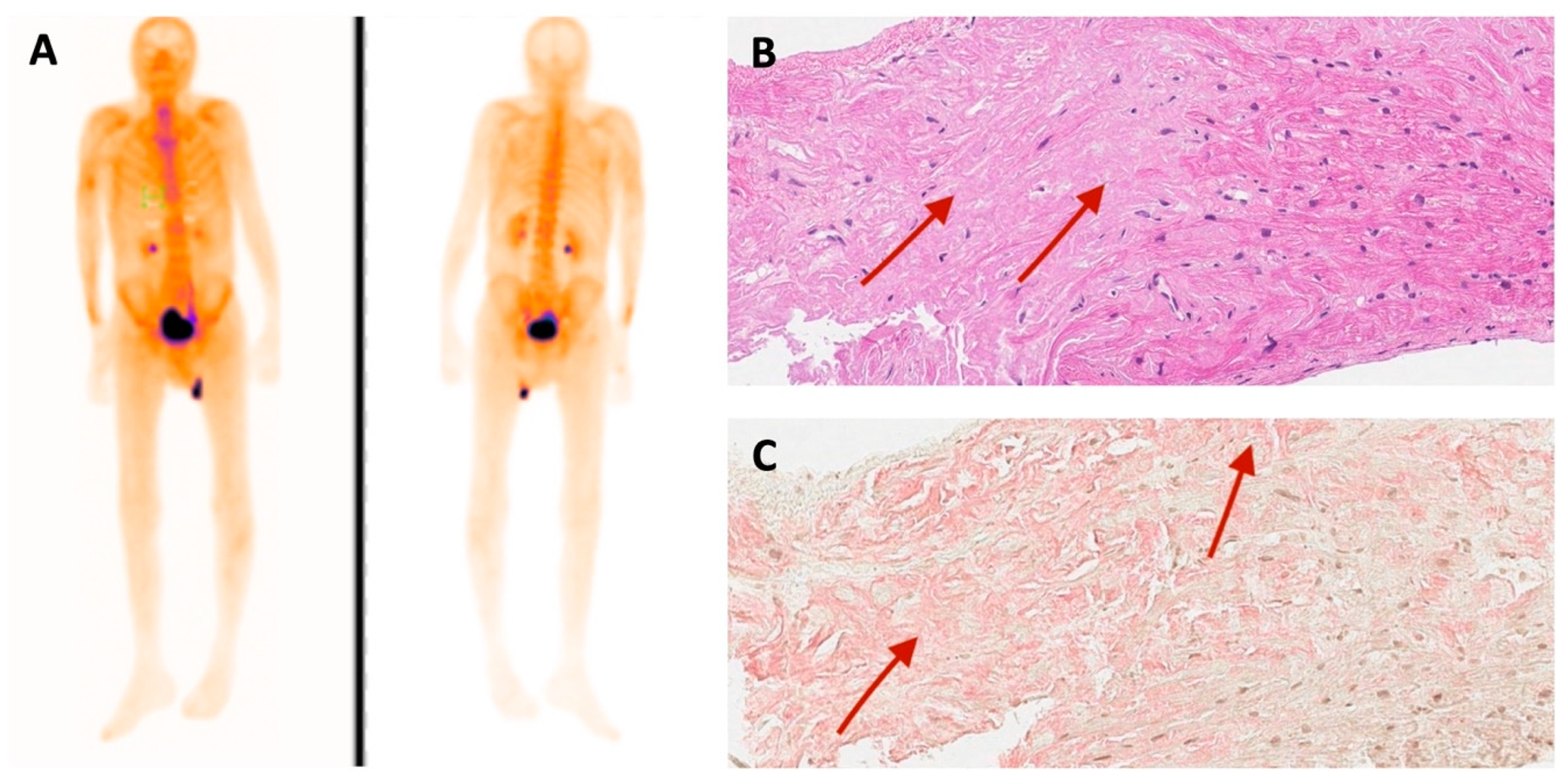
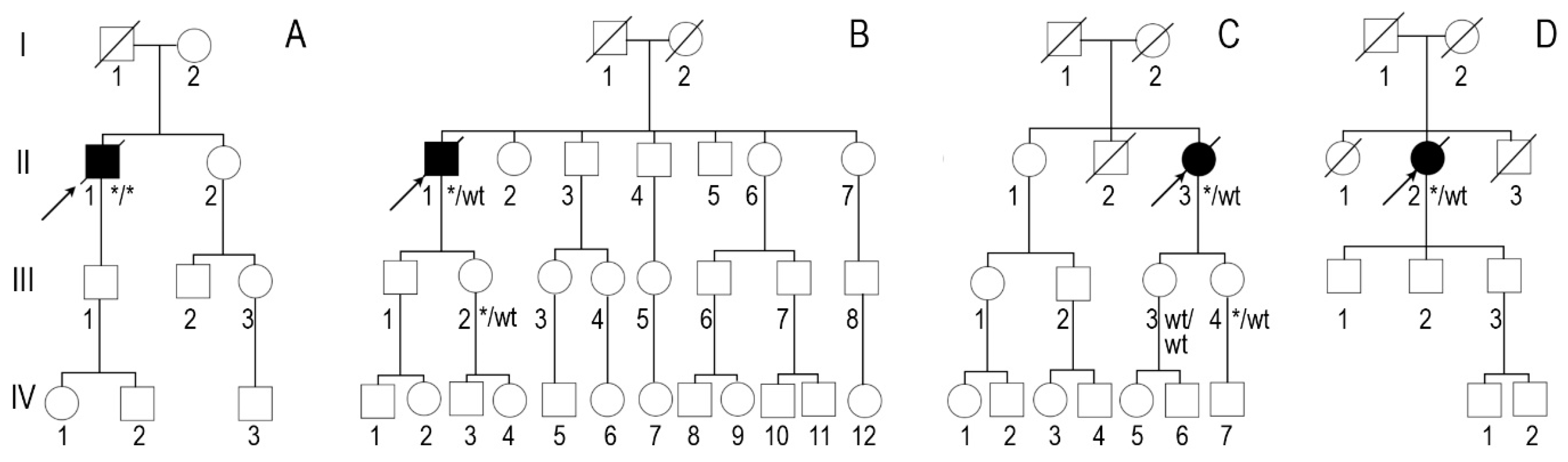
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manganelli, F.; Fabrizi, G.M.; Luigetti, M.; Mandich, P.; Mazzeo, A.; Pareyson, D. Hereditary Transthyretin Amyloidosis Overview. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2022, 43, 595–604. [Google Scholar] [CrossRef]
- Sekijima, Y. Hereditary Transthyretin Amyloidosis. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Mutations in Hereditary Amyloidosis. Available online: http://www.amyloidosismutations.com/ (accessed on 11 February 2023).
- Maurer, M.S.; Bokhari, S.; Damy, T.; Dorbala, S.; Drachman, B.M.; Fontana, M.; Grogan, M.; Kristen, A.V.; Lousada, I.; Nativi-Nicolau, J.; et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Cardiac ATTR Amyloidosis. Circ. Heart Fail. 2019, 12, e006075. [Google Scholar] [CrossRef]
- Jacobson, D.R.; Alexander, A.A.; Tagoe, C.; Buxbaum, J.N. Prevalence of the Amyloidogenic Transthyretin (TTR) V122I Allele in 14 333 African-Americans. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2015, 22, 171–174. [Google Scholar] [CrossRef]
- Damy, T.; Kristen, A.V.; Suhr, O.B.; Maurer, M.S.; Planté-Bordeneuve, V.; Yu, C.-R.; Ong, M.-L.; Coelho, T.; Rapezzi, C.; THAOS Investigators. Transthyretin Cardiac Amyloidosis in Continental Western Europe: An Insight through the Transthyretin Amyloidosis Outcomes Survey (THAOS). Eur. Heart J. 2019, 43, 391–400. [Google Scholar] [CrossRef]
