
Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease


Abstract

{kind=link}
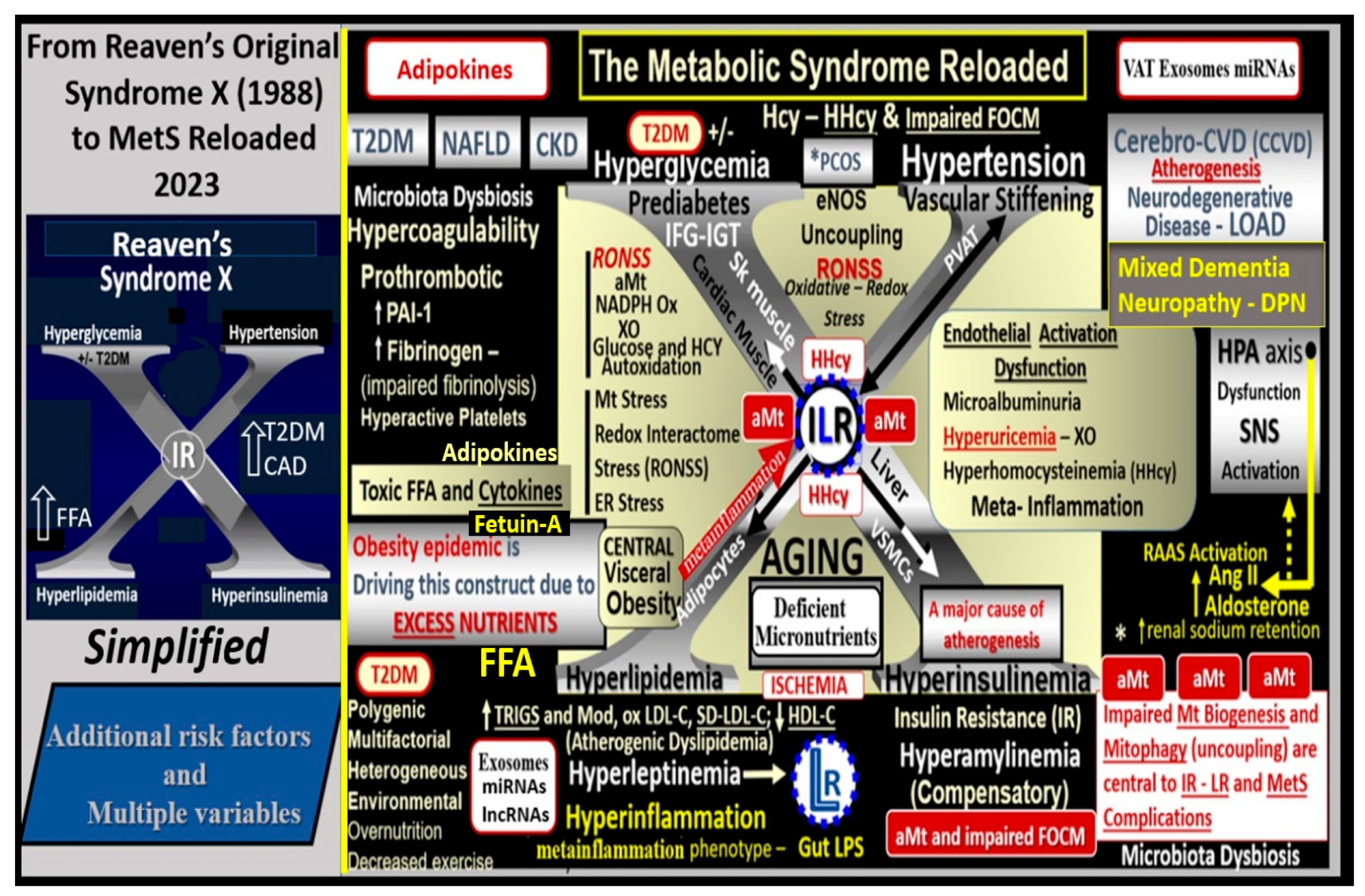{kind=link}
{kind=link}
{kind=link}
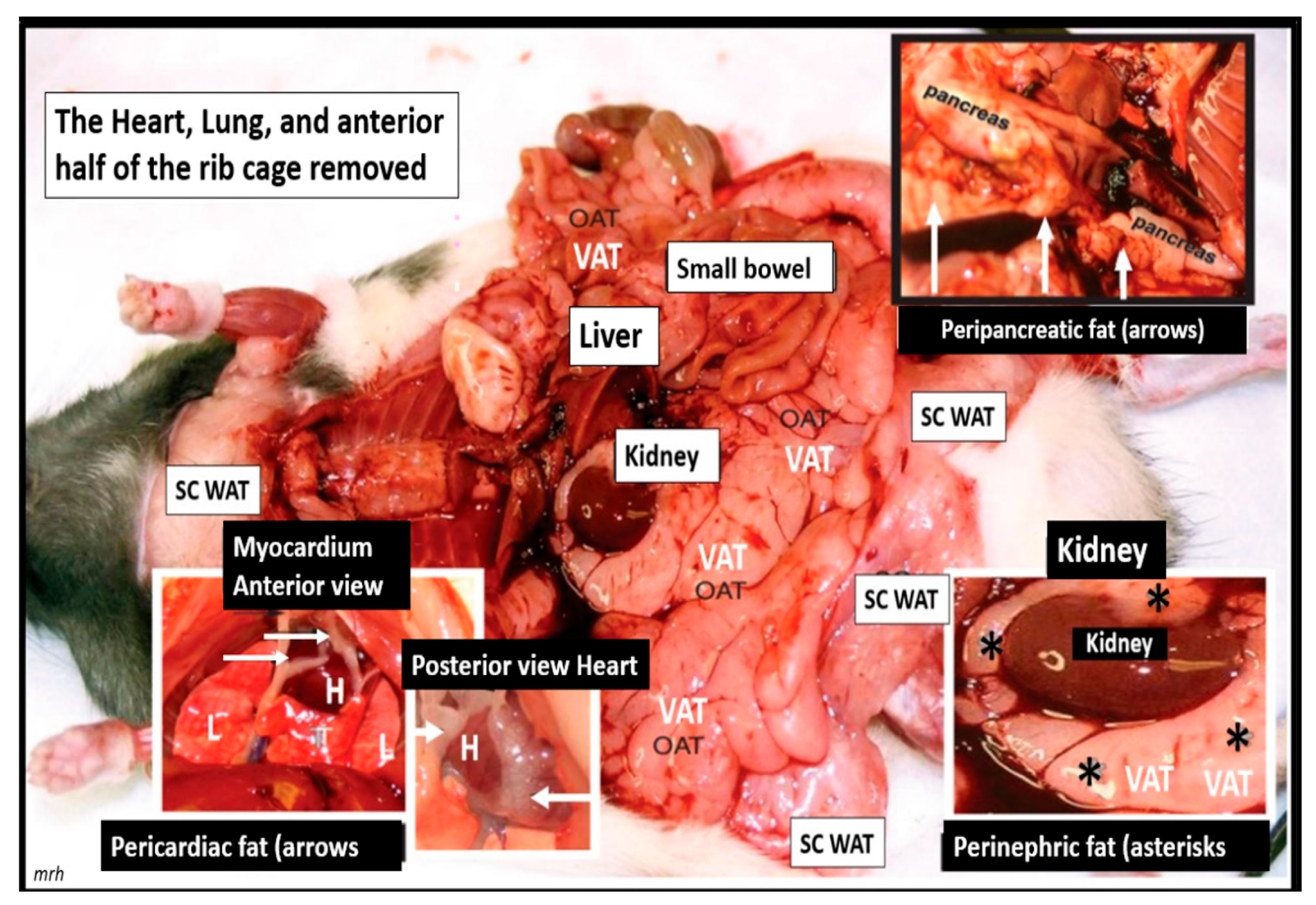{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
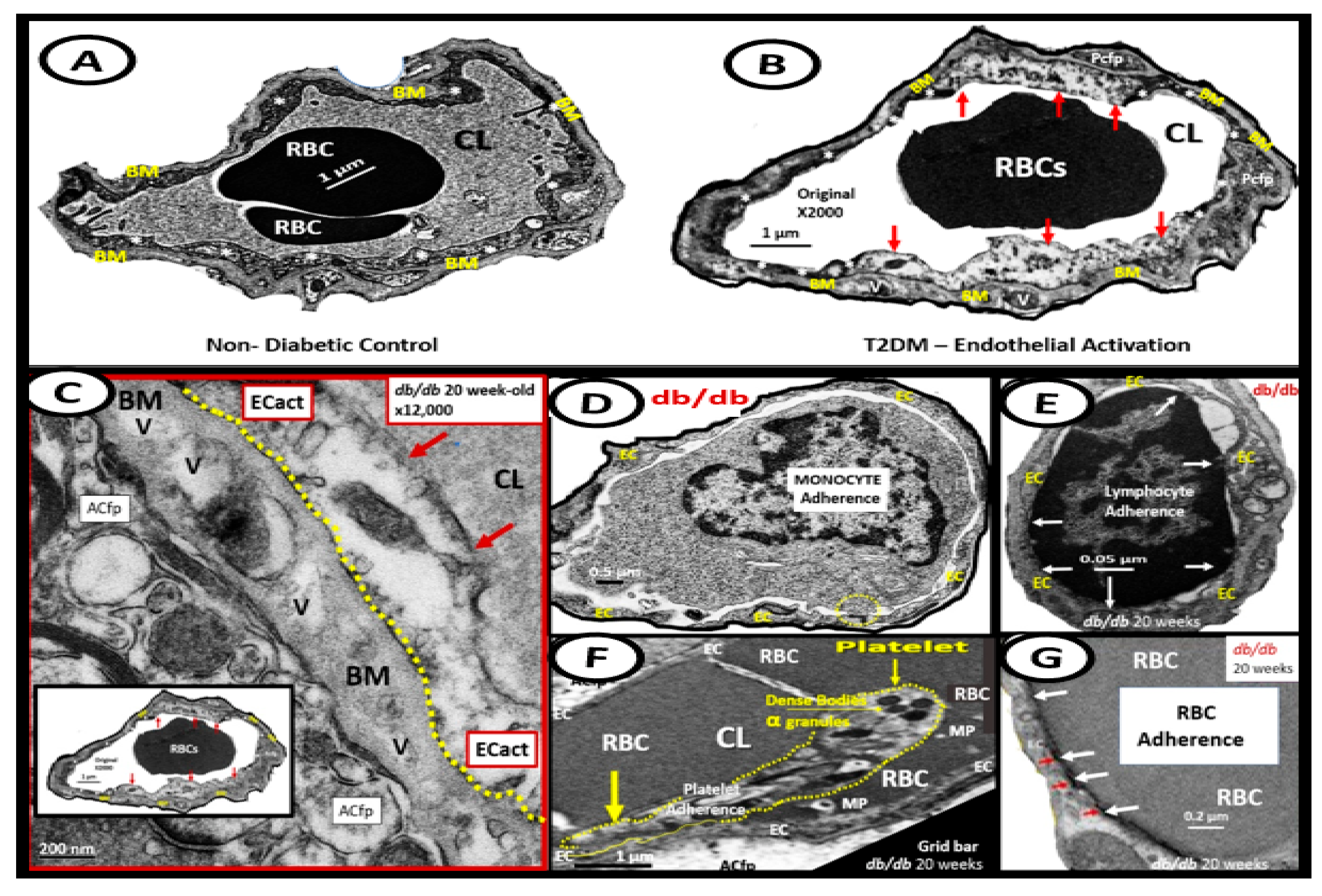{kind=link}
{kind=link}
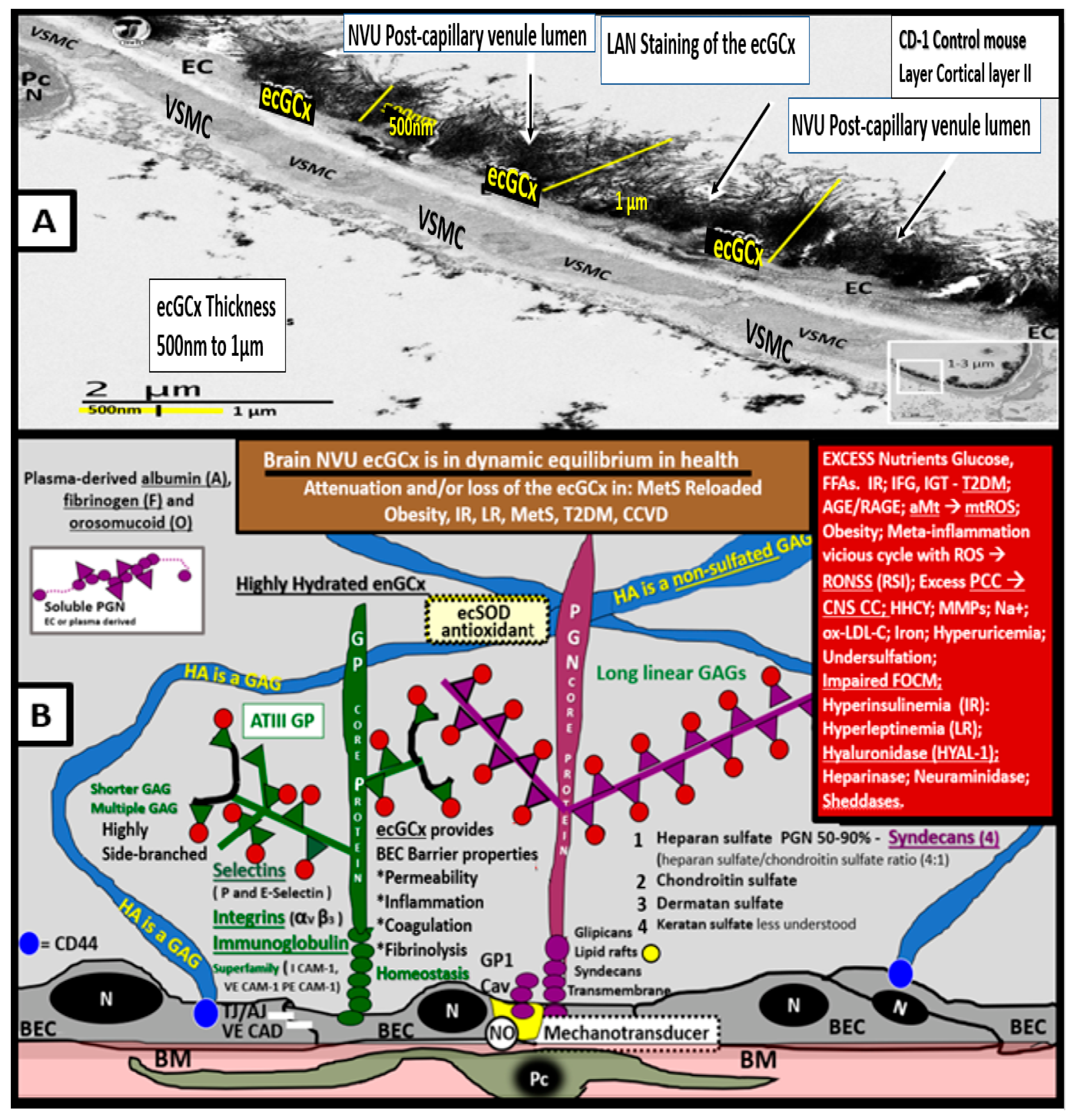{kind=link}
{kind=link}
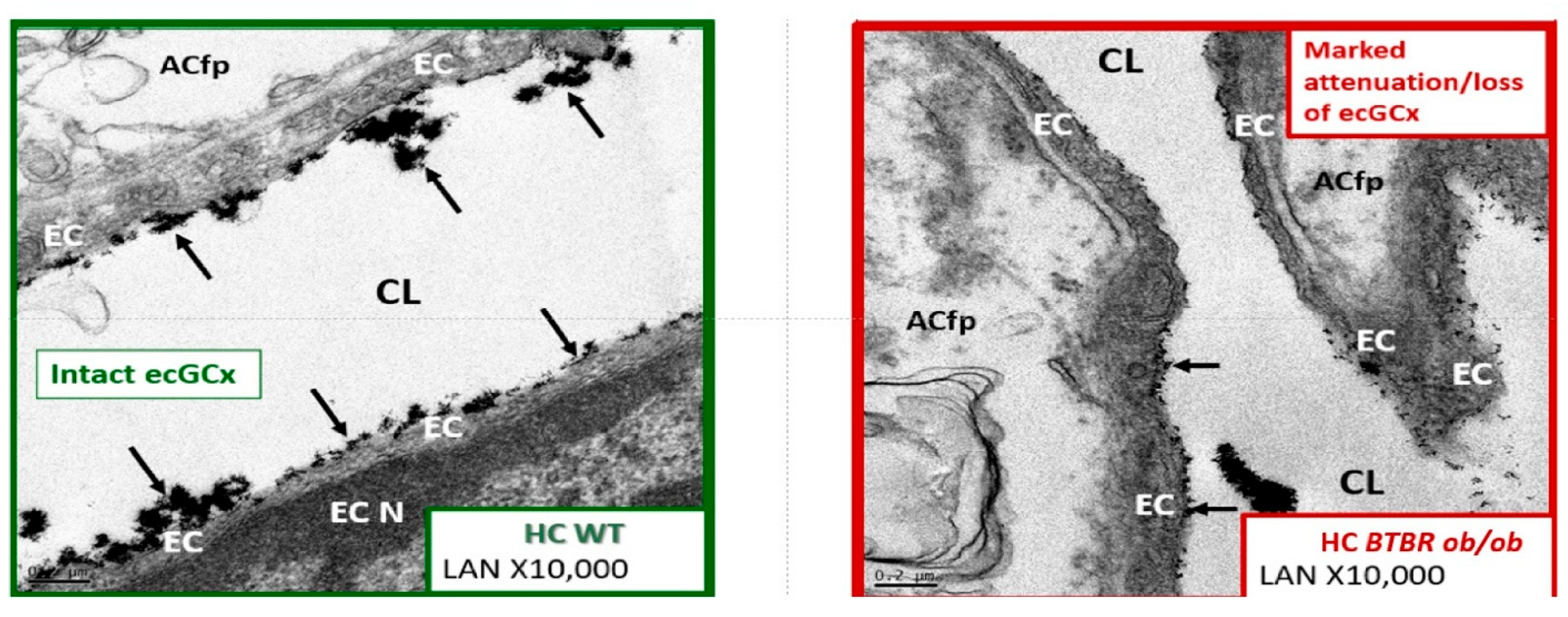{kind=link}
{kind=link}
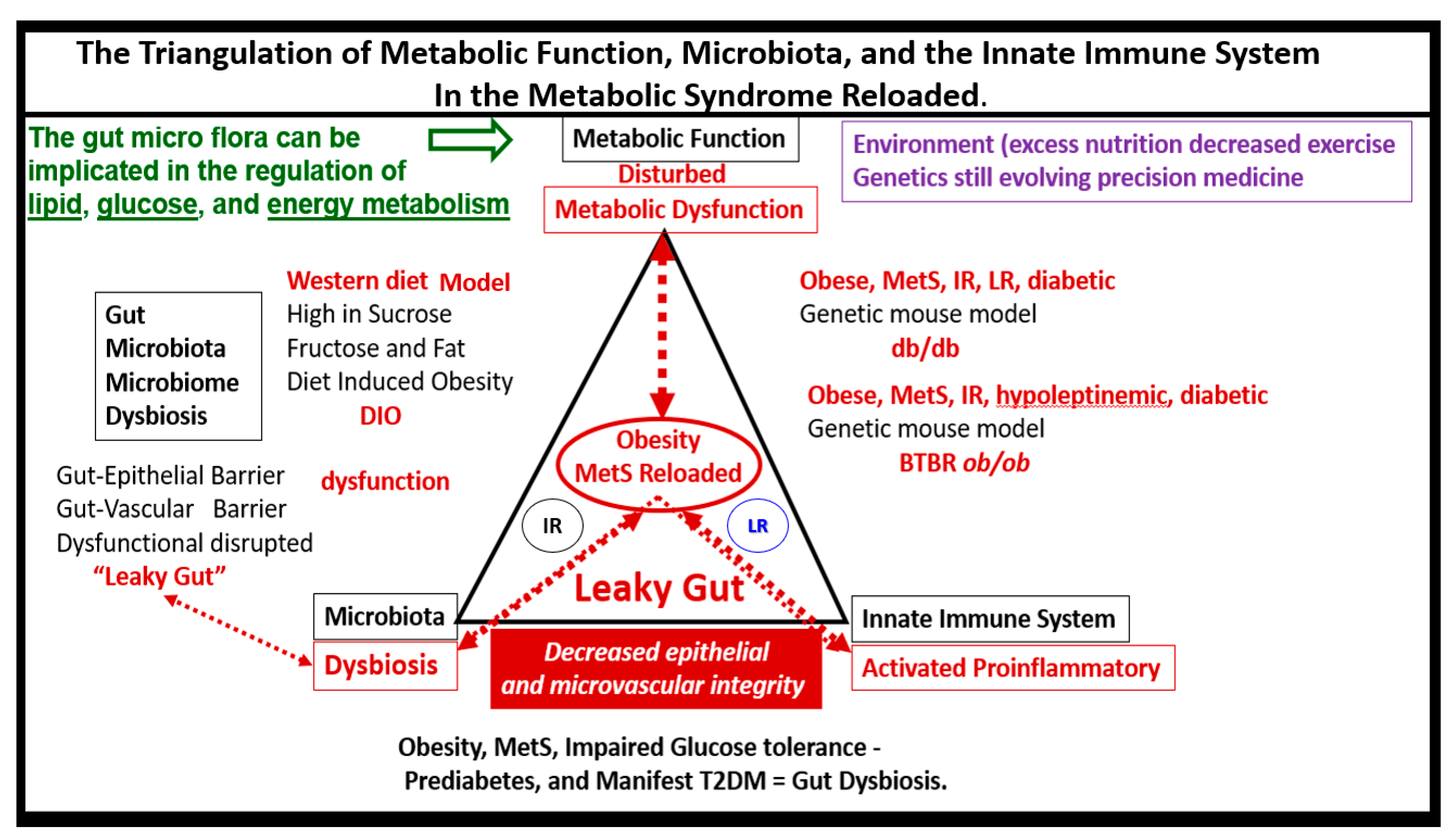{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. MetS Reloaded, Hyperlipidemia—Atherogenic Dyslipidemia
3. MetS Reloaded, Insulin, Hyperinsulinemia, IR, Leptin, Hyperleptinemia, and LR
3.1. MetS Reloaded, Compensatory Hyperinsulinemia, Insulin Resistance (IR), and Compensatory Hyperamylinemia
3.2. MetS Reloaded, Insulin Resistance, Compensatory Hyperamylinemia, and Islet Amyloid Deposition
