
A Possible Case of Centronuclear Myopathy: A Case Report



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- Severe/lethal X-linked recessive form (myotubular myopathy) with a mutation in the MTM1 gene, which encodes myotubularin;
- Sporadic form with variable symptoms or autosomal dominant (AD) form associated with mutation of the DNM2 gene (dynamin);
2. Clinical Case
- Posture: slight right convex scoliosis. Hyperlordosis. Increased base of support;
- Gait: waddling. Weight: 43 kg. Size: 1.71 cm. BMI: 14.72;
- Osteomyoarticular system: both high-arched feet;
- Oral cavity: arch of the high palate;
- Muscle tone: decreased globally (Figure 1).
- -
- Clinical laboratory: normal hematology and blood chemistry. Creatinkinase: 100 UI/L.
- -
- Imaging:
- -
- Abdominal ultrasound: normal.
- -
- Chest X-ray: normal.
- -
- Neurophysiological studies:
2.1. Needle Electromyography
2.2. Neuroconduction
- Right peroneal nerve: Decrease in the amplitudes of the combined motor potential of the muscle in the distal and proximal segments. The reduction in motor potential amplitude could be attributed to signs of muscle hypotrophy in the recording muscles;
- Right posterior tibial nerve: Axonal damage in distal and proximal motor fibers;
- Left posterior tibial nerve: Myelin damage in proximal motor fibers.
- -
- Neurogenetics. DNA study: Molecular study for spinal muscular atrophy, with PCR-RFLP technique. Direct detection. Result: No exon 7 gene SMN1/SMN2 deletion.
- -
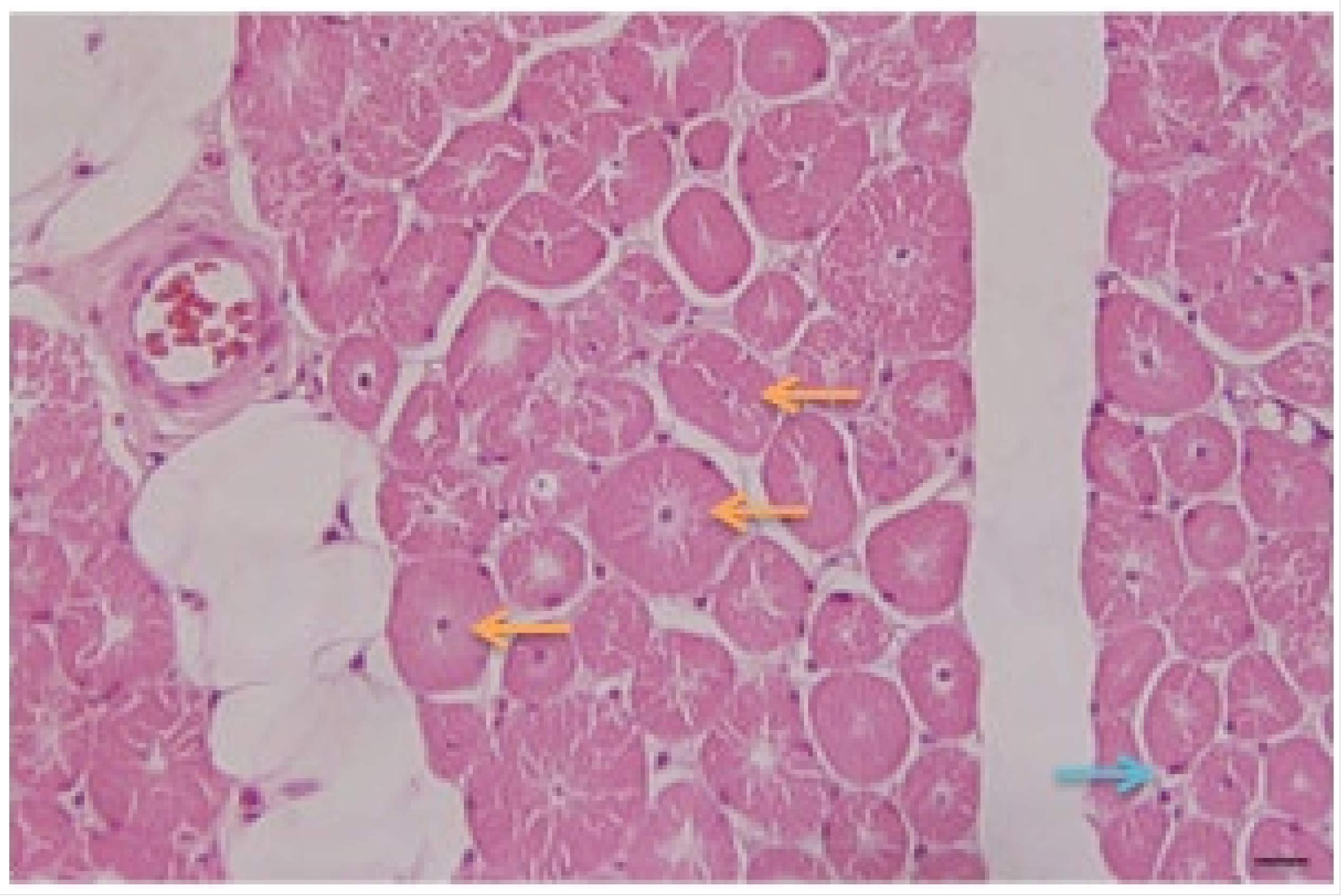
- Pathology: Biopsy of left deltoid striated muscle fragment was performed. The microscopic study of the studied fragments stained with hematoxylin–eosin (H/E) (Figure 3) and Masson’s trichrome showed:
- Presence of fibers with central nuclei;
- Presence of atrophic fibers;
- Fibers of normal diameters and some hypertrophic ones;
- Presence of clear perinuclear spaces and others with multiple central tunnels as an expression of myofilament loss;
- No inflammation or macrophage phenomena were observed;
- No increase in connective tissue is appreciated;
- Discrete fatty infiltration in the perimysial center.
