
Pituitary Tumorigenesis—Implications for Management


Abstract
1. Introduction
2. Cell Signaling Pathways in Pituitary Tumorigenesis
2.1. Gsa/Protein Kinase A/c AMP Signaling Pathway
2.1.1. GNAS Mutations
2.1.2. Protein Kinase A Mutations
2.1.3. AIP
2.1.4. GPR101
2.2. MAPK/ERK and PI3K/Akt Pathways
Ubiquitin-Specific Protease 8 (USP8) and Other Deubiquitinases
2.3. Hippo Pathway
2.4. Wnt Pathway
3. Tumor Suppressor Genes/Oncogenes
3.1. Menin Gene
3.2. CDKN1B Gene
3.3. CABLES1 (CDK5 and ABL Enzyme Substrate 1)
3.4. PitNETs Related to Succinate Dehydrogenase (SDHx) Mutations
3.5. DICER1, Ribonuclease III
4. Stem Cells in the Pituitary Gland and Tumorigenesis
5. MicroRNAs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Banskota, S.; Adamson, D.C. Pituitary Adenomas: From Diagnosis to Therapeutics. Biomedicines 2021, 9, 494. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.F.; Beckers, A. The Epidemiology of Pituitary Adenomas. Endocrinol. Metab. Clin. 2020, 49, 347–355. [Google Scholar] [CrossRef]
- Asa, S.L.; Mete, O.; Perry, A.; Osamura, R.Y. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr. Pathol. 2022, 33, 6–26. [Google Scholar] [CrossRef] [PubMed]
- Teramoto, A.; Hirakawa, K.; Sanno, N.; Osamura, Y. Incidental pituitary lesions in 1,000 unselected autopsy specimens. Radiology 1994, 193, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Vallecillos, F.J.T.; Fernández, S.O. Histopathological features of post-mortem pituitaries: A retrospective analysis. Rev. Assoc. Med. Bras. 2016, 62, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, O.M.; Karavitaki, N.; Pereira, A.M. The epidemiology of aggressive pituitary tumors (and its challenges). Rev. Endocr. Metab. Disord. 2020, 21, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Molitch, M.E. Diagnosis and treatment of pituitary adenomas: A review. JAMA J. Am. Med. Assoc. 2017, 317, 516–524. [Google Scholar] [CrossRef]
- Biermasz, N.R. The burden of disease for pituitary patients. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101309. [Google Scholar] [CrossRef]
- Ershadinia, N.; Dsc, N.A.T. Diagnosis and Treatment of Acromegaly: An Update. Mayo Clin. Proc. 2022, 97, 333–346. [Google Scholar] [CrossRef]
- Lasolle, H.; Ferriere, A.; Vasiljevic, A.; Eimer, S.; Nunes, M.L.; Tabarin, A. Pasireotide-LAR in acromegaly patients treated with a combination therapy: A real-life study. Endocr. Connect. 2019, 8, 1383–1394. [Google Scholar] [CrossRef]
- Inder, W.J.; Jang, C. Treatment of Prolactinoma. Medicina 2022, 58, 1095. [Google Scholar] [CrossRef]
- Yavropoulou, M.P.; Yavropoulou, M.P. The natural history and treatment of non-functioning pituitary adenomas (non-functioning PitNETs). Endocr.-Relat. Cancer 2020, 27, R375–R390. [Google Scholar] [CrossRef]
- Daly, A.F.; Bch, M.B.; Tichomirowa, M.A.; Beckers, A. Best Practice & Research Clinical Endocrinology & Metabolism The epidemiology and genetics of pituitary adenomas. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 543–554. [Google Scholar] [CrossRef]
- Dénes, J.; Korbonits, M. The clinical aspects of pituitary tumour genetics. Endocrine 2021, 71, 663–674. [Google Scholar] [CrossRef]
- Peculis, R.; Niedra, H.; Rovite, V. Large scale molecular studies of pituitary neuroendocrine tumors: Novel markers, mechanisms and translational perspectives. Cancers 2021, 13, 1395. [Google Scholar] [CrossRef]
- Xekouki, P.; Azevedo, M.; Stratakis, C.A. Anterior pituitary adenomas: Inherited syndromes, novel genes and molecular pathways. Expert Rev. Endocrinol. Metab. 2010, 5, 697–709. [Google Scholar] [CrossRef]
- Xekouki, P.; Lodge, E.J.; Matschke, J.; Santambrogio, A.; Apps, J.R.; Sharif, A.; Jacques, T.S.; Aylwin, S.; Prevot, V.; Li, R.; et al. Non-secreting pituitary tumours characterised by enhanced expression of YAP/TAZ. Endocr. Relat. Cancer 2019, 26, 215–225. [Google Scholar] [CrossRef]
- Russell, J.P.; Lodge, E.J.; Andoniadou, C.L. Basic Research Advances on Pituitary Stem Cell Function and Regulation. Neuroendocrinology 2018, 107, 196–203. [Google Scholar] [CrossRef]
- Donati, S.; Aurilia, C.; Palmini, G.; Miglietta, F.; Falsetti, I.; Iantomasi, T.; Brandi, M.L. Micrornas as potential biomarkers in pituitary adenomas. Non-Coding RNA 2021, 7, 55. [Google Scholar] [CrossRef]
- SUTHERLAND, E.W.; RALL, T.W. Fractionation and characterization of a cyclic adenine ribonucleotide formed by tissue particles. J. Biol. Chem. 1958, 232, 1077–1091. [Google Scholar] [CrossRef]
- Milligan, G.; Kostenis, E. Heterotrimeric G-proteins: A short history. Br. J. Pharmacol. 2006, 147, S46–S55. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ramírez, L.C.; Trivellin, G.; Stratakis, C.A. Cyclic 3′,5′-adenosine monophosphate (cAMP) signaling in the anterior pituitary gland in health and disease. Mol. Cell. Endocrinol. 2018, 463, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Goricanec, D.; Stehle, R.; Egloff, P.; Grigoriu, S.; Plückthun, A.; Wagner, G.; Hagn, F. Conformational dynamics of a G-protein α subunit is tightly regulated by nucleotide binding. Proc. Natl. Acad. Sci. USA 2016, 113, E3629–E3638. [Google Scholar] [CrossRef]
- Eckstein, F.; Romaniuk, P.J.; Heideman, W.; Storm, D.R. Stereochemistry of the mammalian adenylate cyclase reaction. J. Biol. Chem. 1981, 256, 9118–9120. [Google Scholar] [CrossRef]
- Shabb, J.B. Physiological substrates of cAMP-dependent protein kinase. Chem. Rev. 2001, 101, 2381–2411. [Google Scholar] [CrossRef]
- Stratakis, C.A. Cyclic AMP, protein kinase A, and phosphodiesterases: Proceedings of an international workshop. Horm. Metab. Res. 2012, 44, 713–715. [Google Scholar] [CrossRef]
- Sun, Y.; McGarrigle, D.; Huang, X.Y. When a G protein-coupled receptor does not couple to a G protein. Mol. Biosyst. 2007, 3, 849–854. [Google Scholar] [CrossRef]
- Pertuit, M.; Barlier, A.; Enjalbert, A.; Gérard, C. Signalling pathway alterations in pituitary adenomas: Involvement of Gsα, cAMP and mitogen-activated protein kinases. J. Neuroendocrinol. 2009, 21, 869–877. [Google Scholar] [CrossRef]
- Vallar, L.; Spada, A.; Giannattasio, G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature 1987, 330, 566–568. [Google Scholar] [CrossRef]
- Mantovani, G.; Ballare, E.; Giammona, E.; Beck-Peccoz, P.; Spada, A. The Gsα gene: Predominant maternal origin of transcription in human thyroid gland and gonads. J. Clin. Endocrinol. Metab. 2002, 87, 4736–4740. [Google Scholar] [CrossRef]
- Bastepe, M.; Jüppner, H. GNAS locus and pseudohypoparathyroidism. Horm. Res. 2005, 63, 65–74. [Google Scholar] [CrossRef]
- Campbell, R.; Gosden, C.M.; Bonthron, D.T. Parental origin of transcription from the human GNAS1 gene. J. Med. Genet. 1994, 31, 607–614. [Google Scholar] [CrossRef]
- Landis, C.A.; Masters, S.B.; Spada, A.; Pace, A.M.; Bourne, H.R.; Vallar, L. GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 1989, 340, 692–696. [Google Scholar] [CrossRef]
- Riminucci, M.; Collins, M.T.; Lala, R.; Corsi, A.; Matarazzo, P.; Robey, P.G.; Bianco, P. An R201H activating mutation of the GNAS1 (Gsα) gene in a corticotroph pituitary adenoma. J. Clin. Pathol.-Mol. Pathol. 2002, 55, 58–60. [Google Scholar] [CrossRef]
- Taboada, G.F.; Tabet, A.L.O.; Naves, L.A.; Carvalho, D.P.; Gadelha, M.R. Prevalence of gsp oncogene in somatotropinomas and clinically non-functioning pituitary adenomas: Our experience. Pituitary 2009, 12, 165–169. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Stalla, G.K. Somatostatin receptors: From signaling to clinical practice. Front. Neuroendocrinol. 2013, 34, 228–252. [Google Scholar] [CrossRef]
- Landis, C.A.; Harsh, G.; Lyons, J.; Davis, R.L.; Mc Cormick, F.; Bourne, H.R. Clinical characteristics of acromegalic patients whose pituitary tumors contain mutant Gs protein. J. Clin. Endocrinol. Metab. 1990, 71, 1416–1420. [Google Scholar] [CrossRef]
- Larkin, S.; Reddy, R.; Karavitaki, N.; Cudlip, S.; Wass, J.; Ansorge, O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naïve patients with somatotroph adenomas. Eur. J. Endocrinol. 2013, 168, 491–499. [Google Scholar] [CrossRef]
- Freda, P.U.; Chung, W.K.; Matsuoka, N.; Walsh, J.E.; Kanibir, M.N.; Kleinman, G.; Wang, Y.; Bruce, J.N.; Post, K.D. Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: Correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary 2007, 10, 275–282. [Google Scholar] [CrossRef]
- Efstathiadou, Z.A.; Bargiota, A.; Chrisoulidou, A.; Kanakis, G.; Papanastasiou, L.; Theodoropoulou, A.; Tigas, S.K.; Vassiliadi, D.A.; Alevizaki, M.; Tsagarakis, S. Impact of gsp mutations in somatotroph pituitary adenomas on growth hormone response to somatostatin analogs: A meta-analysis. Pituitary 2015, 18, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Romanet, P.; Galluso, J.; Kamenicky, P.; Hage, M.; Theodoropoulou, M.; Roche, C.; Graillon, T.; Etchevers, H.C.; De Murat, D.; Mougel, G.; et al. Somatotroph tumors and the epigenetic status of the GNAS locus. Int. J. Mol. Sci. 2021, 22, 7570. [Google Scholar] [CrossRef] [PubMed]
- Foltran, R.K.; Amorim, P.V.G.H.; Duarte, F.H.; Grande, I.P.P.; Freire, A.C.T.B.; Frassetto, F.P.; Dettoni, J.B.; Alves, V.A.; Castro, I.; Trarbach, E.B.; et al. Study of major genetic factors involved in pituitary tumorigenesis and their impact on clinical and biological characteristics of sporadic somatotropinomas and non-functioning pituitary adenomas. Braz. J. Med. Biol. Res. 2018, 51, e7427. [Google Scholar] [CrossRef] [PubMed]
- Albright, F.; Butler, A.M.; Hampton, A.O.; Smith, P. Disseminata, Areas of Pigmentation and Endocrine. N. Engl. J. Med. 1937, 216, 721–746. [Google Scholar]
- Dumitrescu, C.E.; Collins, M.T. McCune-Albright syndrome. Orphanet J. Rare Dis. 2008, 3, 12. [Google Scholar] [CrossRef]
- Weinstein, L.S.; Shenker, A.; Gejman, P.V.; Merino, M.J.; Friedman, E.; Spiegel, A.M. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N. Engl. J. Med. 1991, 325, 1688–1695. [Google Scholar] [CrossRef]
- Salenave, S.; Boyce, A.M.; Collins, M.T.; Chanson, P. Acromegaly and mccune-albright syndrome. J. Clin. Endocrinol. Metab. 2014, 99, 1955–1969. [Google Scholar] [CrossRef]
- Akintoye, S.O.; Kelly, M.H.; Brillante, B.; Cherman, N.; Turner, S.; Butman, J.A.; Robey, P.G.; Collins, M.T. Pegvisomant for the treatment of gsp-mediated growth hormone excess in patients with McCune-Albright syndrome. J. Clin. Endocrinol. Metab. 2006, 91, 2960–2966. [Google Scholar] [CrossRef]
- Liu, F.; Li, W.; Yao, Y.; Li, G.; Yang, Y.; Dou, W.; Zhong, D.; Wang, L.; Zhu, X.; Hu, H.; et al. A case of McCune-Albright syndrome associated with pituitary GH adenoma: Therapeutic process and autopsy. J. Pediatr. Endocrinol. Metab. 2011, 24, 283–287. [Google Scholar] [CrossRef]