- Koike, H.; Misu, K.; Ikeda, S.; Ando, Y.; Nakazato, M.; Ando, E.; Yamamoto, M.; Hattori, N.; Sobue, G. Study Group for Hereditary Neuropathy in Japan Type I (Transthyretin Met30) Familial Amyloid Polyneuropathy in Japan: Early- vs Late-Onset Form. Arch. Neurol. 2002, 59, 1771–1776. [Google Scholar] [CrossRef]
- Ruberg, F.L.; Grogan, M.; Hanna, M.; Kelly, J.W.; Maurer, M.S. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 2872–2891. [Google Scholar] [CrossRef]
- Vidal, R.; Garzuly, F.; Budka, H.; Lalowski, M.; Linke, R.P.; Brittig, F.; Frangione, B.; Wisniewski, T. Meningocerebrovascular Amyloidosis Associated with a Novel Transthyretin Mis-Sense Mutation at Codon 18 (TTRD 18G). Am. J. Pathol. 1996, 148, 361–366. [Google Scholar]
- Gawor, M.; Holcman, K.; Franaszczyk, M.; Lipowska, M.; Michałek, P.; Teresińska, A.; Bilińska, Z.T.; Rubiś, P.; Kostkiewicz, M.; Szot, W.; et al. Spectrum of Transthyretin Gene Mutations and Clinical Characteristics of Polish Patients with Cardiac Transthyretin Amyloidosis. Cardiol. J. 2020, 29, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Rowczenio, D.M.; Noor, I.; Gillmore, J.D.; Lachmann, H.J.; Whelan, C.; Hawkins, P.N.; Obici, L.; Westermark, P.; Grateau, G.; Wechalekar, A.D. Online Registry for Mutations in Hereditary Amyloidosis Including Nomenclature Recommendations. Hum. Mutat. 2014, 35, E2403–E2412. [Google Scholar] [CrossRef] [PubMed]