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Benito, D.N.; Ortez, C.; Carrera Garcia, L.; Expósito, J.; Bobadilla, E.; Nascimento, A. Diagnotico y tratamiento de las miopatías congénitas. Medicina 2019, 79 (Suppl. III), 82–86. [Google Scholar]
- Zarranz Imirizaldu, J.J. Neurologia, 6th ed.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Rodríguez-Zambrano, L.; Orjuela-Rolón, C.; Ortiz-Corredor, F.; Espinosa García, E. Evaluación funcional en paciente con miopatía congénita centronuclear asociada a Diamina 2. In Fisitria; Universidad Nacional de Colombia: Bogota, Colombia, 2017; Available online: http://fisiatria.unal.edu.co/casos-clinicos/evaluacion-funcional-en-un-paciente-con-miopatia-congenita-centronuclear-asociada-a-diamina-2/ (accessed on 31 May 2023).
- Ortega, X.; Corral, G.; Rojas, G.; Carrizo, J.; Suárez, B.; Castiglioni, C. Magnetic resonance of complete body for muscle study and quantification of fatty fraction in pediatric patients with congenital myophaties. Rev. Médica Clínica Condes 2018, 29, 654–662. [Google Scholar] [CrossRef]
- Pelin, K.; Wallgren-Pettersson, C. Update on the Genetics of Congenital Myopathies. Semin. Pediatr. Neurol. 2019, 29, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Haddad, M. The Impact of CB1 Receptor on Nuclear Receptors in Skeletal Muscle Cells. Pathophysiology 2021, 28, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Brigida, A.L.; Schultz, S.; Cascone, M.; Antonucci, N.; Siniscalco, D. Endocannabinod Signal Dysregulation in Autism Spectrum Disorders: A Correlation Link between Inflammatory State and Neuro-Immune Alterations. Int. J. Mol. Sci. 2017, 18, 1425. [Google Scholar] [CrossRef] [PubMed]
- Haddad, M. The Impact of CB1 Receptor on Inflammation in Skeletal Muscle Cells. J. Inflamm. Res. 2021, 14, 3959–3967. [Google Scholar] [CrossRef] [PubMed]
- Dowling, J.J.; Lawlor, M.W.; Das, S. X-Linked Myotubular Myopathy; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; GeneReviews: Seattle, WA, USA; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Dynacure. 2020. Available online: https://www.dynacure.com/pipeline/ (accessed on 23 January 2023).