- Zhai, X.; Duan, L.; Yao, Y.; Xing, B.; Deng, K.; Wang, L.; Feng, F.; Liang, Z.; You, H.; Yang, H.; et al. Clinical Characteristics and Management of Patients with McCune-Albright Syndrome with GH Excess and Precocious Puberty: A Case Series and Literature Review. Front. Endocrinol. 2021, 12, 672394. [Google Scholar] [CrossRef]
- Correa, R.; Salpea, P.; Stratakis, C.A. Carney complex: An update. Eur. J. Endocrinol. 2015, 173, M85–M97. [Google Scholar] [CrossRef]
- Bouys, L.; Bertherat, J. Carney complex: Clinical and genetic update 20 years after the identification of the CNC1 (PRKAR1A) gene. Eur. J. Endocrinol. 2021, 184, R99–R109. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Carney, J.A.; Lin, J.P.; Papanicolaou, D.A.; Karl, M.; Kastner, D.L.; Pras, E.; Chrousos, G.P. Carney complex, a familial multiple neoplasia and lentiginosis syndrome: Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J. Clin. Investig. 1996, 97, 699–705. [Google Scholar] [CrossRef]
- Forlino, A.; Vetro, A.; Garavelli, L.; Ciccone, R.; London, E.; Stratakis, C.A.; Zuffardi, O. PRKACB and Carney complex. N. Engl. J. Med. 2014, 370, 1065–1067. [Google Scholar] [CrossRef]
- Courcoutsakis, N.A.; Tatsi, C.; Patronas, N.J.; Lee, C.C.R.; Prassopoulos, P.K.; Stratakis, C.A. The complex of myxomas, spotty skin pigmentation and endocrine overactivity (Carney complex): Imaging findings with clinical and pathological correlation. Insights Imaging 2013, 4, 119–133. [Google Scholar] [CrossRef]
- Stergiopoulos, S.G.; Abu-Asab, M.S.; Tsokos, M.; Stratakis, C.A. Pituitary pathology in Carney complex patients. Pituitary 2004, 7, 73–82. [Google Scholar] [CrossRef]
- Kirschner, L.S. PRKAR1A and the evolution of pituitary tumors. Mol. Cell. Endocrinol. 2010, 326, 3–7. [Google Scholar] [CrossRef]
- Kiefer, F.W.; Winhofer, Y.; Iacovazzo, D.; Korbonits, M.; Wolfsberger, S.; Knosp, E.; Trautinger, F.; Höftberger, R.; Krebs, M.; Luger, A.; et al. PRKAR1A mutation causing pituitary-dependent Cushing disease in a patient with Carney complex. Eur. J. Endocrinol. 2017, 177, K7–K12. [Google Scholar] [CrossRef]
- Hernández-Ramírez, L.C.; Tatsi, C.; Lodish, M.B.; Faucz, F.R.; Pankratz, N.; Chittiboina, P.; Lane, J.; Kay, D.M.; Valdés, N.; Dimopoulos, A.; et al. Corticotropinoma as a component of carney complex. J. Endocr. Soc. 2017, 1, 918–925. [Google Scholar] [CrossRef]
- Pepe, S.; Korbonits, M.; Iacovazzo, D. Germline and mosaic mutations causing pituitary tumours: Genetic and molecular aspects. J. Endocrinol. 2019, 240, R21–R45. [Google Scholar] [CrossRef]
- Salpea, P.; Horvath, A.; London, E.; Faucz, F.R.; Vetro, A.; Levy, I.; Gourgari, E.; Dauber, A.; Holm, I.A.; Morrison, P.J.; et al. Deletions of the PRKAR1A locus at 17q24.2-q24.3 in carney complex: Genotype-phenotype correlations and implications for genetic testing. J. Clin. Endocrinol. Metab. 2014, 99, E183–E188. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hernández-Ramírez, L.C.; Gabrovska, P.; Dénes, J.; Stals, K.; Trivellin, G.; Tilley, D.; Ferraù, F.; Evanson, J.; Ellard, S.; Grossman, A.B.; et al. Landscape of familial isolated and young-onset pituitary adenomas: Prospective diagnosis in AIP mutation carriers. J. Clin. Endocrinol. Metab. 2015, 100, E1242–E1254. [Google Scholar] [CrossRef] [PubMed]
- Tiryakioǧlu, Ö.; Caneroǧlu, N.Ü.; Yilmaz, E.; Gazioǧlu, N.; Kadioǧlu, P.; Açbay, Ö.; Gündoǧdu, S. Familial acromegaly: A familial report and review of the literature. Endocr. Res. 2004, 30, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Vierimaa, O.; Georgitsi, M.; Lehtonen, R.; Vahteristo, P.; Kokko, A.; Raitila, A.; Tuppurainen, K.; Ebeling, T.M.L.; Salmela, P.I.; Paschke, R.; et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science 2006, 312, 1228–1230. [Google Scholar] [CrossRef]
- Daly, A.F.; Tichomirowa, M.A.; Petrossians, P.; Heliövaara, E.; Jaffrain-Rea, M.L.; Barlier, A.; Naves, L.A.; Ebeling, T.; Karhu, A.; Raappana, A.; et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: An international collaborative study. J. Clin. Endocrinol. Metab. 2010, 95, 373–383. [Google Scholar] [CrossRef]
- Daly, A.F.; Beckers, A. The role of AIP mutations in pituitary adenomas: 10 years on. Endocrine 2016, 55, 333–335. [Google Scholar] [CrossRef][Green Version]
- Xekouki, P.; Mastroyiannis, S.A.; Avgeropoulos, D.; De La Luz Sierra, M.; Trivellin, G.; Gourgari, E.A.; Lyssikatos, C.; Quezado, M.; Patronas, N.; Kanaka-Gantenbein, C.; et al. Familial pituitary apoplexy as the only presentation of a novel AIP mutation. Endocr. Relat. Cancer 2013, 20, 11–14. [Google Scholar] [CrossRef]
- Trivellin, G.; Korbonits, M. AIP and its interacting partners. J. Endocrinol. 2011, 210, 137–155. [Google Scholar] [CrossRef]
- Formosa, R.; Xuereb-Anastasi, A.; Vassallo, J. Aip regulates cAMP signalling and GH secretion in GH3 cells. Endocr. Relat. Cancer 2013, 20, 495–505. [Google Scholar] [CrossRef]
- Hernández-Ramírez, L.C.; Trivellin, G.; Stratakis, C.A. Role of Phosphodiesterases on the Function of Aryl Hydrocarbon Receptor-Interacting Protein (AIP) in the Pituitary Gland and on the Evaluation of AIP Gene Variants. Horm. Metab. Res. 2017, 49, 286–295. [Google Scholar] [CrossRef]
- Ritvonen, E.; Pitkänen, E.; Karppinen, A.; Vehkavaara, S.; Demir, H.; Paetau, A.; Schalin-Jäntti, C.; Karhu, A. Impact of AIP and inhibitory G protein alpha 2 proteins on clinical features of sporadic GH-secreting pituitary adenomas. Eur. J. Endocrinol. 2017, 176, 243–252. [Google Scholar] [CrossRef]
- Schernthaner-Reiter, M.H.; Trivellin, G.; Stratakis, C.A. Interaction of AIP with protein kinase A (cAMP-dependent protein kinase). Hum. Mol. Genet. 2018, 27, 2604–2613. [Google Scholar] [CrossRef]
- Jaffrain-Rea, M.-L.; Beckers, A. The Role of Aryl Hydrocarbon Receptor (AHR) and AHR-Interacting Protein (AIP) in the Pathogenesis of Pituitary Adenomas. In Tumors of the Central Nervous System; Hayat, M.A., Ed.; Springer: Dordrecht, The Netherlands, 2013; Volume 10, pp. 189–201. [Google Scholar] [CrossRef]
- Lecoq, A.L.; Viengchareun, S.; Hage, M.; Bouligand, J.; Young, J.; Boutron, A.; Zizzari, P.; Lombès, M.; Chanson, P.; Kamenicky, P. AIP mutations impair AhR signaling in pituitary adenoma patients fibroblasts and in GH3 cells. Endocr. Relat. Cancer 2016, 23, 433–443. [Google Scholar] [CrossRef]
- Formosa, R.; Borg, J.; Vassallo, J. Aryl hydrocarbon receptor (AHR) is a potential tumour suppressor in pituitary adenomas. Endocr. Relat. Cancer 2017, 24, 445–457. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Stalla, G.K.; Spengler, D. ZAC1 target genes and pituitary tumorigenesis. Mol. Cell. Endocrinol. 2010, 326, 60–65. [Google Scholar] [CrossRef]
- De Pinho, L.K.J.; Neto, L.V.; Wildemberg, L.E.A.; Gasparetto, E.L.; Marcondes, J.; De Almeida Nunes, B.; Takiya, C.M.; Gadelha, M.R. Low aryl hydrocarbon receptor-interacting protein expression is a better marker of invasiveness in somatotropinomas than Ki-67 and p53. Neuroendocrinology 2011, 94, 39–48. [Google Scholar] [CrossRef]
- Chahal, H.S.; Trivellin, G.; Leontiou, C.A.; Alband, N.; Fowkes, R.C.; Tahir, A.; Igreja, S.C.; Chapple, J.P.; Jordan, S.; Lupp, A.; et al. Somatostatin analogs modulate AIP in somatotroph adenomas: The role of the ZAC1 pathway. J. Clin. Endocrinol. Metab. 2012, 97, E1411–E1420. [Google Scholar] [CrossRef]
- Dutta, P.; Reddy, K.S.; Rai, A.; Madugundu, A.K.; Solanki, H.S.; Bhansali, A.; Radotra, B.D.; Kumar, N.; Collier, D.; Iacovazzo, D.; et al. Surgery, Octreotide, Temozolomide, Bevacizumab, Radiotherapy, and Pegvisomant Treatment of an AIP Mutation—Positive Child. J. Clin. Endocrinol. Metab. 2019, 104, 3539–3544. [Google Scholar] [CrossRef]
- Williams, F.; Hunter, S.; Bradley, L.; Chahal, H.S.; Storr, H.L.; Akker, S.A.; Kumar, A.V.; Orme, S.M.; Evanson, J.; Abid, N.; et al. Clinical experience in the screening and management of a large kindred with familial isolated pituitary adenoma due to an aryl hydrocarbon receptor interacting protein (AIP) mutation. J. Clin. Endocrinol. Metab. 2014, 99, 1122–1131. [Google Scholar] [CrossRef]
- Mangupli, R.; Rostomyan, L.; Castermans, E.; Caberg, J.H.; Camperos, P.; Krivoy, J.; Cuauro, E.; Bours, V.; Daly, A.F.; Beckers, A. Combined treatment with octreotide LAR and pegvisomant in patients with pituitary gigantism: Clinical evaluation and genetic screening. Pituitary 2016, 19, 507–514. [Google Scholar] [CrossRef]
- Ferraù, F.; Romeo, P.D.; Puglisi, S.; Ragonese, M.; Spagnolo, F.; Salpietro, C.; Ientile, R.; Currò, M.; Visalli, G.; Alibrandi, A.; et al. GSTP1 gene methylation and AHR rs2066853 variant predict resistance to first generation somatostatin analogs in patients with acromegaly. J. Endocrinol. Investig. 2019, 42, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Daly, A.F.; Rostomyan, L.; Betea, D.; Bonneville, J.F.; Villa, C.; Pellegata, N.S.; Waser, B.; Reubi, J.C.; Stephan, C.W.; Christ, E.; et al. Aip-mutated acromegaly resistant to first-generation somatostatin analogs: Long-term control with pasireotide lar in two patients. Endocr. Connect. 2019, 8, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, M.D.; Fleseriu, M.; Neggers, S.; Colao, A.; Sheppard, M.; Gu, F.; Shen, C.C.; Gadelha, M.; Farrall, A.J.; Reséndiz, K.H.; et al. Switching patients with acromegaly from octreotide to pasireotide improves biochemical control: Crossover extension to a randomized, double-blind, Phase III study. BMC Endocr. Disord. 2016, 16, 16. [Google Scholar] [CrossRef]
- Garcia-Rendueles, A.R.; Chenlo, M.; Oroz-Gonjar, F.; Solomou, A.; Mistry, A.; Barry, S.; Gaston-Massuet, C.; Garcia-Lavandeira, M.; Perez-Romero, S.; Suarez-Fariña, M.; et al. RET signalling provides tumorigenic mechanism and tissue specificity for AIP-related somatotrophinomas. Oncogene 2021, 40, 6354–6368. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.; Carlsen, E.; Marques, P.; Stiles, C.E.; Gadaleta, E.; Berney, D.M.; Roncaroli, F.; Chelala, C.; Solomou, A.; Herincs, M.; et al. Tumor microenvironment defines the invasive phenotype of AIP-mutation-positive pituitary tumors. Oncogene 2019, 38, 5381–5395. [Google Scholar] [CrossRef]
- Marques, P.; Caimari, F.; Hernández-Ramírez, L.C.; Collier, A.; Iacovazzo, D.; Ronaldson, A.; Magid, K.; Lim, C.T.; Stals, K.; Ellard, S.; et al. Significant benefits of AIP testing and clinical screening in familial isolated and young-onset pituitary tumors. J. Clin. Endocrinol. Metab. 2020, 105, 2247–2260. [Google Scholar] [CrossRef]
- Trivellin, G.; Daly, A.F.; Faucz, F.R.; Yuan, B.; Rostomyan, L.; Larco, D.O.; Schernthaner-Reiter, M.H.; Szarek, E.; Leal, L.F.; Caberg, J.-H.; et al. Gigantism and Acromegaly Due to Xq26 Microduplications and GPR101 Mutation. N. Engl. J. Med. 2014, 371, 2363–2374. [Google Scholar] [CrossRef]