- Rowczenio, D.; Quarta, C.C.; Fontana, M.; Whelan, C.J.; Martinez-Naharro, A.; Trojer, H.; Baginska, A.; Ferguson, S.M.; Gilbertson, J.; Rezk, T.; et al. Analysis of the TTR Gene in the Investigation of Amyloidosis: A 25-Year Single UK Center Experience. Hum. Mutat. 2019, 40, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Koh, J.S.; Saini, M.; Tay, K.S.S.; Jayne Tan, Y.; Chai, J.Y.H.; Fam, S.R.; Juraidah, A.R.; Lim, P.K.; Ng, A.S.L.; et al. Hereditary Transthyretin Amyloidosis-Clinical and Genetic Characteristics of a Multiracial South-East Asian Cohort in Singapore. J. Neuromuscul. Dis. 2021, 8, 723–733. [Google Scholar] [CrossRef]
- Grigaitė, J.; Šiaurytė, K.; Audronytė, E.; Preikšaitienė, E.; Burnytė, B.; Pranckevičienė, E.; Ekkert, A.; Utkus, A.; Jatužis, D. Novel In-Frame Deletion in HTRA1 Gene, Responsible for Stroke at a Young Age and Dementia-A Case Study. Genes 2021, 12, 1955. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kõressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating Masking of Template Sequence with Primer Design Software. Bioinforma. Oxf. Engl. 2018, 34, 1937–1938. [Google Scholar] [CrossRef] [PubMed]
- VCV000495842.10—ClinVar—NCBI. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/495842/?oq=TTR[gene]+AND+c.302C%3ET[varname]+&m=NM_000371.4(TTR):c.302C%3ET%20(p.Ala101Val) (accessed on 13 April 2023).