3.3. MetS Reloaded, Leptin, Hyperleptinemia, and LR
4. MetS Reloaded, Essential Hypertension (HTN), and Vascular Stiffening
5. MetS Reloaded, Hyperglycemia, Mitochondrial Dysfunction, and Importance of the Reactive Oxygen Species Interactome (RSI)
5.1. MetS Reloaded and Impaired Folate-Mediated One-Carbon Metabolism (FOCM) in the Metabolic Syndrome Reloaded
6. MetS Reloaded and Endothelial Activation (ECact) and Dysfunction (ECdys)
7. MetS Reloaded and Metainflammation
7.1. MetS Reloaded and Adipokines/Hormones, Cytokines/Chemokines, and Toxic Free Fatty Acids (FFA)
7.2. PVAT Adipocyte and Macrophage-Derived Exosome microRNAs (miRNAs) and Long-Non-Coding RNAs (lncRNAs)
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- National Cholesterol Education Program (NCEP); Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2002, 285, 2486–2497. [Google Scholar] [CrossRef]
- Grundy, S.M.; Brewer, H.B., Jr.; Cleeman, J.I.; Smith, S.C., Jr.; Lenfant, C.; American Heart Association; National Heart, Lung, and Blood Institute. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 2004, 109, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Kylin, E. Studien ueber das Hypertonie-Hyperglyka “mie-Hyperurika” miesyndrom. Zent. Fuer Inn. Med. 1923, 44, 105–127. [Google Scholar]
- Himsworth, H.P.; Kerr, R.B. Insulin-sensitive and insulin-insensitive types of diabetes mellitus. Clin. Sci. 1939, 4, 119–152. [Google Scholar]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Banks, W.A.; Shah, G.N.; Gu, Z.; Sowers, J.R. Cardiorenal metabolic syndrome and diabetic cognopathy. Cardiorenal Med. 2013, 3, 265–282. [Google Scholar] [CrossRef]