- Trivellin, G.; Hernández-Ramírez, L.C.; Swan, J.; Stratakis, C.A. An orphan G-protein-coupled receptor causes human gigantism and/or acromegaly: Molecular biology and clinical correlations. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 125–140. [Google Scholar] [CrossRef]
- Daly, A.F.; Yuan, B.; Fina, F.; Caberg, J.H.; Trivellin, G.; Rostomyan, L.; De Herder, W.W.; Naves, L.A.; Metzger, D.; Cuny, T.; et al. Somatic mosaicism underlies X-linked acrogigantism syndrome in sporadic male subjects. Endocr. Relat. Cancer 2016, 23, 221–233. [Google Scholar] [CrossRef]
- Iacovazzo, D.; Caswell, R.; Bunce, B.; Jose, S.; Yuan, B.; Hernández-Ramírez, L.C.; Kapur, S.; Caimari, F.; Evanson, J.; Ferraù, F.; et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: A clinico-pathological and genetic study. Acta Neuropathol. Commun. 2016, 4, 56. [Google Scholar] [CrossRef]
- Daly, A.F.; Lysy, P.A.; Desfilles, C.; Rostomyan, L.; Mohamed, A.; Caberg, J.H.; Raverot, V.; Castermans, E.; Marbaix, E.; Maiter, D.; et al. GHRH excess and blockade in X-LAG syndrome. Endocr. Relat. Cancer 2016, 23, 161–170. [Google Scholar] [CrossRef]
- Beckers, A.; Lodish, M.B.; Trivellin, G.; Rostomyan, L.; Lee, M.; Faucz, F.R.; Yuan, B.; Choong, C.S.; Caberg, J.H.; Verrua, E.; et al. X-linked acrogigantism syndrome: Clinical profile and therapeutic responses. Endocr. Relat. Cancer 2015, 22, 353–367. [Google Scholar] [CrossRef]
- Alexander, S.P.H.; Christopoulos, A.; Davenport, A.P.; Kelly, E.; Mathie, A.; Peters, J.A.; Veale, E.L.; Armstrong, J.F.; Faccenda, E.; Harding, S.D.; et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: G protein-coupled receptors. Br. J. Pharmacol. 2019, 176, S21–S141. [Google Scholar] [CrossRef]
- Trivellin, G.; Bjelobaba, I.; Daly, A.F.; Larco, D.O.; Palmeira, L.; Faucz, F.R.; Thiry, A.; Leal, L.F.; Rostomyan, L.; Quezado, M.; et al. Characterization of GPR101 transcript structure and expression patterns. J. Mol. Endocrinol. 2016, 57, 97–111. [Google Scholar] [CrossRef]
- Trivellin, G.; Faucz, F.R.; Daly, A.F.; Beckers, A.; Stratakis, C.A. Hereditary endocrine tumours: Current state-of-the-art and research opportunities: GPR101, an orphan GPCR with roles in growth and pituitary tumorigenesis. Endocr. Relat. Cancer 2020, 27, T87–T97. [Google Scholar] [CrossRef]
- Martin, A.L.; Steurer, M.A.; Aronstam, R.S. Constitutive Activity among Orphan Class-A G Protein Coupled Receptors. PLoS ONE 2015, 10, e0138463. [Google Scholar] [CrossRef]
- Abboud, D.; Daly, A.F.; Dupuis, N.; Bahri, M.A.; Inoue, A.; Chevigné, A.; Ectors, F.; Plenevaux, A.; Pirotte, B.; Beckers, A.; et al. GPR101 drives growth hormone hypersecretion and gigantism in mice via constitutive activation of Gs and Gq/11. Nat. Commun. 2020, 11, 4752. [Google Scholar] [CrossRef]
- Cho-Clark, M.; Larco, D.O.; Zahn, B.R.; Mani, S.K.; Wu, T.J. GnRH-(1-5) activates matrix metallopeptidase-9 to release epidermal growth factor and promote cellular invasion. Mol. Cell. Endocrinol. 2015, 415, 114–125. [Google Scholar] [CrossRef]
- Vasilev, V.; Daly, A.F.; Trivellin, G.; Stratakis, C.A.; Zacharieva, S.; Beckers, A. Hereditary endocrine tumours: Current state-of-the-art and research opportunities: The roles of AIP and GPR101 in familial isolated pituitary adenomas (FIPA). Endocr. Relat. Cancer 2020, 27, T77–T86. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Cakir, M.; Grossman, A.B. Targeting MAPK (Ras/ERK) and PI3K/Akt pathways in pituitary tumorigenesis. Expert Opin. Ther. Targets 2009, 13, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.Z.; Liu, W.; Fan, H.T.; Liu, B.; Pang, B.; Xin, T.; Xu, S.C.; Pang, Q. Expression of hepatocyte growth factor and its receptor c-Met in human pituitary adenomas. Neuro. Oncol. 2010, 12, 799. [Google Scholar] [CrossRef] [PubMed]
- Casar-Borota, O.; Fougner, S.L.; Bollerslev, J.; Nesland, J.M. KIT protein expression and mutational status of KIT gene in pituitary adenomas. Virchows Arch. 2012, 460, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.; Onofri, C.; Colaco, T.; Stalla, G.K.; Renner, U. Platelet-derived growth factor (PDGF) and PDGF receptor expression and function in folliculostellate pituitary cells. Exp. Clin. Endocrinol. Diabetes 2010, 118, 113–120. [Google Scholar] [CrossRef]
- Monsalves, E.; Juraschka, K.; Tateno, T.; Agnihotri, S.; Asa, S.L.; Ezzat, S.; Zadeh, G. The PI3K/AKT/mTOR pathway in the pathophysiology and treatment of pituitary adenomas. Endocr. Relat. Cancer 2014, 21, R331–R344. [Google Scholar] [CrossRef]
- Lodge, E.J.; Santambrogio, A.; Russell, J.P.; Xekouki, P.; Jacques, T.S.; Johnson, R.L.; Thavaraj, S.; Bornstein, S.R.; Andoniadou, C.L. Homeostatic and tumourigenic activity of SOX2 + pituitary stem cells is controlled by the LATS/YAP/TAZ cascade. Elife 2019, 8, e43996. [Google Scholar] [CrossRef]
- Gaston-Massuet, C.; Andoniadou, C.L.; Signore, M.; Jayakody, S.A.; Charolidi, N.; Kyeyune, R.; Vernay, B.; Jacques, T.S.; Taketo, M.M.; Le Tissier, P.; et al. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 11482–11487. [Google Scholar] [CrossRef]
- Cox, B.; Roose, H.; Vennekens, A.; Vankelecom, H. Pituitary stem cell regulation: Who is pulling the strings? J. Endocrinol. 2017, 234, R135–R158. [Google Scholar] [CrossRef][Green Version]
- Gomes, D.C.; Jamra, S.A.; Leal, L.F.; Colli, L.M.; Campanini, M.L.; Oliveira, R.S.; Martinelli, C.E.; Moreira, A.C.; Machado, H.R.; Saggioro, F.; et al. Sonic Hedgehog pathway is upregulated in adamantinomatous craniopharyngiomas. Eur. J. Endocrinol. 2015, 172, 603–608. [Google Scholar] [CrossRef]
- Karga, H.J.; Alexander, J.M.; Hedley-Whyte, E.T.; Klibanski, A.; Jameson, J.L. Ras mutations in human pituitary tumors. J. Clin. Endocrinol. Metab. 1992, 74, 914–919. [Google Scholar] [CrossRef]
- Cai, W.Y.; Alexander, J.M.; Hedley-Whyte, E.T.; Scheithauer, B.W.; Jameson, J.L.; Zervas, N.T.; Klibanski, A. Ras mutations in human prolactinomas and pituitary carcinomas. J. Clin. Endocrinol. Metab. 1994, 78, 89–93. [Google Scholar] [CrossRef]
- Ewing, I.; Pedder-Smith, S.; Franchi, G.; Ruscica, M.; Emery, M.; Vax, V.; Garcia, E.; Czirják, S.; Hanzély, Z.; Kola, B.; et al. A mutation and expression analysis of the oncogene BRAF in pituitary adenomas. Clin. Endocrinol. 2007, 66, 348–352. [Google Scholar] [CrossRef]
- Dworakowska, D.; Wlodek, E.; Leontiou, C.A.; Igreja, S.; Cakir, M.; Teng, M.; Prodromou, N.; Góth, M.I.; Grozinsky-Glasberg, S.; Gueorguiev, M.; et al. Activation of RAF/MEK/ERK and PI3K/AKT/mTOR pathways in pituitary adenomas and their effects on downstream effectors. Endocr. Relat. Cancer 2009, 16, 1329–1338. [Google Scholar] [CrossRef]
- Chaturvedi, K.; Sarkar, D.K. Mediation of basic fibroblast growth factor-induced lactotropic cell proliferation by Src-Ras-mitogen-activated protein kinase p44/42 signaling. Endocrinology 2005, 146, 1948–1955. [Google Scholar] [CrossRef][Green Version]
- Oomizu, S.; Chaturvedi, K.; Sarkar, D.K. Folliculostellate cells determine the susceptibility of lactotropes to estradiol’s mitogenic action. Endocrinology 2004, 145, 1473–1480. [Google Scholar] [CrossRef]
- Booth, A.; Trudeau, T.; Gomez, C.; Scott Lucia, M.; Gutierrez-Hartmann, A. Persistent ERK/MAPK activation promotes lactotrope differentiation and diminishes tumorigenic phenotype. Mol. Endocrinol. 2014, 28, 1999–2011. [Google Scholar] [CrossRef]
- Woodmansee, W.W.; Kerr, J.M.; Tucker, E.A.; Mitchell, J.R.; Haakinson, D.J.; Gordon, D.F.; Ridgway, E.C.; Wood, W.M. The proliferative status of thyrotropes is dependent on modulation of specific cell cycle regulators by thyroid hormone. Endocrinology 2006, 147, 272–282. [Google Scholar] [CrossRef]
- Pombo, C.M.; Zalvide, J.; Gaylinn, B.D.; Diéguez, C. Growth hormone-releasing hormone stimulates mitogen-activated protein kinase. Endocrinology 2000, 141, 2113–2119. [Google Scholar] [CrossRef]
- Lania, A.; Filopanti, M.; Corbetta, S.; Losa, M.; Ballaré, E.; Beck-Peccoz, P.; Spada, A. Effects of hypothalamic neuropeptides on extracellular signal-regulated kinase (ERK1 and ERK2) cascade in human tumoral pituitary cells. J. Clin. Endocrinol. Metab. 2003, 88, 1692–1696. [Google Scholar] [CrossRef]
- Cuny, T.; Gerard, C.; Saveanu, A.; Barlier, A.; Enjalbert, A. Physiopathology of somatolactotroph cells: From transduction mechanisms to cotargeting therapy. Ann. N. Y. Acad. Sci. 2011, 1220, 60–70. [Google Scholar] [CrossRef]
- Hubina, E.; Nanzer, A.M.; Hanson, M.R.; Ciccarelli, E.; Losa, M.; Gaia, D.; Papotti, M.; Terreni, M.R.; Khalaf, S.; Jordan, S.; et al. Somatostatin analogues stimulate p27 expression and inhibit the MAP kinase pathway in pituitary tumours. Eur. J. Endocrinol. 2006, 155, 371–379. [Google Scholar] [CrossRef]
- Liu, F.; Austin, D.A.; Mellon, P.L.; Olefsky, J.M.; Webster, N.J.G. GnRH activates ERK1/2 leading to the induction of c-fos and LHbeta protein expression in LbetaT2 cells. Mol. Endocrinol. 2002, 16, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Rubinfeld, H.; Shimon, I. PI3K/Akt/mTOR and Raf/MEK/ERK signaling pathways perturbations in non-functioning pituitary adenomas. Endocrine 2012, 42, 285–291. [Google Scholar] [CrossRef]
- Zhang, D.; Bergsneider, M.; Wang, M.B.; Heaney, A.P. Targeting the ERK pathway for the treatment of Cushing’s disease. Oncotarget 2016, 7, 69149–69158. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Taylor-Weiner, A.; Manley, P.E.; Jones, R.T.; Dias-Santagata, D.; Thorner, A.R.; Lawrence, M.S.; Rodriguez, F.J.; Bernardo, L.A.; Schubert, L.; et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat. Genet. 2014, 46, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Larkin, S.J.; Preda, V.; Karavitaki, N.; Grossman, A.; Ansorge, O. BRAF V600E mutations are characteristic for papillary craniopharyngioma and may coexist with CTNNB1-mutated adamantinomatous craniopharyngioma. Acta Neuropathol. 2014, 127, 927–929. [Google Scholar] [CrossRef]
- Haston, S.; Pozzi, S.; Carreno, G.; Manshaei, S.; Panousopoulos, L.; Gonzalez-Meljem, J.M.; Apps, J.R.; Virasami, A.; Thavaraj, S.; Gutteridge, A.; et al. MAPK pathway control of stem cell proliferation and differentiation in the embryonic pituitary provides insights into the pathogenesis of papillary craniopharyngioma. Development 2017, 144, 2141–2152. [Google Scholar] [CrossRef]
- Aylwin, S.J.B.; Bodi, I.; Beaney, R. Pronounced response of papillary craniopharyngioma to treatment with vemurafenib, a BRAF inhibitor. Pituitary 2016, 19, 544–546. [Google Scholar] [CrossRef]
- Chik, C.L.; Van Landeghem, F.K.H.; Easaw, J.C.; Mehta, V. Aggressive Childhood-onset Papillary Craniopharyngioma Managed With Vemurafenib, a BRAF Inhibitor. J. Endocr. Soc. 2021, 5, bvab043. [Google Scholar] [CrossRef]