- Benson, M.D.; Kincaid, J.C. The Molecular Biology and Clinical Features of Amyloid Neuropathy. Muscle Nerve 2007, 36, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Munar-Qués, M.; López Domínguez, J.M.; Viader-Farré, C.; Moreira, P.; Saraiva, M.J. Two Spanish Sibs with Familial Amyloidotic Polyneuropathy Homozygous for the V30M-TTR Gene. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2001, 8, 121–123. [Google Scholar] [CrossRef] [PubMed]
- Jacob, E.K.; Edwards, W.D.; Zucker, M.; D’Cruz, C.; Seshan, S.V.; Crow, F.W.; Highsmith, W.E. Homozygous Transthyretin Mutation in an African American Male. J. Mol. Diagn. JMD 2007, 9, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Reddi, H.V.; Jenkins, S.; Theis, J.; Thomas, B.C.; Connors, L.H.; Van Rhee, F.; Highsmith, W.E., Jr. Homozygosity for the V122I Mutation in Transthyretin Is Associated with Earlier Onset of Cardiac Amyloidosis in the African American Population in the Seventh Decade of Life. J. Mol. Diagn. JMD 2014, 16, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, D.R.; Gorevic, P.D.; Buxbaum, J.N. A Homozygous Transthyretin Variant Associated with Senile Systemic Amyloidosis: Evidence for a Late-Onset Disease of Genetic Etiology. Am. J. Hum. Genet. 1990, 47, 127–136. [Google Scholar] [PubMed]
- Hamour, I.M.; Lachmann, H.J.; Goodman, H.J.B.; Petrou, M.; Burke, M.M.; Hawkins, P.N.; Banner, N.R. Heart Transplantation for Homozygous Familial Transthyretin (TTR) V122I Cardiac Amyloidosis. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2008, 8, 1056–1059. [Google Scholar] [CrossRef]