- Firoz, C.K.; Jabir, N.R.; Khan, M.S.; Mahmoud, M.; Shakil, S.; Damanhouri, G.A.; Zaidi, S.K.; Tabrez, S.; Kamal, M.A. An overview on the correlation of neurological disorders with cardiovascular disease. Saudi J. Biol. Sci. 2015, 22, 19–23. [Google Scholar] [CrossRef]
- Hayden, M.R. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262. [Google Scholar] [CrossRef]
- Paoletti, F.P.; Simoni, S.; Parnetti, L.; Gaetani, L. The Contribution of Small Vessel Disease to Neurodegeneration: Focus on Alzheimer’s Disease, Parkinson’s Disease and Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 4958. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Sowers, J.R. Arterial Stiffness: A Nexus between Cardiac and Renal Disease. Cardiorenal Med. 2014, 4, 60–71. [Google Scholar] [CrossRef]
- Hayden, M.R. The Mighty Mitochondria Are Unifying Organelles and Metabolic Hubs in Multiple Organs of Obesity, Insulin Resistance, Metabolic Syndrome, and Type 2 Diabetes: An Observational Ultrastructure Study. Int. J. Mol. Sci. 2022, 23, 4820. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Intimal redox stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovasc. Diabetol. 2002, 1, 3. [Google Scholar] [CrossRef]
- Aroor, A.R.; Das, N.A.; Carpenter, A.J.; Habibi, J.; Jia, G.; Ramirez-Perez, F.I.; Martinez-Lemus, L.; Manrique-Acevedo, C.M.; Hayden, M.R.; Duta, C.; et al. Glycemic control by the SGLT2 inhibitor empagliflozin decreases aortic stiffness, renal resistivity index and kidney injury. Cardiovasc. Diabetol. 2018, 17, 108. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Is type 2 diabetes mellitus a vascular disease (atheroscleropathy) with hyperglycemia a late manifestation? The role of NOS, NO, and redox stress. Cardiovasc. Diabetol. 2003, 2, 2. [Google Scholar] [CrossRef]