- Himes, B.T.; Ruff, M.W.; Van Gompel, J.J.; Park, S.S.; Galanis, E.; Kaufmann, T.J.; Uhm, J.H. Recurrent papillary craniopharyngioma with BRAF V600E mutation treated with dabrafenib: Case report. J. Neurosurg. 2018, 130, 1299–1303. [Google Scholar] [CrossRef]
- Rao, M.; Bhattacharjee, M.; Shepard, S.; Hsu, S. Newly diagnosed papillary craniopharyngioma with BRAF V600E mutation treated with single-agent selective BRAF inhibitor dabrafenib: A case report. Oncotarget 2019, 10, 6038–6042. [Google Scholar] [CrossRef]
- Bernstein, A.; Mrowczynski, O.D.; Greene, A.; Ryan, S.; Chung, C.; Zacharia, B.E.; Glantz, M. Dual BRAF/MEK therapy in BRAF V600E-mutated primary brain tumors: A case series showing dramatic clinical and radiographic responses and a reduction in cutaneous toxicity. J. Neurosurg. 2019, 133, 1704–1709. [Google Scholar] [CrossRef]
- Khaddour, K.; Chicoine, M.R.; Huang, J.; Dahiya, S.; Ansstas, G. Successful Use of BRAF/MEK Inhibitors as a Neoadjuvant Approach in the Definitive Treatment of Papillary Craniopharyngioma. J. Natl. Compr. Cancer Netw. 2020, 18, 1590–1595. [Google Scholar] [CrossRef]
- Di Stefano, A.L.; Guyon, D.; Sejean, K.; Feuvret, L.; Villa, C.; Berzero, G.; Desforges Bullet, V.; Halimi, E.; Boulin, A.; Baussart, B.; et al. Medical debulking with BRAF/MEK inhibitors in aggressive BRAF-mutant craniopharyngioma. Neuro-Oncol. Adv. 2020, 2, vdaa141. [Google Scholar] [CrossRef]
- Calvanese, F.; Jacquesson, T.; Manet, R.; Vasiljevic, A.; Lasolle, H.; Ducray, F.; Raverot, G.; Jouanneau, E. Neoadjuvant B-RAF and MEK Inhibitor Targeted Therapy for Adult Papillary Craniopharyngiomas: A New Treatment Paradigm. Front. Endocrinol. 2022, 13, 882381. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Twohy, E.; Geyer, S.M.; Gerstner, E.R.; Kaufmann, T.J.; Ruff, M.; Bota, D.A.; Reardon, D.A.; Cohen, A.L.; La Fuente, M.I.D.; et al. Alliance A071601: Phase II trial of BRAF/MEK inhibition in newly diagnosed papillary craniopharyngiomas. J. Clin. Oncol. 2021, 39, 2000. [Google Scholar] [CrossRef]
- Stork, P.J.S.; Schmitt, J.M. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002, 12, 258–266. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Pergameno, M.; Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: Role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012, 16 (Suppl. 2), S17–S27. [Google Scholar] [CrossRef]
- Carracedo, A.; Pandolfi, P.P. The PTEN-PI3K pathway: Of feedbacks and cross-talks. Oncogene 2008, 27, 5527–5541. [Google Scholar] [CrossRef]
- Chen, R.; Duan, J.; Li, L.; Ma, Q.; Sun, Q.; Ma, J.; Li, C.; Zhou, X.; Chen, H.; Jing, Y.; et al. mTOR promotes pituitary tumor development through activation of PTTG1. Oncogene 2017, 36, 979–988. [Google Scholar] [CrossRef]
- Lin, S.J.; Leng, Z.G.; Guo, Y.H.; Cai, L.; Cai, Y.; Li, N.; Shang, H.B.; Le, W.D.; Zhao, W.G.; Wu, Z.B. Suppression of mTOR pathway and induction of autophagy-dependent cell death by cabergoline. Oncotarget 2015, 6, 39329–39341. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Willingham, M.C.; Furuya, F.; Cheng, S.Y. Activation of phosphatidylinositol 3-kinase signaling promotes aberrant pituitary growth in a mouse model of thyroid-stimulating hormone-secreting pituitary tumors. Endocrinology 2008, 149, 3339–3345. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Labeur, M.; Wölfel, B.; Stalla, J.; Stalla, G.K. TMEFF2 is an endogenous inhibitor of the CRH signal transduction pathway. J. Mol. Endocrinol. 2015, 54, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Muşat, M.; Korbonits, M.; Kola, B.; Borboli, N.; Hanson, M.R.; Nanzer, A.M.; Grigson, J.; Jordan, S.; Morris, D.G.; Gueorguiev, M.; et al. Enhanced protein kinase B/Akt signalling in pituitary tumours. Endocr. Relat. Cancer 2005, 12, 423–433. [Google Scholar] [CrossRef]
- Sajjad, E.A.; Zieliński, G.; Maksymowicz, M.; Hutnik, Ł.; Bednarczuk, T.; Włodarski, P. mTOR is frequently active in GH-secreting pituitary adenomas without influencing their morphopathological features. Endocr. Pathol. 2013, 24, 11–19. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, X.; Shen, Y.; Li, M.; Ma, H.; Xing, M.; Lu, Y. Frequent mutations and amplifications of the PIK3CA gene in pituitary tumors. Endocr. Relat. Cancer 2009, 16, 301–310. [Google Scholar] [CrossRef]
- Murat, C.B.; Braga, P.B.S.; Fortes, M.A.H.Z.; Bronstein, M.D.; Corrêa-Giannella, M.L.C.; Giorgi, R.R. Mutation and genomic amplification of the PIK3CA proto-oncogene in pituitary adenomas. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Med. Biol. 2012, 45, 851–855. [Google Scholar] [CrossRef]
- Reubi, J.C.; Laissue, J.A. Multiple actions of somatostatin in neoplastic disease. Trends Pharmacol. Sci. 1995, 16, 110–115. [Google Scholar] [CrossRef]
- Bruns, C.; Lewis, I.; Briner, U.; Meno-Tetang, G.; Weckbecker, G. SOM230: A novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur. J. Endocrinol. 2002, 146, 707–716. [Google Scholar] [CrossRef]
- Weckbecker, G.; Briner, U.; Lewis, I.; Bruns, C. SOM230: A new somatostatin peptidomimetic with potent inhibitory effects on the growth hormone/insulin-like growth factor-I axis in rats, primates, and dogs. Endocrinology 2002, 143, 4123–4130. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Zhang, J.; Laupheimer, S.; Paez-Pereda, M.; Erneux, C.; Florio, T.; Pagotto, U.; Stalla, G.K. Octreotide, a somatostatin analogue, mediates its antiproliferative action in pituitary tumor cells by altering phosphatidylinositol 3-kinase signaling and inducing Zac1 expression. Cancer Res. 2006, 66, 1576–1582. [Google Scholar] [CrossRef]
- Liu, J.C.; Baker, R.E.; Sun, C.; Sundmark, V.C.; Elsholtz, H.P. Activation of Go-coupled dopamine D2 receptors inhibits ERK1/ERK2 in pituitary cells. A key step in the transcriptional suppression of the prolactin gene. J. Biol. Chem. 2002, 277, 35819–35825. [Google Scholar] [CrossRef]
- Iaccarino, C.; Samad, T.A.; Mathis, C.; Kercret, H.; Picetti, R.; Borrelli, E. Control of lactotrop proliferation by dopamine: Essential role of signaling through D2 receptors and ERKs. Proc. Natl. Acad. Sci. USA 2002, 99, 14530–14535. [Google Scholar] [CrossRef]
- Radl, D.; De Mei, C.; Chen, E.; Lee, H.; Borrelli, E. Each individual isoform of the dopamine D2 receptor protects from lactotroph hyperplasia. Mol. Endocrinol. 2013, 27, 953–965. [Google Scholar] [CrossRef]
- Rocheville, M.; Lange, D.C.; Kumar, U.; Patel, S.C.; Patel, R.C.; Patel, Y.C. Receptors for dopamine and somatostatin: Formation of hetero-oligomers with enhanced functional activity. Science 2000, 288, 154–157. [Google Scholar] [CrossRef]
- Saveanu, A.; Lavaque, E.; Gunz, G.; Barlier, A.; Kim, S.; Taylor, J.E.; Culler, M.D.; Enjalbert, A.; Jaquet, P. Demonstration of enhanced potency of a chimeric somatostatin-dopamine molecule, BIM-23A387, in suppressing growth hormone and prolactin secretion from human pituitary somatotroph adenoma cells. J. Clin. Endocrinol. Metab. 2002, 87, 5545–5552. [Google Scholar] [CrossRef]
- Jaquet, P.; Gunz, G.; Saveanu, A.; Dufour, H.; Taylor, J.; Dong, J.; Kim, S.; Moreau, J.P.; Enjalbert, A.; Culler, M.D. Efficacy of chimeric molecules directed towards multiple somatostatin and dopamine receptors on inhibition of GH and prolactin secretion from GH-secreting pituitary adenomas classified as partially responsive to somatostatin analog therapy. Eur. J. Endocrinol. 2005, 153, 135–141. [Google Scholar] [CrossRef]
- Culler, M.D. Somatostatin-dopamine chimeras: A novel approach to treatment of neuroendocrine tumors. Horm. Metab. Res. 2011, 43, 854–857. [Google Scholar] [CrossRef]
- Vázquez-Borrego, M.C.; L-López, F.; Gálvez-Moreno, M.A.; Fuentes-Fayos, A.C.; Venegas-Moreno, E.; Herrera-Martínez, A.D.; Blanco-Acevedo, C.; Solivera, J.; Landsman, T.; Gahete, M.D.; et al. A New Generation Somatostatin-Dopamine Analogue Exerts Potent Antitumoral Actions on Pituitary Neuroendocrine Tumor Cells. Neuroendocrinology 2020, 110, 70–82. [Google Scholar] [CrossRef]
- Kim, J.; Oh, J.H.; Harlem, H.; Culler, M.D.; Ku, C.R.; Lee, E.J. Therapeutic Effect of a Novel Chimeric Molecule Targeting Both Somatostatin and Dopamine Receptors on Growth Hormone-Secreting Pituitary Adenomas. Endocrinol. Metab. 2020, 35, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.D.; Lasolle, H.; Raverot, G. Emerging and Novel Treatments for Pituitary Tumors. J. Clin. Med. 2019, 8, 1107. [Google Scholar] [CrossRef]
- Beuvink, I.; Boulay, A.; Fumagalli, S.; Zilbermann, F.; Ruetz, S.; O’Reilly, T.; Natt, F.; Hall, J.; Lane, H.A.; Thomas, G. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell 2005, 120, 747–759. [Google Scholar] [CrossRef]
- Gorshtein, A.; Rubinfeld, H.; Kendler, E.; Theodoropoulou, M.; Cerovac, V.; Stalla, G.K.; Cohen, Z.R.; Hadani, M.; Shimon, I. Mammalian target of rapamycin inhibitors rapamycin and RAD001 (everolimus) induce anti-proliferative effects in GH-secreting pituitary tumor cells in vitro. Endocr. Relat. Cancer 2009, 16, 1017–1027. [Google Scholar] [CrossRef]
- Zatelli, M.C.; Minoia, M.; Filieri, C.; Tagliati, F.; Buratto, M.; Ambrosio, M.R.; Lapparelli, M.; Scanarini, M.; Degli Uberti, E.C. Effect of everolimus on cell viability in nonfunctioning pituitary adenomas. J. Clin. Endocrinol. Metab. 2010, 95, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Cerovac, V.; Monteserin-Garcia, J.; Rubinfeld, H.; Buchfelder, M.; Losa, M.; Florio, T.; Paez-Pereda, M.; Stalla, G.K.; Theodoropoulou, M. The somatostatin analogue octreotide confers sensitivity to rapamycin treatment on pituitary tumor cells. Cancer Res. 2010, 70, 666–674. [Google Scholar] [CrossRef]
- Chanal, M.; Chevallier, P.; Raverot, V.; Fonteneau, G.; Lucia, K.; Garcia, J.L.M.; Rachwan, A.; Jouanneau, E.; Trouillas, J.; Honnorat, J.; et al. Differential Effects of PI3K and Dual PI3K/mTOR Inhibition in Rat Prolactin-Secreting Pituitary Tumors. Mol. Cancer Ther. 2016, 15, 1261–1270. [Google Scholar] [CrossRef]
- Pivonello, C.; Patalano, R.; Solari, D.; Auriemma, R.S.; Frio, F.; Vitulli, F.; Grasso, L.F.S.; Di Cera, M.; De Martino, M.C.; Cavallo, L.M.; et al. Effect of combined treatment with a pan-PI3K inhibitor or an isoform-specific PI3K inhibitor and everolimus on cell proliferation in GH-secreting pituitary tumour in an experimental setting. Endocrine 2018, 62, 663–680. [Google Scholar] [CrossRef]
- Dai, C.; Zhang, B.; Liu, X.; Ma, S.; Yang, Y.; Yao, Y.; Feng, M.; Bao, X.; Li, G.; Wang, J.; et al. Inhibition of PI3K/AKT/mTOR pathway enhances temozolomide-induced cytotoxicity in pituitary adenoma cell lines in vitro and xenografted pituitary adenoma in female nude mice. Endocrinology 2013, 154, 1247–1259. [Google Scholar] [CrossRef][Green Version]
- Mishra, R.; Patel, H.; Alanazi, S.; Kilroy, M.K.; Garrett, J.T. PI3K Inhibitors in Cancer: Clinical Implications and Adverse Effects. Int. J. Mol. Sci. 2021, 22, 3464. [Google Scholar] [CrossRef]