- Lopes, L.R.; Futema, M.; Akhtar, M.M.; Lorenzini, M.; Pittman, A.; Syrris, P.; Elliott, P.M. Prevalence of TTR Variants Detected by Whole-Exome Sequencing in Hypertrophic Cardiomyopathy. Amyloid Int. J. Exp. Clin. Investig. Off. J. Int. Soc. Amyloidosis 2019, 26, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, G.; Lundgren, E.; Kangawa, K.; Kurihara, T.; Matsukura, S.; Matsukura, H.; Nakazato, M.; Steen, L. Diagnostic Radioimmunoassay and DNA-Analysis in Swedish and Japanese Patients with Familial Amyloidotic Polyneuropathy. Homozygosity for the TTR Met30 Gene. Acta Neurol. Scand. 1993, 87, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Li, Z.; Araki, K.; Haruna, K.; Yamaguchi, K.; Araki, M.; Takeya, M.; Ando, Y.; Yamamura, K. Inconsistency between Hepatic Expression and Serum Concentration of Transthyretin in Mice Humanized at the Transthyretin Locus. Genes Cells Devoted Mol. Cell. Mech. 2008, 13, 1257–1268. [Google Scholar] [CrossRef]
- Shawky, R.M. Reduced Penetrance in Human Inherited Disease. Egypt. J. Med. Hum. Genet. 2014, 15, 103–111. [Google Scholar] [CrossRef]
- Gillmore, J.D.; Maurer, M.S.; Falk, R.H.; Merlini, G.; Damy, T.; Dispenzieri, A.; Wechalekar, A.D.; Berk, J.L.; Quarta, C.C.; Grogan, M.; et al. Nonbiopsy Diagnosis of Cardiac Transthyretin Amyloidosis. Circulation 2016, 133, 2404–2412. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Rapezzi, C.; Adler, Y.; Arad, M.; Basso, C.; Brucato, A.; Burazor, I.; Caforio, A.L.P.; Damy, T.; Eriksson, U.; et al. Diagnosis and Treatment of Cardiac Amyloidosis: A Position Statement of the ESC Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2021, 42, 1554–1568. [Google Scholar] [CrossRef]
- Pilebro, B.; Suhr, O.B.; Näslund, U.; Westermark, P.; Lindqvist, P.; Sundström, T. (99m)Tc-DPD Uptake Reflects Amyloid Fibril Composition in Hereditary Transthyretin Amyloidosis. Ups. J. Med. Sci. 2016, 121, 17–24. [Google Scholar] [CrossRef]