- Ropper, A.H.; Samuels, M.A.; Klein, J.P.; Prasad, S. (Eds.) Adams and Victor’s Principles of Neurology, 11th ed.; McGraw Hill: New York, NY, USA, 2019; Available online: https://accessmedicine.mhmedical.com/content.aspx?bookid=1477§ionid=215135193 (accessed on 31 May 2023).
- Preston, D.C.; Shapiro, B.E. Electromyography and Neuromuscular Disorders, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Malfatti, E. Miopatías congénitas. Rev. Médica Clínica Condes 2018, 29, 636–642. [Google Scholar] [CrossRef]
- Kumar, V.; Abbas, A.K.; Aster, J.C. Robbins & Cotran Pathologic Basis of Disease, 9th ed.; Elsevier: Philadelphia, PA, USA, 2015. [Google Scholar]
- Quintana-Vega, V.; Barragán-Pérez, E.J.; Alarcón-De la Luz, E.; Alarcón-Cabrera, E.; Sadowinski-Pine, S.; Aguirre-Hernández, J. Fenotipo de miopatía congénita central core autosómica dominante con alteraciones del gen RYR1. A propósito de un caso clínico. Acta Pediatr. Mex. 2022, 43, 353–357. [Google Scholar] [CrossRef]
- Agrawal, P.B.; Pierson, C.R.; Joshi, M.; Liu, X.; Ravenscroft, G.; Moghadaszadeh, B.; Talabere, T.; Viola, M.; Swanson, L.C.; Haliloğlu, G.; et al. SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am. J. Hum. Genet. 2014, 95, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Witting, N.; Werlauff, U.; Duno, M.; Vissing, J. Phenotypes, genotypes, and prevalence of congenital myopathies older than 5 years in Denmark. Neurol. Genet. 2017, 3, e140. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo-Ferrán, N.; Junco-Rodriguez, J.M.; Lestayo-O’Farrill, Z.; Robinson-Agramonte, M.d.l.A.; Camejo-León, Z.; Gómez-Suárez, H.J.; Salinas-Olivares, M.; Antiguas-Valdez, E.; Falcón-Lamazares, E.; Siniscalco, D. A Possible Case of Centronuclear Myopathy: A Case Report. Medicina 2023, 59, 1112. https://doi.org/10.3390/medicina59061112
Castillo-Ferrán N, Junco-Rodriguez JM, Lestayo-O’Farrill Z, Robinson-Agramonte MdlA, Camejo-León Z, Gómez-Suárez HJ, Salinas-Olivares M, Antiguas-Valdez E, Falcón-Lamazares E, Siniscalco D. A Possible Case of Centronuclear Myopathy: A Case Report. Medicina. 2023; 59(6):1112. https://doi.org/10.3390/medicina59061112
Chicago/Turabian StyleCastillo-Ferrán, Narjara, Juan Mario Junco-Rodriguez, Zurina Lestayo-O’Farrill, María de los Angeles Robinson-Agramonte, Zoilo Camejo-León, Héctor Jesús Gómez-Suárez, Mercedes Salinas-Olivares, Evelyn Antiguas-Valdez, Elizabeth Falcón-Lamazares, and Dario Siniscalco. 2023. "A Possible Case of Centronuclear Myopathy: A Case Report" Medicina 59, no. 6: 1112. https://doi.org/10.3390/medicina59061112
APA StyleCastillo-Ferrán, N., Junco-Rodriguez, J. M., Lestayo-O’Farrill, Z., Robinson-Agramonte, M. d. l. A., Camejo-León, Z., Gómez-Suárez, H. J., Salinas-Olivares, M., Antiguas-Valdez, E., Falcón-Lamazares, E., & Siniscalco, D. (2023). A Possible Case of Centronuclear Myopathy: A Case Report. Medicina, 59(6), 1112. https://doi.org/10.3390/medicina59061112