- Ben-Shlomo, A.; Cooper, O. Role of tyrosine kinase inhibitors in the treatment of pituitary tumours: From bench to bedside. Curr. Opin. Endocrinol. Diabetes Obes. 2017, 24, 301–305. [Google Scholar] [CrossRef]
- Asari, Y.; Kageyama, K.; Sugiyama, A.; Kogawa, H.; Niioka, K.; Daimon, M. Lapatinib decreases the ACTH production and proliferation of corticotroph tumor cells. Endocr. J. 2019, 66, 515–522. [Google Scholar] [CrossRef]
- Fukuoka, H.; Cooper, O.; Mizutani, J.; Tong, Y.; Ren, S.G.; Bannykh, S.; Melmed, S. HER2/ErbB2 receptor signaling in rat and human prolactinoma cells: Strategy for targeted prolactinoma therapy. Mol. Endocrinol. 2011, 25, 92–103. [Google Scholar] [CrossRef]
- Cooper, O.; Mamelak, A.; Bannykh, S.; Carmichael, J.; Bonert, V.; Lim, S.; Cook-Wiens, G.; Ben-Shlomo, A. Prolactinoma ErbB receptor expression and targeted therapy for aggressive tumors. Endocrine 2014, 46, 318–327. [Google Scholar] [CrossRef]
- Cooper, O.; Bonert, V.S.; Rudnick, J.; Pressman, B.D.; Lo, J.; Salvatori, R.; Yuen, K.C.J.; Fleseriu, M.; Melmed, S. EGFR/ErbB2-Targeting Lapatinib Therapy for Aggressive Prolactinomas. J. Clin. Endocrinol. Metab. 2021, 106, E917–E925. [Google Scholar] [CrossRef] [PubMed]
- Naviglio, S.; Matteucci, C.; Matoskova, B.; Nagase, T.; Nomura, N.; Di Fiore, P.P.; Draetta, G.F. UBPY: A growth-regulated human ubiquitin isopeptidase. EMBO J. 1998, 17, 3241–3250. [Google Scholar] [CrossRef]
- Islam, M.T.; Chen, F.; Chen, H. The oncogenic role of ubiquitin specific peptidase (USP8) and its signaling pathways targeting for cancer therapeutics. Arch. Biochem. Biophys. 2021, 701, 108811. [Google Scholar] [CrossRef]
- Reincke, M.; Sbiera, S.; Hayakawa, A.; Theodoropoulou, M.; Osswald, A.; Beuschlein, F.; Meitinger, T.; Mizuno-Yamasaki, E.; Kawaguchi, K.; Saeki, Y.; et al. Mutations in the deubiquitinase gene USP8 cause Cushing’s disease. Nat. Genet. 2015, 47, 31–38. [Google Scholar] [CrossRef]
- Theodoropoulou, M.; Reincke, M.; Fassnacht, M.; Komada, M. Decoding the genetic basis of Cushing’s disease: USP8 in the spotlight. Eur. J. Endocrinol. 2015, 173, M73–M83. [Google Scholar] [CrossRef]
- Perez-Rivas, L.G.; Theodoropoulou, M.; Ferraù, F.; Nusser, C.; Kawaguchi, K.; Stratakis, C.A.; Rueda Faucz, F.; Wildemberg, L.E.; Assié, G.; Beschorner, R.; et al. The Gene of the Ubiquitin-Specific Protease 8 Is Frequently Mutated in Adenomas Causing Cushing’s Disease. J. Clin. Endocrinol. Metab. 2015, 100, E997–E1004. [Google Scholar] [CrossRef] [PubMed]
- Albani, A.; Pérez-Rivas, L.G.; Dimopoulou, C.; Zopp, S.; Colón-Bolea, P.; Roeber, S.; Honegger, J.; Flitsch, J.; Rachinger, W.; Buchfelder, M.; et al. The USP8 mutational status may predict long-term remission in patients with Cushing’s disease. Clin. Endocrinol. 2018, 89, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Inoshita, N.; Kawaguchi, K.; Ardisasmita, A.I.; Suzuki, H.; Fukuhara, N.; Okada, M.; Nishioka, H.; Takeuchi, Y.; Komada, M.; et al. The USP8 mutational status may predict drug susceptibility in corticotroph adenomas of Cushing’s disease. Eur. J. Endocrinol. 2016, 174, 213–226. [Google Scholar] [CrossRef]
- Ma, Z.Y.; Song, Z.J.; Chen, J.H.; Wang, Y.F.; Li, S.Q.; Zhou, L.F.; Mao, Y.; Li, Y.M.; Hu, R.G.; Zhang, Z.Y.; et al. Recurrent gain-of-function USP8 mutations in Cushing’s disease. Cell Res. 2015, 25, 306–317. [Google Scholar] [CrossRef]
- Pérez-Rivas, L.G.; Theodoropoulou, M.; Puar, T.H.; Fazel, J.; Stieg, M.R.; Ferraù, F.; Assié, G.; Gadelha, M.R.; Deutschbein, T.; Fragoso, M.C.; et al. Somatic USP8 mutations are frequent events in corticotroph tumor progression causing Nelson’s tumor. Eur. J. Endocrinol. 2018, 178, 57–63. [Google Scholar] [CrossRef]
- Cohen, M.; Persky, R.; Stegemann, R.; Hernández-Ramírez, L.C.; Zeltser, D.; Lodish, M.B.; Chen, A.; Keil, M.F.; Tatsi, C.; Faucz, F.R.; et al. Germline USP8 Mutation Associated With Pediatric Cushing Disease and Other Clinical Features: A New Syndrome. J. Clin. Endocrinol. Metab. 2019, 104, 4676–4682. [Google Scholar] [CrossRef]
- Kageyama, K.; Asari, Y.; Sugimoto, Y.; Niioka, K.; Daimon, M. Ubiquitin-specific protease 8 inhibitor suppresses adrenocorticotropic hormone production and corticotroph tumor cell proliferation. Endocr. J. 2020, 67, 177–184. [Google Scholar] [CrossRef]
- Treppiedi, D.; Di Muro, G.; Marra, G.; Barbieri, A.M.; Mangili, F.; Catalano, R.; Serban, A.; Ferrante, E.; Locatelli, M.; Lania, A.G.; et al. USP8 inhibitor RA-9 reduces ACTH release and cell growth in tumor corticotrophs. Endocr. Relat. Cancer 2021, 28, 573–582. [Google Scholar] [CrossRef]
- Albani, A.; Perez-Rivas, L.G.; Tang, S.; Simon, J.; Lucia, K.E.; Colón Bolea, P.; Schopohl, J.; Roeber, S.; Buchfelder, M.; Rotermund, R.; et al. Improved pasireotide response in USP8 mutant corticotroph tumours in vitro. Endocr. Relat. Cancer 2022, 29, 503–511. [Google Scholar] [CrossRef]
- Treppiedi, D.; Marra, G.; Di Muro, G.; Esposito, E.; Barbieri, A.M.; Catalano, R.; Mangili, F.; Bravi, F.; Locatelli, M.; Lania, A.G.; et al. P720R USP8 Mutation Is Associated with a Better Responsiveness to Pasireotide in ACTH-Secreting PitNETs. Cancers 2022, 14, 2455. [Google Scholar] [CrossRef]
- Chen, J.; Jian, X.; Deng, S.; Ma, Z.; Shou, X.; Shen, Y.; Zhang, Q.; Song, Z.; Li, Z.; Peng, H.; et al. Identification of recurrent USP48 and BRAF mutations in Cushing’s disease. Nat. Commun. 2018, 9, 3171. [Google Scholar] [CrossRef] [PubMed]
- Sbiera, S.; Perez-Rivas, L.G.; Taranets, L.; Weigand, I.; Flitsch, J.; Graf, E.; Monoranu, C.M.; Saeger, W.; Hagel, C.; Honegger, J.; et al. Driver mutations in USP8 wild-type Cushing’s disease. Neuro-Oncol. 2019, 21, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Lodge, E.J.; Russell, J.P.; Patist, A.L.; Francis-West, P.; Andoniadou, C.L. Expression analysis of the Hippo cascade indicates a role in pituitary stem cell development. Front. Physiol. 2016, 7, 114. [Google Scholar] [CrossRef]
- Zheng, Y.; Pan, D. The Hippo Signaling Pathway in Development and Disease. Dev. Cell 2019, 50, 264–282. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef]
- Basu-Roy, U.; Bayin, N.S.; Rattanakorn, K.; Han, E.; Placantonakis, D.G.; Mansukhani, A.; Basilico, C. Sox2 antagonizes the Hippo pathway to maintain stemness in cancer cells. Nat. Commun. 2015, 6, 6411. [Google Scholar] [CrossRef]
- St John, M.A.R.; Tao, W.; Fei, X.; Fukumoto, R.; Carcangiu, M.L.; Brownstein, D.G.; Parlow, A.F.; McGrath, J.; Xu, T. Mice deficient of Lats1 develop soft-tissue sarcomas, ovarian tumours and pituitary dysfunction. Nat. Genet. 1999, 21, 182–186. [Google Scholar] [CrossRef]
- Lalonde-Larue, A.; Boyer, A.; Dos Santos, E.C.; Boerboom, D.; Bernard, D.J.; Zamberlam, G. The Hippo Pathway Effectors YAP and TAZ Regulate LH Release by Pituitary Gonadotrope Cells in Mice. Endocrinology 2022, 163, bqab238. [Google Scholar] [CrossRef]
- Lee, T.F.; Tseng, Y.C.; Nguyen, P.A.; Li, Y.C.; Ho, C.C.; Wu, C.W. Enhanced YAP expression leads to EGFR TKI resistance in lung adenocarcinomas. Sci. Rep. 2018, 8, 271. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, S.; Asahina, H.; Chen, T.; Guo, S.; Quiceno, L.G.; Cavanaugh, J.D.; Merlino, A.A.; Tange, S.; Terai, H.; Kim, J.W.; et al. Overcoming Resistance to Dual Innate Immune and MEK Inhibition Downstream of KRAS. Cancer Cell 2018, 34, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.S.; Xia, H.W.; Zhou, S.; Liu, Q.; Tang, Q.L.; Bi, N.X.; Zhou, J.T.; Gong, Q.Y.; Nie, Y.Z.; Bi, F. Inhibition of YAP reverses primary resistance to EGFR inhibitors in colorectal cancer cells. Oncol. Rep. 2018, 40, 2171–2182. [Google Scholar] [CrossRef]
- Nakatani, K.; Maehama, T.; Nishio, M.; Goto, H.; Kato, W.; Omori, H.; Miyachi, Y.; Togashi, H.; Shimono, Y.; Suzuki, A. JB Review Targeting the Hippo signalling pathway for cancer treatment. J. Biochem. 2017, 161, 237–244. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Ciamporcero, E.; Shen, H.; Ramakrishnan, S.; Yu Ku, S.; Chintala, S.; Shen, L.; Adelaiye, R.; Miles, K.M.; Ullio, C.; Pizzimenti, S.; et al. YAP activation protects urothelial cell carcinoma from treatment-induced DNA damage. Oncogene 2016, 35, 1541–1553. [Google Scholar] [CrossRef]
- Song, S.; Ajani, J.A.; Honjo, S.; Maru, D.M.; Chen, Q.; Scott, A.W.; Heallen, T.R.; Xiao, L.; Hofstetter, W.L.; Weston, B.; et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014, 74, 4170–4182. [Google Scholar] [CrossRef]
- Dey, A.; Varelas, X.; Guan, K.L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef]
- Picozzi, V.; Alseidi, A.; Winter, J.; Pishvaian, M.; Mody, K.; Glaspy, J.; Larson, T.; Matrana, M.; Carney, M.; Porter, S.; et al. Gemcitabine/nab-paclitaxel with pamrevlumab: A novel drug combination and trial design for the treatment of locally advanced pancreatic cancer. ESMO Open 2020, 5, e000668. [Google Scholar] [CrossRef]
- Iglesias, P. Targeted therapies in the medical management of craniopharyngioma. Pituitary 2022, 25, 383–392. [Google Scholar] [CrossRef]
- Miyakoshi, T.; Takei, M.; Kajiya, H.; Egashira, N.; Takekoshi, S.; Teramoto, A.; Osamura, R.Y. Expression of Wnt4 in human pituitary adenomas regulates activation of the beta-catenin-independent pathway. Endocr. Pathol. 2008, 19, 261–273. [Google Scholar] [CrossRef]
- Formosa, R.; Gruppetta, M.; Falzon, S.; Santillo, G.; DeGaetano, J.; Xuereb-Anastasi, A.; Vassallo, J. Expression and clinical significance of Wnt players and survivin in pituitary tumours. Endocr. Pathol. 2012, 23, 123–131. [Google Scholar] [CrossRef]
- Semba, S.; Han, S.-Y.; Ikeda, H.; Horii, A. Frequent Nuclear Accumulation of-Catenin in Pituitary Adenoma. Cancer 2001, 91, 42–48. [Google Scholar] [CrossRef]
- Demarchi, G.; Valla, S.; Perrone, S.; Chimento, A.; Bonadeo, N.; Vitale, D.L.; Spinelli, F.M.; Cervio, A.; Sevlever, G.; Alaniz, L.; et al. β-Catenin is reduced in membranes of human prolactinoma cells and it is inhibited by temozolomide in prolactin secreting tumor models. Tumour Biol. 2022, 44, 85–105. [Google Scholar] [CrossRef]
- Elston, M.S.; Gill, A.J.; Conaglen, J.V.; Clarkson, A.; Shaw, J.M.; Law, A.J.J.; Cook, R.J.; Little, N.S.; Clifton-Bligh, R.J.; Robinson, B.G.; et al. Wnt pathway inhibitors are strongly down-regulated in pituitary tumors. Endocrinology 2008, 149, 1235–1242. [Google Scholar] [CrossRef]
- Sekine, S.; Shibata, T.; Kokubu, A.; Morishita, Y.; Noguchi, M.; Nakanishi, Y.; Sakamoto, M.; Hirohashi, S. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am. J. Pathol. 2002, 161, 1997–2001. [Google Scholar] [CrossRef]
- Hofmann, B.M.; Kreutzer, J.; Saeger, W.; Buchfelder, M.; Blümcke, I.; Fahlbusch, R.; Buslei, R. Nuclear beta-catenin accumulation as reliable marker for the differentiation between cystic craniopharyngiomas and rathke cleft cysts: A clinico-pathologic approach. Am. J. Surg. Pathol. 2006, 30, 1595–1603. [Google Scholar] [CrossRef]