- Bergström, J.; Gustavsson, A.; Hellman, U.; Sletten, K.; Murphy, C.L.; Weiss, D.T.; Solomon, A.; Olofsson, B.-O.; Westermark, P. Amyloid Deposits in Transthyretin-Derived Amyloidosis: Cleaved Transthyretin Is Associated with Distinct Amyloid Morphology. J. Pathol. 2005, 206, 224–232. [Google Scholar] [CrossRef]
- Ihse, E.; Ybo, A.; Suhr, O.; Lindqvist, P.; Backman, C.; Westermark, P. Amyloid Fibril Composition Is Related to the Phenotype of Hereditary Transthyretin V30M Amyloidosis. J. Pathol. 2008, 216, 253–261. [Google Scholar] [CrossRef]
- Musumeci, M.B.; Cappelli, F.; Russo, D.; Tini, G.; Canepa, M.; Milandri, A.; Bonfiglioli, R.; Di Bella, G.; My, F.; Luigetti, M.; et al. Low Sensitivity of Bone Scintigraphy in Detecting Phe64Leu Mutation-Related Transthyretin Cardiac Amyloidosis. JACC Cardiovasc. Imaging 2020, 13, 1314–1321. [Google Scholar] [CrossRef]
- Dasari, S.; Theis, J.D.; Vrana, J.A.; Rech, K.L.; Dao, L.N.; Howard, M.T.; Dispenzieri, A.; Gertz, M.A.; Hasadsri, L.; Highsmith, W.E.; et al. Amyloid Typing by Mass Spectrometry in Clinical Practice: A Comprehensive Review of 16,175 Samples. Mayo Clin. Proc. 2020, 95, 1852–1864. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proband No. | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Zygosity of NM_000371.3:c.302C>T, NP_000362.1:p.(Ala101Val) variant in TTR | Homozygous | Heterozygous | Heterozygous | Heterozygous |
| Sex | Male | Male | Female | Female |
| Age of onset of symptoms (years) | 44 | 74 | 50 | 72 |
| Age at diagnosis (years) | 49 | 77 | 57 | 74 |
| NYHA class of heart failure | III | III | III | III |
| Positive family history | Mother at an older age and aunt on mother‘s side had heart disease | Brother had heart disease | Mother had heart disease, grandmother on mother‘s side had sudden death | Mother and brother died of stroke at an older age |
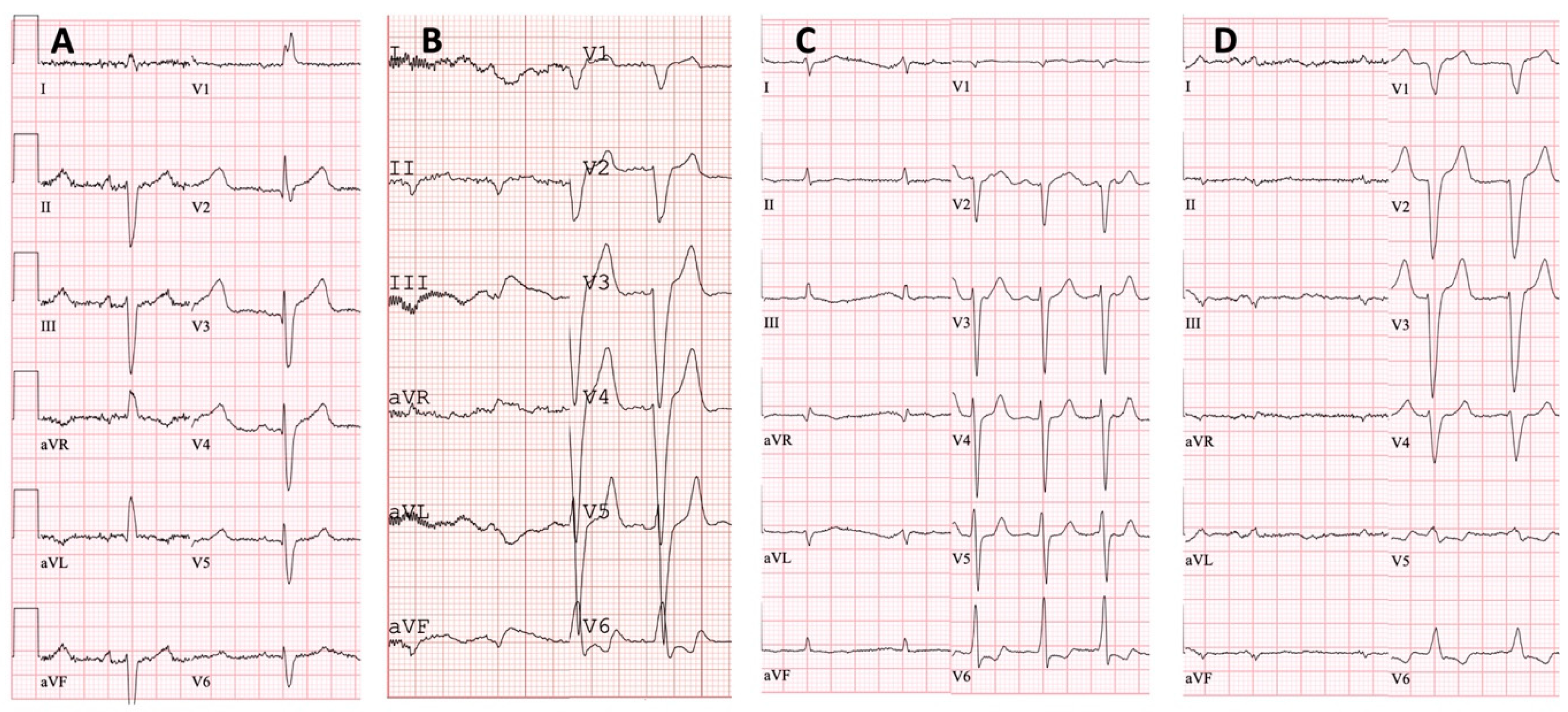
| Low QRS voltage | - | + | + | + |
| Pseudoinfarct pattern on ECG | - | - | - | - |
| Conduction disturbances | RBBB, LAFB | LBBB | - | LBBB |
| Atrial fibrillation | - | + | + | - |
| LV hypertrophy | Concentric | Concentric | Concentric | Concentric |
| Maximal wall thickness (mm) | 13 | 21 | 14 | 19 |
| LVEF (%) | 67 | 10 | 55 | 40 |
| Restrictive LV filling pattern | - | + | + | + |
| Increased RV wall thickness | + | + | - | + |
| Pericardial effusion | - | + | - | - |
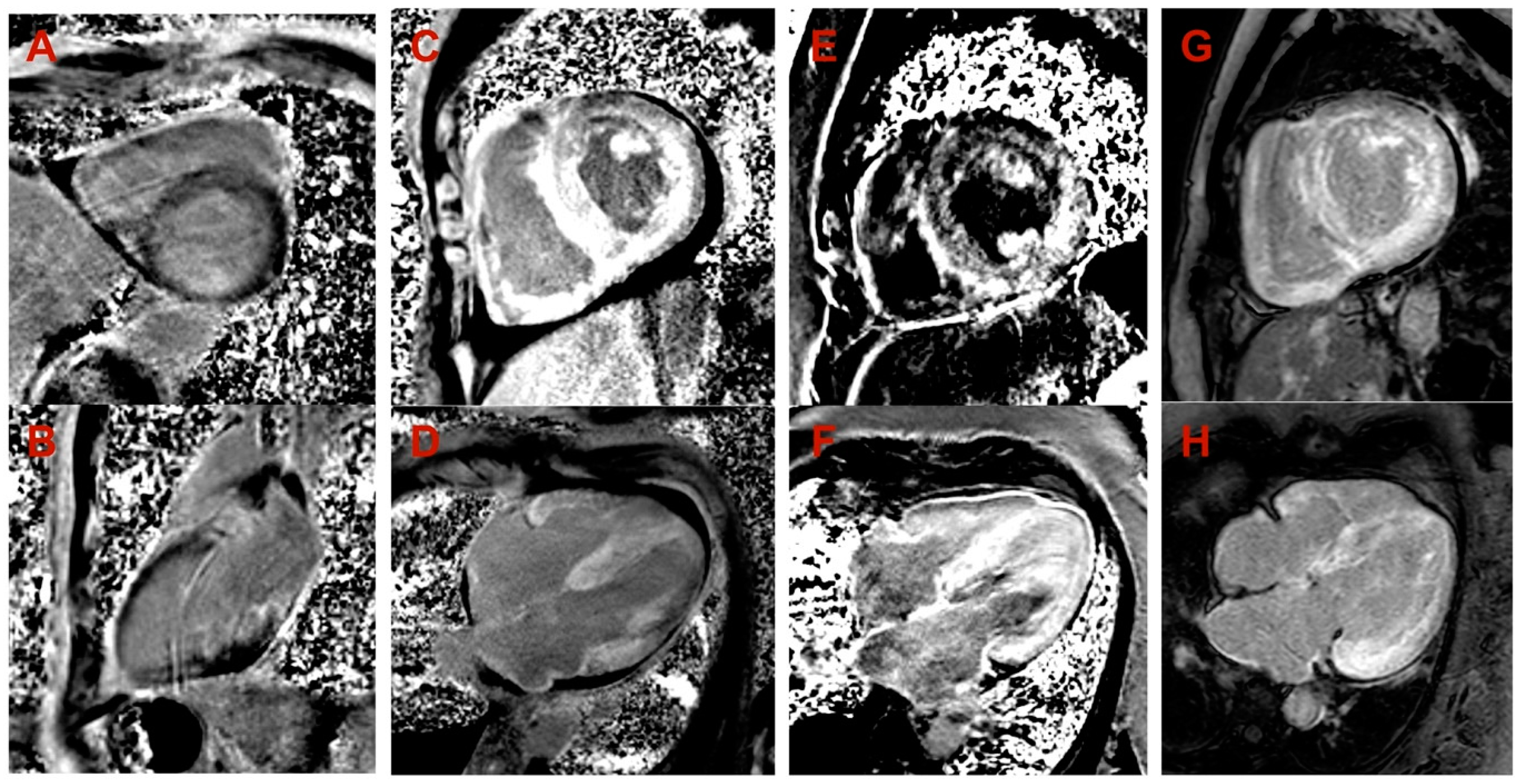
| Cardiac MRI LV hypertrophy | Asymmetric (predominantly in transventricular septum) | Symmetric | Symmetric | Symmetric |
| Cardiac MRI maximal wall thickness (mm) | 16 | 19 | 14 | 20 |
| Cardiac MRI LVEF (%) | 77 | 45 | 50 | 44 |
| Cardiac MRI LGE | Midmyocardial LGE in LV septum and inferior wall | Diffuse subendocardial LGE in LV and RV | Diffuse midmyocardial LGE in LV and RV | Diffuse subendocardial LGE in LV and RV |
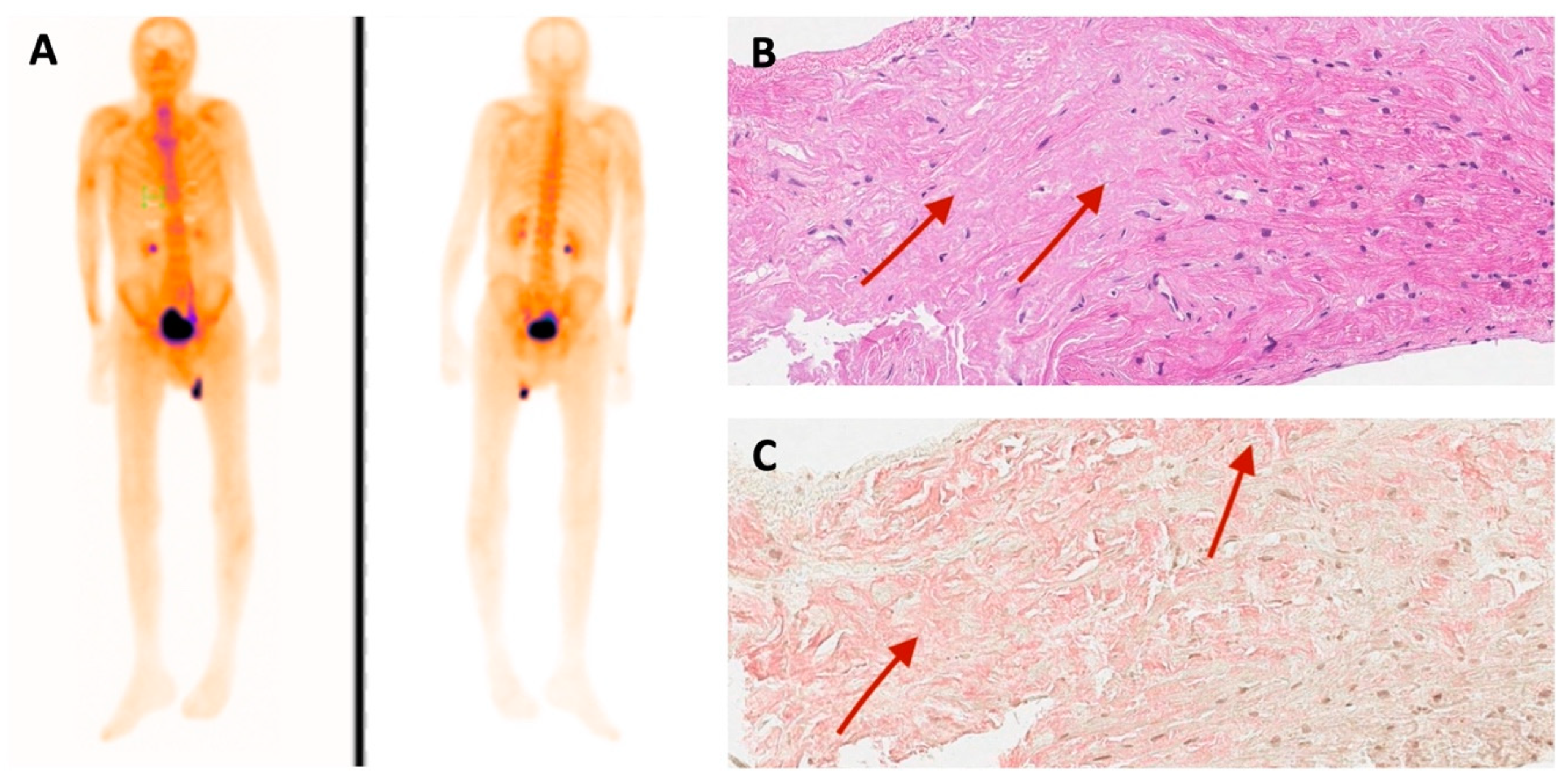
| 99mTc-PYP bone scintigraphy | Grade 0 | - | - | Grade 3 |
| Histological confirmation | Amyloid deposits, likely non-specific reaction to transthyretin on immunohistochemistry in bone marrow trepanobiopsy and endomyocardial biopsy | TTR amyloid deposition in adipose tissue biopsy | TTR amyloid deposition in endomyocardial biopsy | - |
| NT-proBNP (pg/mL) * | 474 | 11401 | 3368 | 2471 |
| Troponin I (ng/L) * | 111 | 95 | 65 | 45 |
| Polyneuropathy | + | - | - | + |
| Chronic kidney disease | - | + | + | - |
| Gastrointestinal manifestation | + | + | - | - |
| Carpal tunnel syndrome | - | - | + | + |
| Biceps tendon rupture | - | NA | - | - |