- Campanini, M.L.; Colli, L.M.; Paixao, B.M.C.; Cabral, T.P.F.; Amaral, F.C.; Machado, H.R.; Neder, L.S.; Saggioro, F.; Moreira, A.C.; Antonini, S.R.R.; et al. CTNNB1 gene mutations, pituitary transcription factors, and MicroRNA expression involvement in the pathogenesis of adamantinomatous craniopharyngiomas. Horm. Cancer 2010, 1, 187–196. [Google Scholar] [CrossRef]
- Cani, C.M.G.; Matushita, H.; Carvalho, L.R.S.; Soares, I.C.; Brito, L.P.; Almeida, M.Q.; Mendonça, B.B. PROP1 and CTNNB1 expression in adamantinomatous craniopharyngiomas with or without β-catenin mutations. Clinics 2011, 66, 1849–1854. [Google Scholar] [CrossRef] [PubMed]
- Jucá, C.E.B.; Colli, L.M.; Martins, C.S.; Campanini, M.L.; Paixão, B.; Jucá, R.V.; Saggioro, F.P.; De Oliveira, R.S.; Moreira, A.C.; Machado, H.R.; et al. Impact of the Canonical Wnt Pathway Activation on the Pathogenesis and Prognosis of Adamantinomatous Craniopharyngiomas. Horm. Metab. Res. 2018, 50, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Andoniadou, C.L.; Gaston-Massuet, C.; Reddy, R.; Schneider, R.P.; Blasco, M.A.; Le Tissier, P.; Jacques, T.S.; Pevny, L.H.; Dattani, M.T.; Martinez-Barbera, J.P. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol. 2012, 124, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Boult, J.K.R.; Apps, J.R.; Hölsken, A.; Hutchinson, J.C.; Carreno, G.; Danielson, L.S.; Smith, L.M.; Bäuerle, T.; Buslei, R.; Buchfelder, M.; et al. Preclinical transgenic and patient-derived xenograft models recapitulate the radiological features of human adamantinomatous craniopharyngioma. Brain Pathol. 2018, 28, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Lee, K.J.; Chang, W.C.L.; Chen, X.; Valiyaveettil, J.; Ramirez-Alcantara, V.; Gavin, E.; Musiyenko, A.; Da Silva, L.M.; Annamdevula, N.S.; Leavesley, S.J.; et al. Suppression of Colon Tumorigenesis in Mutant Apc Mice by a Novel PDE10 Inhibitor that Reduces Oncogenic β-Catenin. Cancer Prev. Res. 2021, 14, 995–1008. [Google Scholar] [CrossRef]
- Hu, J.; Wang, Z.; Chen, J.; Yu, Z.; Zhang, J.; Li, W.; Lin, M.; Yang, X.; Liu, H. Overexpression of ICAT Inhibits the Progression of Colorectal Cancer by Binding with β-Catenin in the Cytoplasm. Technol. Cancer Res. Treat. 2021, 20, 15330338211041253. [Google Scholar] [CrossRef]
- Grob, S.; Mirsky, D.M.; Donson, A.M.; Dahl, N.; Foreman, N.K.; Hoffman, L.M.; Hankinson, T.C.; Levy, J.M.M. Targeting IL-6 Is a Potential Treatment for Primary Cystic Craniopharyngioma. Front. Oncol. 2019, 9, 791. [Google Scholar] [CrossRef]
- Carreno, G.; Boult, J.K.R.; Apps, J.; Gonzalez-Meljem, J.M.; Haston, S.; Guiho, R.; Stache, C.; Danielson, L.S.; Koers, A.; Smith, L.M.; et al. SHH pathway inhibition is protumourigenic in adamantinomatous craniopharyngioma. Endocr. Relat. Cancer 2019, 26, 355–366. [Google Scholar] [CrossRef]
- Alexandraki, K.I.; Xekouki, P. Medical Therapy for Craniopharyngiomas. TouchREVIEWS Endocrinol. 2021, 17, 121–132. [Google Scholar] [CrossRef]
- Apps, J.R.; Carreno, G.; Gonzalez-Meljem, J.M.; Haston, S.; Guiho, R.; Cooper, J.E.; Manshaei, S.; Jani, N.; Hölsken, A.; Pettorini, B.; et al. Tumour compartment transcriptomics demonstrates the activation of inflammatory and odontogenic programmes in human adamantinomatous craniopharyngioma and identifies the MAPK/ERK pathway as a novel therapeutic target. Acta Neuropathol. 2018, 135, 757–777. [Google Scholar] [CrossRef]
- Patel, K.; Allen, J.; Zagzag, D.; Wisoff, J.; Radmanesh, A.; Gindin, T.; Nicolaides, T. Radiologic response to MEK inhibition in a patient with a WNT-activated craniopharyngioma. Pediatr. Blood Cancer 2021, 68, e28753. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ incorporation in the β-catenin destruction complex orchestrates the Wnt response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef]
- Pan, J.X.; Xiong, L.; Zhao, K.; Zeng, P.; Wang, B.; Tang, F.L.; Sun, D.; Guo, H.H.; Yang, X.; Cui, S.; et al. YAP promotes osteogenesis and suppresses adipogenic differentiation by regulating β-catenin signaling. Bone Res. 2018, 6, 18. [Google Scholar] [CrossRef]
- Varelas, X.; Miller, B.W.; Sopko, R.; Song, S.; Gregorieff, A.; Fellouse, F.A.; Sakuma, R.; Pawson, T.; Hunziker, W.; McNeill, H.; et al. The Hippo pathway regulates Wnt/beta-catenin signaling. Dev. Cell 2010, 18, 579–591. [Google Scholar] [CrossRef]
- Thakker, R.V.; Newey, P.J.; Walls, G.V.; Bilezikian, J.; Dralle, H.; Ebeling, P.R.; Melmed, S.; Sakurai, A.; Tonelli, F.; Brandi, M.L. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J. Clin. Endocrinol. Metab. 2012, 97, 2990–3011. [Google Scholar] [CrossRef]
- Wu, Y.; Gao, L.; Guo, X.; Wang, Z.; Lian, W.; Deng, K.; Lu, L.; Xing, B.; Zhu, H. Pituitary adenomas in patients with multiple endocrine neoplasia type 1: A single-center experience in China. Pituitary 2019, 22, 113–123. [Google Scholar] [CrossRef]
- Cohen-cohen, S.; Brown, D.A.; Himes, B.T.; Wheeler, L.P.; Ruff, M.W.; Major, B.T.; Ospina, N.M.S.; Atkinson, J.L.D.; Meyer, F.B.; Bancos, I.; et al. Pituitary adenomas in the setting of multiple endocrine neoplasia type 1: A single-institution experience. J. Neurosurg. 2021, 134, 1132–1138. [Google Scholar] [CrossRef]
- Kamilaris, C.D.C.; Stratakis, C.A. Multiple endocrine neoplasia type 1 (MEN1): An update and the significance of early genetic and clinical diagnosis. Front. Endocrinol. 2019, 10, 1–7. [Google Scholar] [CrossRef]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Concolino, P.; Costella, A.; Capoluongo, E. Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet. 2016, 209, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Matkar, S.; Thiel, A.; Hua, X. Menin: A scaffold protein that controls gene expression and cell signaling. Trends Biochem. Sci. 2013, 38, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Gurung, B.; Wan, B.; Matkar, S.; Veniaminova, N.A.; Wan, K.; Merchant, J.L.; Hua, X.; Lei, M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 2012, 482, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; LiVolsi, V.A.; Gibbs, D.; et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 749–754. [Google Scholar] [CrossRef]
- Gillam, M.P.; Nimbalkar, D.; Sun, L.; Christov, K.; Ray, D.; Kaldis, P.; Liu, X.; Kiyokawa, H. MEN1 tumorigenesis in the pituitary and pancreatic islet requires CDK4 but not Cdk2. Oncogene 2015, 34, 932–938. [Google Scholar] [CrossRef]
- Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol. Cell. Endocrinol. 2014, 386, 2–15. [Google Scholar] [CrossRef]
- Giusti, F.; Cianferotti, L.; Boaretto, F.; Cetani, F.; Cioppi, F.; Colao, A.; Davì, M.V.; Faggiano, A.; Fanciulli, G.; Ferolla, P.; et al. Multiple endocrine neoplasia syndrome type 1: Institution, management, and data analysis of a nationwide multicenter patient database. Endocrine 2017, 58, 349–359. [Google Scholar] [CrossRef]
- Chi, S.G.; Minami, Y. Emerging Targeted Therapy for Specific Genomic Abnormalities in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2022, 23, 2362. [Google Scholar] [CrossRef]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Murai, M.J.; Belcher, A.M.; Hartley, T.; et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar] [CrossRef]
- Pellegata, N.S.; Quintanilla-Martinez, L.; Siggelkow, H.; Samson, E.; Bink, K.; Höfler, H.; Fend, F.; Graw, J.; Atkinson, M.J. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 15558–15563. [Google Scholar] [CrossRef]
- Lee, M.; Pellegata, N.S. Multiple endocrine neoplasia type 4. Front. Horm. Res. 2013, 41, 63–78. [Google Scholar] [CrossRef]
- Frederiksen, A.; Rossing, M.; Hermann, P.; Ejersted, C.; Thakker, R.V.; Frost, M. Clinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases. J. Clin. Endocrinol. Metab. 2019, 104, 3637–3646. [Google Scholar] [CrossRef]
- Roussel-gervais, A.; Couture, C.; Langlais, D.; Takayasu, S.; Balsalobre, A.; Rueda, B.R.; Zukerberg, L.R.; Figarella-branger, D.; Brue, T.; Drouin, J. The Cables1 Gene in Glucocorticoid Regulation of Pituitary Corticotrope Growth and Cushing Disease. J. Clin. Endocrinol. Metab. 2016, 101, 513–522. [Google Scholar] [CrossRef]
- Matsuoka, M.; Matsuura, Y.; Semba, K.; Nishimoto, I. Molecular cloning of a cyclin-like protein associated with cyclin-dependent kinase 3 (cdk 3) in vivo. Biochem. Biophys. Res. Commun. 2000, 273, 442–447. [Google Scholar] [CrossRef]
- Hernández-ramírez, L.C.; Gam, R.; Valdés, N.; Lodish, M.B.; Pankratz, N.; Balsalobre, A.; Gauthier, Y.; Faucz, F.R. Loss-of-function mutations in the CABLES1 gene are a novel cause of Cushing ’ s disease. Endocr.-Relat. Cancer 2017, 24, 379–392. [Google Scholar] [CrossRef]
- Zukerberg, L.R.; Patrick, G.N.; Nikolic, M.; Humbert, S.; Wu, C.L.; Lanier, L.M.; Gertler, F.B.; Vidal, M.; Van Etten, R.A.; Tsai, L.H. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron 2000, 26, 633–646. [Google Scholar] [CrossRef]
- Alrezk, R.; Suarez, A.; Tena, I.; Pacak, K. Update of Pheochromocytoma Syndromes: Genetics, Biochemical Evaluation, and Imaging. Front. Endocrinol. 2018, 9, 515. [Google Scholar] [CrossRef]
- Xekouki, P.; Pacak, K.; Almeida, M.; Wassif, C.A.; Rustin, P.; Nesterova, M.; De La Luz Sierra, M.; Matro, J.; Ball, E.; Azevedo, M.; et al. Succinate dehydrogenase (SDH) D subunit (SDHD) inactivation in a growth-hormone-producing pituitary tumor: A new association for SDH? J. Clin. Endocrinol. Metab. 2012, 97, 357–366. [Google Scholar] [CrossRef]
- Xekouki, P.; Szarek, E.; Bullova, P.; Giubellino, A.; Quezado, M.; Mastroyannis, S.A.; Mastorakos, P.; Wassif, C.A.; Raygada, M.; Rentia, N.; et al. Pituitary adenoma with paraganglioma/pheochromocytoma (3PAs) and succinate dehydrogenase defects in humans and mice. J. Clin. Endocrinol. Metab. 2015, 100, E710–E719. [Google Scholar] [CrossRef]
- Papathomas, T.G.; Gaal, J.; Corssmit, E.P.M.; Oudijk, L.; Korpershoek, E.; Heimdal, K.; Bayley, J.P.; Morreau, H.; Van Dooren, M.; Papaspyrou, K.; et al. Non-pheochromocytoma (PCC)/paraganglioma (PGL) tumors in patients with succinate dehydrogenase-related PCC-PGL syndromes: A clinicopathological and molecular analysis. Eur. J. Endocrinol. 2014, 170, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, B.; Zhu, X.L.; Yang, W.C.; Yang, G.F. Molecular pathogenesis of tumorigenesis caused by succinate dehydrogenase defect. Eur. J. Cell Biol. 2020, 99, 151057. [Google Scholar] [CrossRef] [PubMed]
- Moosavi, B.; Berry, E.A.; Zhu, X.L.; Yang, W.C.; Yang, G.F. The assembly of succinate dehydrogenase: A key enzyme in bioenergetics. Cell. Mol. Life Sci. 2019, 76, 4023–4042. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.A.; Maxwell, P.H. HIF, a missing link between metabolism and cancer. Nat. Med. 2005, 11, 1047–1048. [Google Scholar] [CrossRef]