| Follow-up after diagnosis (years) | 3 | 2 | 3 | 1 |
| Outcome | Death at age 52 due to pneumonia complications | Death at age 79 due to colon adenocarcinoma | Death at age 60 due to heart failure decompensation | Death at age 75 due to heart failure decompensation |
| Family segregation analysis | NA | Variant identified in phenotypically negative 47-year-old daughter | Variant identified in phenotypically negative 33-year-old daughter | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Žebrauskienė, D.; Sadauskienė, E.; Masiulienė, R.; Aidietienė, S.; Šiaudinienė, A.; Pečeliūnas, V.; Žukauskaitė, G.; Žurauskas, E.; Valevičienė, N.; Barysienė, J.; et al. Rare c.302C>T TTR Variant Associated with Transthyretin Amyloidosis. Medicina 2024, 60, 237. https://doi.org/10.3390/medicina60020237
Žebrauskienė D, Sadauskienė E, Masiulienė R, Aidietienė S, Šiaudinienė A, Pečeliūnas V, Žukauskaitė G, Žurauskas E, Valevičienė N, Barysienė J, et al. Rare c.302C>T TTR Variant Associated with Transthyretin Amyloidosis. Medicina. 2024; 60(2):237. https://doi.org/10.3390/medicina60020237
Chicago/Turabian StyleŽebrauskienė, Dovilė, Eglė Sadauskienė, Rūta Masiulienė, Sigita Aidietienė, Agnė Šiaudinienė, Valdas Pečeliūnas, Gabrielė Žukauskaitė, Edvardas Žurauskas, Nomeda Valevičienė, Jūratė Barysienė, and et al. 2024. "Rare c.302C>T TTR Variant Associated with Transthyretin Amyloidosis" Medicina 60, no. 2: 237. https://doi.org/10.3390/medicina60020237
APA StyleŽebrauskienė, D., Sadauskienė, E., Masiulienė, R., Aidietienė, S., Šiaudinienė, A., Pečeliūnas, V., Žukauskaitė, G., Žurauskas, E., Valevičienė, N., Barysienė, J., & Preikšaitienė, E. (2024). Rare c.302C>T TTR Variant Associated with Transthyretin Amyloidosis. Medicina, 60(2), 237. https://doi.org/10.3390/medicina60020237