- Tretter, L.; Patocs, A.; Chinopoulos, C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1086–1101. [Google Scholar] [CrossRef]
- Dénes, J.; Swords, F.; Rattenberry, E.; Stals, K.; Owens, M.; Cranston, T.; Xekouki, P.; Moran, L.; Kumar, A.; Wassif, C.; et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: Results from a large patient cohort. J. Clin. Endocrinol. Metab. 2015, 100, E531–E541. [Google Scholar] [CrossRef]
- Kluckova, K.; Tennant, D.A. Metabolic implications of hypoxia and pseudohypoxia in pheochromocytoma and paraganglioma. Cell Tissue Res. 2018, 372, 367–378. [Google Scholar] [CrossRef]
- Xekouki, P.; Brennand, A.; Whitelaw, B.; Pacak, K.; Stratakis, C.A. The 3PAs: An Update on the Association of Pheochromocytomas, Paragangliomas, and Pituitary Tumors. Horm. Metab. Res. 2019, 51, 419–436. [Google Scholar] [CrossRef]
- Guerrero-Pérez, F.; Fajardo, C.; Torres Vela, E.; Giménez-Palop, O.; Lisbona Gil, A.; Martín, T.; González, N.; Díez, J.J.; Iglesias, P.; Robledo, M.; et al. 3P association (3PAs): Pituitary adenoma and pheochromocytoma/paraganglioma. A heterogeneous clinical syndrome associated with different gene mutations. Eur. J. Intern. Med. 2019, 69, 14–19. [Google Scholar] [CrossRef]
- Daly, A.F.; Castermans, E.; Oudijk, L.; Guitelman, M.A.; Beckers, P.; Potorac, I.; Neggers, S.J.C.M.M.; Sacre, N.; van der Lely, A.J.; Bours, V.; et al. Pheochromocytomas and pituitary adenomas in three patients with MAX exon deletions. Endocr. Relat. Cancer 2018, 25, L37–L42. [Google Scholar] [CrossRef]
- Comino-méndez, I.; Gracia-aznárez, F.J.; Schiavi, F.; Landa, I.; Leandro-garcía, L.J.; Letón, R.; Honrado, E.; Ramos-medina, R.; Caronia, D.; Pita, G.; et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat. Genet. 2011, 43, 663–667. [Google Scholar] [CrossRef]
- Pang, Y.; Liu, Y.; Pacak, K.; Yang, C. Pheochromocytomas and paragangliomas: From genetic diversity to targeted therapies. Cancers 2019, 11, 436. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Priest, J.R.; Duchaine, T.F. DICER1: Mutations, microRNAs and mechanisms. Nat. Rev. Cancer 2014, 14, 662–672. [Google Scholar] [CrossRef]
- Song, M.S.; Rossi, J.J. Molecular mechanisms of Dicer: Endonuclease and enzymatic activity. Biochem. J. 2017, 474, 1603–1618. [Google Scholar] [CrossRef]
- Faure, A.; Atkinson, J.; Bouty, A.; Brien, M.O. DICER1 pleuropulmonary blastoma familial tumour predisposition syndrome: What the paediatric urologist needs to know. J. Pediatr. Urol. 2016, 12, 5–10. [Google Scholar] [CrossRef]
- Sahakitrungruang, T.; Srichomthong, C.; Pornkunwilai, S.; Amornfa, J.; Shuangshoti, S.; Kulawonganunchai, S.; Suphapeetiporn, K.; Shotelersuk, V. Germline and Somatic DICER1 Mutations in a Pituitary Blastoma Causing Infantile-Onset Cushing’s Disease. J. Clin. Endocrinol. Metab. 2014, 99, E1487–E1492. [Google Scholar] [CrossRef]
- Martínez de LaPiscina, I.; Hernández-Ramírez, L.C.; Portillo, N.; Gómez-Gila, A.L.; Urrutia, I.; Martínez-Salazar, R.; García-Castaño, A.; Aguayo, A.; Rica, I.; Gaztambide, S.; et al. Rare Germline DICER1 Variants in Pediatric Patients With Cushing’s Disease: What Is Their Role? Front. Endocrinol. 2020, 11, 433. [Google Scholar] [CrossRef]
- Vankelecom, H. Pituitary stem/progenitor cells: Embryonic players in the adult gland? Eur. J. Neurosci. 2010, 32, 2063–2081. [Google Scholar] [CrossRef]
- Davis, S.W.; Mortensen, A.H.; Camper, S.A. Birthdating studies reshape models for pituitary gland cell specification. Dev. Biol. 2011, 352, 215–227. [Google Scholar] [CrossRef]
- Rizzoti, K.; Akiyama, H.; Lovell-Badge, R. Mobilized adult pituitary stem cells contribute to endocrine regeneration in response to physiological demand. Cell Stem Cell 2013, 13, 419–432. [Google Scholar] [CrossRef]
- Andoniadou, C.L.; Matsushima, D.; Mousavy Gharavy, S.N.; Signore, M.; Mackintosh, A.I.; Schaeffer, M.; Gaston-Massuet, C.; Mollard, P.; Jacques, T.S.; Le Tissier, P.; et al. Sox2(+) stem/progenitor cells in the adult mouse pituitary support organ homeostasis and have tumor-inducing potential. Cell Stem Cell 2013, 13, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Lepore, D.A.; Roeszler, K.; Wagner, J.; Ross, S.A.; Bauer, K.; Thomas, P.Q. Identification and enrichment of colony-forming cells from the adult murine pituitary. Exp. Cell Res. 2005, 308, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Gleiberman, A.S.; Michurina, T.; Encinas, J.M.; Roig, J.L.; Krasnov, P.; Balordi, F.; Fishell, G.; Rosenfeld, M.G.; Enikolopov, G. Genetic approaches identify adult pituitary stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 6332–6337. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Kato, T.; Susa, T.; Cai, L.Y.; Nakayama, M.; Kato, Y. PROP1 coexists with SOX2 and induces PIT1-commitment cells. Biochem. Biophys. Res. Commun. 2009, 385, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lavandeira, M.; Quereda, V.; Flores, I.; Saez, C.; Diaz-Rodriguez, E.; Japon, M.A.; Ryan, A.K.; Blasco, M.A.; Dieguez, C.; Malumbres, M.; et al. A GRFa2/Prop1/stem (GPS) cell niche in the pituitary. PLoS ONE 2009, 4, e4815. [Google Scholar] [CrossRef]
- Higuchi, M.; Yoshida, S.; Ueharu, H.; Chen, M.; Kato, T.; Kato, Y. PRRX1 and PRRX2 distinctively participate in pituitary organogenesis and a cell-supply system. Cell Tissue Res. 2014, 357, 323–335. [Google Scholar] [CrossRef]
- Chen, J.; Gremeaux, L.; Fu, Q.; Liekens, D.; Van Laere, S.; Vankelecom, H. Pituitary progenitor cells tracked down by side population dissection. Stem Cells 2009, 27, 1182–1195. [Google Scholar] [CrossRef]
- Fauquier, T.; Rizzoti, K.; Dattani, M.; Lovell-Badge, R.; Robinson, I.C.A.F. SOX2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc. Natl. Acad. Sci. USA 2008, 105, 2907–2912. [Google Scholar] [CrossRef]
- Jayakody, S.A.; Andoniadou, C.L.; Gaston-Massuet, C.; Signore, M.; Cariboni, A.; Bouloux, P.M.; Le Tissier, P.; Pevny, L.H.; Dattani, M.T.; Martinez-Barbera, J.P. SOX2 regulates the hypothalamic-pituitary axis at multiple levels. J. Clin. Investig. 2012, 122, 3635–3646. [Google Scholar] [CrossRef]
- Yoshida, S.; Kato, T.; Yako, H.; Susa, T.; Cai, L.Y.; Osuna, M.; Inoue, K.; Kato, Y. Significant quantitative and qualitative transition in pituitary stem/progenitor cells occurs during the postnatal development of the rat anterior pituitary. J. Neuroendocrinol. 2011, 23, 933–943. [Google Scholar] [CrossRef]
- Horiguchi, K.; Fujiwara, K.; Yoshida, S.; Nakakura, T.; Arae, K.; Tsukada, T.; Hasegawa, R.; Takigami, S.; Ohsako, S.; Yashiro, T.; et al. Isolation and characterisation of CD9-positive pituitary adult stem/progenitor cells in rats. Sci. Rep. 2018, 8, 5533. [Google Scholar] [CrossRef]
- Russell, J.P.; Lim, X.; Santambrogio, A.; Yianni, V.; Kemkem, Y.; Wang, B.; Fish, M.; Haston, S.; Grabek, A.; Hallang, S.; et al. Pituitary stem cells produce paracrine WNT signals to control the expansion of their descendant progenitor cells. Elife 2021, 10, e59142. [Google Scholar] [CrossRef]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Manoranjan, B.; Mahendram, S.; Almenawer, S.A.; Venugopal, C.; McFarlane, N.; Hallett, R.; Vijayakumar, T.; Algird, A.; Murty, N.K.; Sommer, D.D.; et al. The identification of human pituitary adenoma-initiating cells. Acta Neuropathol. Commun. 2016, 4, 125. [Google Scholar] [CrossRef]
- Würth, R.; Barbieri, F.; Pattarozzi, A.; Gaudenzi, G.; Gatto, F.; Fiaschi, P.; Ravetti, J.L.; Zona, G.; Daga, A.; Persani, L.; et al. Phenotypical and Pharmacological Characterization of Stem-Like Cells in Human Pituitary Adenomas. Mol. Neurobiol. 2017, 54, 4879–4895. [Google Scholar] [CrossRef]
- Peverelli, E.; Giardino, E.; Treppiedi, D.; Meregalli, M.; Belicchi, M.; Vaira, V.; Corbetta, S.; Verdelli, C.; Verrua, E.; Serban, A.L.; et al. Dopamine receptor type 2 (DRD2) and somatostatin receptor type 2 (SSTR2) agonists are effective in inhibiting proliferation of progenitor/stem-like cells isolated from nonfunctioning pituitary tumors. Int. J. Cancer 2017, 140, 1870–1880. [Google Scholar] [CrossRef]
- Mertens, F.; Gremeaux, L.; Chen, J.; Fu, Q.; Willems, C.; Roose, H.; Govaere, O.; Roskams, T.; Cristina, C.; Becú-Villalobos, D.; et al. Pituitary tumors contain a side population with tumor stem cell-associated characteristics. Endocr. Relat. Cancer 2015, 22, 481–504. [Google Scholar] [CrossRef]
- Xu, Q.; Yuan, X.; Tunici, P.; Liu, G.; Fan, X.; Xu, M.; Hu, J.; Hwang, J.Y.; Farkas, D.L.; Black, K.L.; et al. Isolation of tumour stem-like cells from benign tumours. Br. J. Cancer 2009, 101, 303–311. [Google Scholar] [CrossRef]
- Chen, L.; Ye, H.; Wang, X.; Tang, X.; Mao, Y.; Zhao, Y.; Wu, Z.; Mao, X.O.; Xie, L.; Jin, K.; et al. Evidence of brain tumor stem progenitor-like cells with low proliferative capacity in human benign pituitary adenoma. Cancer Lett. 2014, 349, 61–66. [Google Scholar] [CrossRef]
- Zubeldía-Brenner, L.; De Winne, C.; Perrone, S.; Rodríguez-Seguí, S.A.; Willems, C.; Ornstein, A.M.; Lacau-Mengido, I.; Vankelecom, H.; Cristina, C.; Becu-Villalobos, D. Inhibition of Notch signaling attenuates pituitary adenoma growth in Nude mice. Endocr. Relat. Cancer 2019, 26, 13–29. [Google Scholar] [CrossRef]
- Wierinckx, A.; Roche, M.; Legras-lachuer, C. MicroRNAs in pituitary tumors. Mol. Cell. Endocrinol. 2017, 456, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Bottoni, A.; Piccin, D.; Tagliati, F.; Luchin, A.; Zatelli, M.C.; Uberti, E.C.D. miR-15a and miR-16-1 down-regulation in pituitary adenomas. J. Cell. Phys. 2005, 204, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Butz, H. Circulating Noncoding RNAs in Pituitary Neuroendocrine Tumors—Two Sides of the Same Coin. Int. J. Mol. Sci. 2022, 23, 5122. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Wang, L. The Involvement of miRNAs in Pituitary Adenomas Pathogenesis and the Clinical Implications. Eur. Neurol. 2022, 85, 171–176. [Google Scholar] [CrossRef]
- Butz, H.; Patócs, A. MicroRNAs in endocrine tumors. EJIFCC 2019, 30, 146. [Google Scholar]
- Zhang, R.; Yang, F.; Fan, H.; Wang, H.; Wang, Q.; Yang, J.; Song, T. Long non-coding RNA TUG1/microRNA-187-3p/TESC axis modulates progression of pituitary adenoma via regulating the NF- κ B signaling pathway. Cell Death Dis. 2021, 12, 524. [Google Scholar] [CrossRef]
- Zhou, K.; Zhang, T.; Fan, Y.; Du, G.; Wu, P. MicroRNA-106b promotes pituitary tumor cell proliferation and invasion through PI3K/AKT signaling pathway by targeting PTEN. Tumor Biol. 2016, 37, 13469–13477. [Google Scholar] [CrossRef]
- Renjie, W.; Haiqian, L. MiR-132, miR-15a and miR-16 synergistically inhibit pituitary tumor cell proliferation, invasion and migration by targeting Sox5. Cancer Lett. 2015, 356, 568–578. [Google Scholar] [CrossRef]
- Adrian, E.B.; Sebastian, F.D.; Auli, G.; Irmler, M.; Beckers, J.; Mohr, H.; Beckers, A.; Pellegata, N.S. miR-34a is upregulated in AIP- mutated somatotropinomas and promotes octreotide resistance. Int. J. Cancer 2020, 147, 3523–3538. [Google Scholar] [CrossRef]
- Zhao, P.; Cheng, J.; Li, B.; Nie, D.; Li, C.; Gui, S.; Wang, H.; Zhang, Y. Up-regulation of the expressions of MiR-149-5p and MiR-99a-3p in exosome inhibits the progress of pituitary adenomas. Cell Biol. Toxicol. 2021, 37, 633–651. [Google Scholar] [CrossRef]
- Gentilin, E.; Uberti, E.; Zatelli, M.C. MicroRNAs in the pituitary. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 629–639. [Google Scholar] [CrossRef]
- Miroshnichenko, S.; Patutina, O. Enhanced Inhibition of Tumorigenesis Using Combinations of miRNA-Targeted Therapeutics. Front. Pharmacol. 2019, 10, 488. [Google Scholar] [CrossRef]



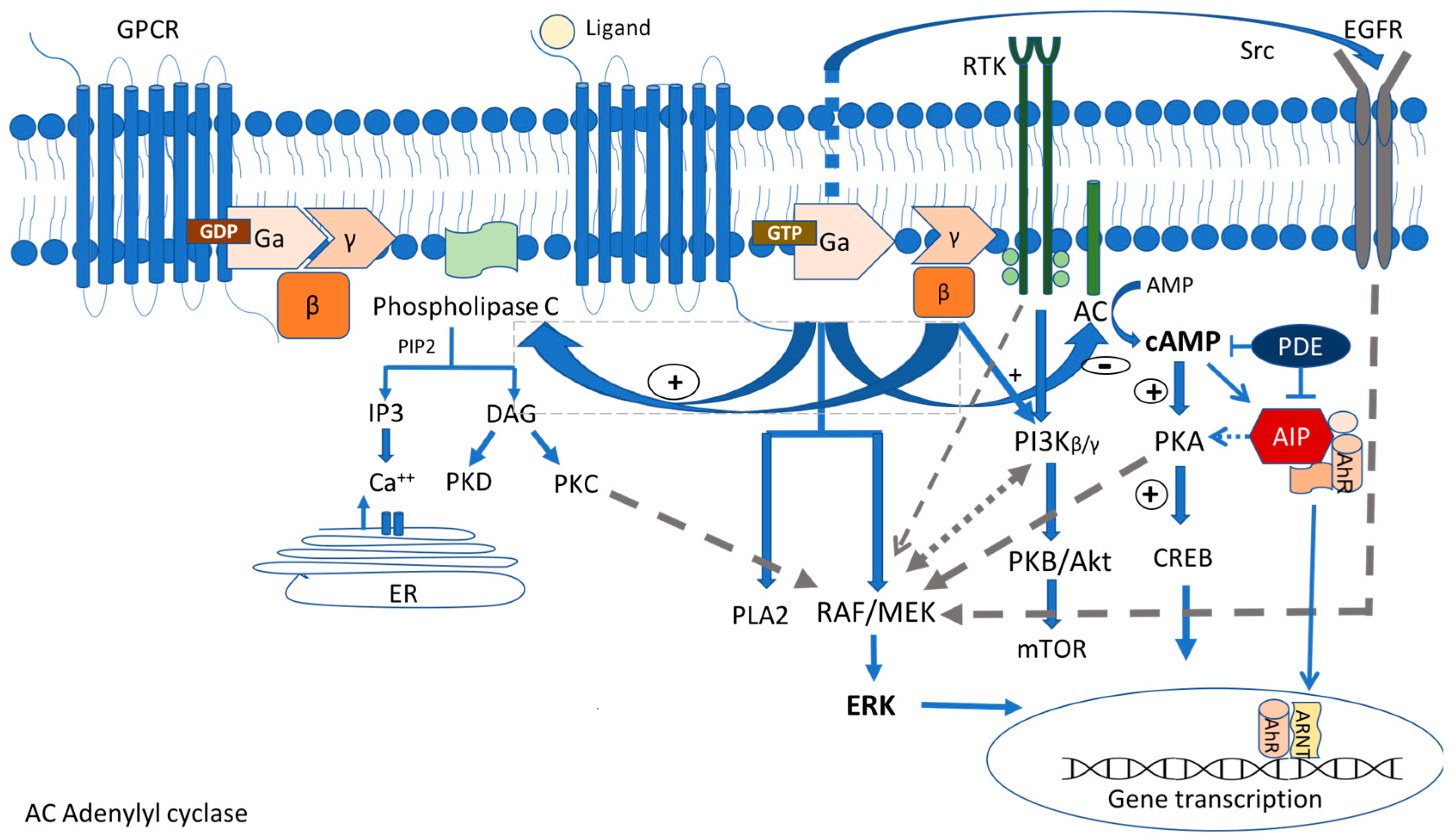{kind=link}
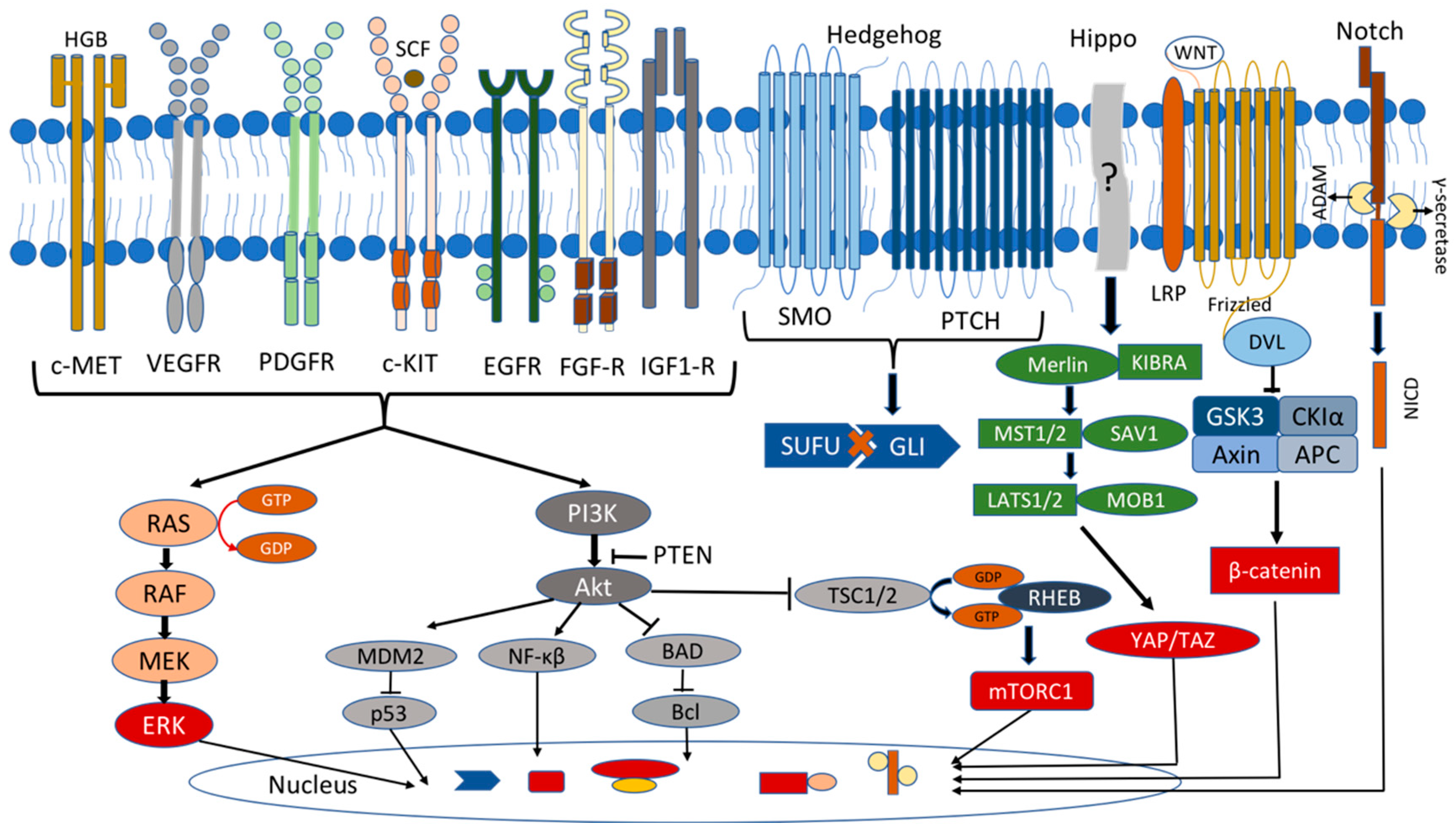{kind=link}
| Molecular Mechanisms | Protein (Gene) | Function | Syndrome | Mutation Origin | Clinical/Pathology Characteristics | Current Treatment Strategies |
|---|---|---|---|---|---|---|
| Gsa/protein kinase A/c AMP signaling pathway | G-coupled stimulatory protein subunit A (GNAS) | Oncogene, c-AMP pathway stimulation | McCune–Albright Sporadic | Mosaic Postzygotic | GH PitNETs, polyostic fibrus dysplasia, café au lait spots. | Surgery, SSAs—partial response |
| - | Somatic | GH-secreting PitNETs in adults, less aggressive behavior, ACTH-, NF-secreting tumors rare. | Surgery, SSAs—most tumors seem to respond better. However, there are some controversial studies. | |||
| Regulatory subunit protein kinase 1A (PRKAR1A) | PKA activity regulation | Carney Familiar/ sporadic | Germline | Pituitary GH/PRL hyperplasia, GH PitNETs, corticotropinomas, lactotroph adenomas, spotty skin pigmentation, PPNAD, myxomas, thyroid, testis, and ovarian tumors. | Surgery, SSAs, dopamine agonists. No systemic medical treatment developed according to the genetic defect or targeting the cAMP/PKA signaling pathway in the Carney complex. | |
| Aryl hydrocarbon receptor-interacting protein (AIP) | Tumor suppressor, co-chaperone protein | AIP-mutated adenomas Familiar/ sporadic | Germlline | Early onset of PitNETs GH, PRL, GH + PRL. NF- and ACTH- rare. Macroadenomas, pituitary apoplexy, aggressive. | Surgery, poor response to first-generation SSAs, and dopamine agonists. Better response to pasireotide. Interplay between AIP and RET pathway-RET inhibitors: potential new therapeutic approach for resistant tumors.Inhibition of CCL5/CCR5 pathway by maravirorik (experimental) as another therapeutic target. | |
| Orphan G-protein-coupled receptor (GPCR) protein (GPR101) | Oncogene, class A, rhodopsin-like orphan GPCR, coupled to Gs subunit, constitutive activation of the cAMP pathway | XLAG Familiar/ sporadic | Germline (females) Somatic (sporadic males) | Early childhood (<5 years old) onset gigantism due to GH-secreting or mixed GH- and PRL-secreting PitNets and/or hyperplasia, acanthosis nigricans, insulin resistance, increased appetite. | Surgery plus pegvisomat, radiotherapy. Resistance to SSA; potential new approach—therapeutic blockade of GHRH secretion (experimental). | |
| MAPK/ERK pathway | Serine/threonine-protein kinase B-raf (BRAF) | B-Raf proto-oncogene, phosphorylate MEK and ERK1/2 kinases | Sporadic | Somatic | PCPs: GOF mutations BRAF V600E. | BRAF inhibitors as monotherapy/plus MEK inhibitors in cases of BRAFV600E mutant PCPs. Only one clinical trial for the treatment of BRAFV600E mutant PCPs: BRAF/MEK inhibitors (vemurafenib/cobimetinib) (NCT03224767). |
| ACPs: BRAF V600E may coexist with CTNNB1-mutated ACPs. | ||||||
| ACTH-secreting. PitNETs: -Ras/ERK signaling activation -BRAF V600E in 16.4% of corticotroph tumors | MEK inhibitor (binimetinib), both in vitro and in vivo, and BRAF inhibitor (vemurafenib) in vitro inhibited corticotroph tumor cell proliferation and ACTH secretion. | |||||
| PI3K/Akt pathway | Epidermal growth factor (EGF) receptor family of receptor tyrosine kinases (RTKs): the main TK target for PitNETs
| RTKs activate the MAPK/ERK and PI3K/Akt pathways leading to pituitary tumorigenesis | Sporadic | Somatic | Pit1 lineage-specific mTOR-activation leads to lactotroph adenomas in mice. Somatic mutations of PIK3CA in human PitNETs. | Everolimus: the only active mTOR inhibitor administered in patients with PitNETs. PI3K/mTOR inhibitors: a greater antiproliferative effect in vitro (no dual PI3K/mTOR inhibitor in clinical practice). |
| HER2/ErbB2 induces PRL and tumorigenic effects in rat prolactin-secreting PitNETs. ErbB2 is mainly associated with aggressive and/or resistant prolactin-secreting PitNET in human studies. | Lapatinib, a dual EGFR and HER2 inhibitor, has a more influential role in prolactin-secreting PitNETs both in vitro and in vivo. A phase 2a clinical trial suggests that lapatinib may be a suitable treatment option for aggressive prolactin-secreting PitNETs. | |||||
| EGFR overexpression in ACTH-secreting PitNETs. | EGFR inhibitors (gefitinib and lapatinib), in both human and mouse corticotroph primary cultures. | |||||
| Ubiquitin-specific protease 8 (USP8) | Deubiquitinase controls the lysosomal trafficking and abundance of EGFR. | Sporadic | Somatic/one case of germline | GOF mutations of USP8 in 20–60% of ACTH-secreting PitNETs. | Treatment with pasireotide: correlation of USP8 mutational status with a higher SSTR5 expression. | |
| HIPPO pathway | Yes-associated protein (YAP) Transcriptional co-activator with PDZ-binding motif (TAZ) | Oncogene, unphosphorylated nuclear YAP/TAZ act as co-activators to TEAD transcription factors. | Sporadic | Somatic | In SOX2 + pituitary stem cells in mice. In fetal and adult human pituitary. Increased expression in NF-PitNETs in humans. | No available YAP/TAZ inhibitors in clinical practice for PitNETs. |
| WNT pathway | b-catenin (CTNNB1) | Oncogene, unphosphorylated nuclear b-catenin acts as a transcription factor for cell proliferation genes. | Sporadic | Somatic | GOF mutations of CTNNB1 in ACPs. | WNT pathway is not considered among the intervention strategies for CPs. Tocilizumab, an IL-6 inhibitor in an open clinical trial for recurrent/progressed ACPs (NCT03970226). |
| Tumor suppressor genes | Menin (MEN1) | Tumor suppressor, nuclear protein with ubiquitous expression | MEN1 Familiar/ sporadic | Germline/ somatic | PitNETs—mostly PRL, followed by NF, GH—secreting rare pituitary carcinoma (macroadenomas, early onset, aggressive), parathyroid hyperplasia, and gastroenteropancreatic neuroendocrine tumors (GEP-NETs). | Surgery, SSAs, DAs, radiotherapy, temozolamide. Possible phenotype–genotype correlation. |
| Cyclin-dependent kinase 1B/protein p27Kip1 (CDKN1B) | Tumor suppressor, CDK inhibitor-uncontrolled cell cycle proliferation | MEN4 Familiar/ sporadic | Germline | PitNEts (somatotroph, corticotroph), adrenal, enteropancreatic tumors, testicular, papillary thyroid cancer, non-endocrine tumors (cervical carcinoma, colon cancer, meningiomas). | Limited experience. Similar to that of non-MEN4 pituitary tumors. | |
| CDK5 and ABL enzyme substrate 1 (CABLES 1) | Tumor suppressor, counteraction of the cell cycle progression activated in corticotroph cells in response to glucocorticoids, regulation of the function of CDKN1B. | Sporadic | Germline | ACTH-secreting PitNETs or silent corticotropinomas, macroadenomas, children or young adults, cushingoid features, mass effect symptoms, high ki-67 proliferative index | Difficult to treat with a tendency to recure. Roscovitine (seliciclib), an inhibitor of the cyclin-dependent kinase cyclin E, effectively decrease the corticotroph cell growth—potential new therapeutic approach (experimental). | |
| Succinate dehydrogenase complex (subunits A,B,C,D) SDH assemply factor (SDHx) | Tumor suppressor, critical role in oxidative phosphorylation and tricarboxylic acid cycle. | 3PAs Familiar/ sporadic (very rare) | Germline | PRL, GH, ACTH-secreting PitNets, aggressive macroadenomas. Pheochromocytomas and/or paragagglioma. | Surgery, poor responses to SSAs, new evolving therapies targeting HIF/pseudohypoxia pathway. | |
| DICER1 protein, ribonuclease (RNase) III (DICER1) | Tumor suppressor, endonouclease, member of the family ribonuclease (RNase) III, microRNA (miRNA) and small interfering RNA (siRNA) | DICER1 syndrome Familiar/ sporadic | Germline or mosaic loss-of-function (LOF) | Pituitary blastoma (ACTH-secreting) aggressive tumors, pleuropulmonary blastoma, cystic nephroma, Wilms tumor, ovarian sex cord-stromal tumor, embryonal rhabdomyosarcomas. | Surgery, polychemotherapy and adjuvant radiotherapy-limited experience. | |
| Stem cells | SOX2/S100β pituitary stem cells | (1) Generate all pituitary cell lineages (2) Self-renewal (3) Clonal expansion | - | - | A representative type of adult PSCs. | - |
| hPASCs express markers of stemness: OCT4, Notch1, 4, CD15, CD90, CD133, NESTIN, NANOG, CXCR4, KLF4 | Sphere-forming potential in cultures that express pituitary-specific markers. | - | - | hPASCs express DRD2, SSTR2 and SSTR5. | Promising results by in vitro activation of DRD2, SSTR2, and SSTR5 using Das and SSAs analogues. | |
| microRNAs | MiR-187-3p, MiR-17-5p, MiR-20a, MiR-106b, MiR-21, MiR200c, and MiR-128, MiR-132, miR-15a, miR-16, miR-34a, miR-149-5p, and miR-99a-3p | Short protein non-coding RNAs control the post-transcriptional expression of specific genes through RNA interference and mRNA destabilization; control up to 50% of all protein-coding genes | - | - | - | Potential novel drug targets. Act as epidrugs or antagomirs, since modulating miRNA activity may restrain the tumor progression or weaken the symptoms associated with aberrant hormonal secretion-experimental. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vamvoukaki, R.; Chrysoulaki, M.; Betsi, G.; Xekouki, P. Pituitary Tumorigenesis—Implications for Management. Medicina 2023, 59, 812. https://doi.org/10.3390/medicina59040812
Vamvoukaki R, Chrysoulaki M, Betsi G, Xekouki P. Pituitary Tumorigenesis—Implications for Management. Medicina. 2023; 59(4):812. https://doi.org/10.3390/medicina59040812
Chicago/Turabian StyleVamvoukaki, Rodanthi, Maria Chrysoulaki, Grigoria Betsi, and Paraskevi Xekouki. 2023. "Pituitary Tumorigenesis—Implications for Management" Medicina 59, no. 4: 812. https://doi.org/10.3390/medicina59040812
APA StyleVamvoukaki, R., Chrysoulaki, M., Betsi, G., & Xekouki, P. (2023). Pituitary Tumorigenesis—Implications for Management. Medicina, 59(4), 812. https://doi.org/10.3390/medicina59040812