- Reaven, G.M. Role of Insulin Resistance in Human Disease (Syndrome X): An Expanded Definition. Annu. Rev. Med. 1993, 44, 121–131. [Google Scholar] [CrossRef]
- Reaven, G.M. The insulin resistance syndrome. Curr. Atheroscler. Rep. 2003, 5, 364–371. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Impaired Folate-Mediated One-Carbon Metabolism in Type 2 Diabetes, Late-Onset Alzheimer’s Disease and Long COVID. Medicina 2021, 58, 16. [Google Scholar] [CrossRef]
- Alberti, K.G.; Zimmet, P.Z. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998, 15, 539–553. [Google Scholar] [CrossRef]
- Friedman, J. Leptin at 20: An overview. J. Endocrinol. 2014, 223, T1–T8. [Google Scholar] [CrossRef]
- Friedman, J. The long road to leptin. J. Clin. Investig. 2016, 126, 4727–4734. [Google Scholar] [CrossRef]
- Ingalls, A.M.; Dickie, M.M.; Snell, G.D. Obese, a new mutation in the house mouse. J. Hered. 1950, 41, 317–318. [Google Scholar] [CrossRef] [PubMed]
- Zucker, L.M.; Zucker, T.F. Fatty, a new mutation in the rat. J. Hered. 1961, 52, 275–278. [Google Scholar] [CrossRef]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a new mutation in the mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.G.; Shaw, W.N.; Neel, M.-A.; Little, L.A.; Eichberg, J. Zucker Diabetic Fatty Rat as a Model for Non-insulin-dependent Diabetes Mellitus. ILAR J. 1990, 32, 16–19. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 1995, 270, 26746–26749. [Google Scholar] [CrossRef]
- Trujillo, M.E.; Scherer, P.E. Adiponectin-journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J. Intern. Med. 2005, 257, 167–175. [Google Scholar] [CrossRef]
- Wang, Z.V.; Scherer, P.E. Adiponectin, the past two decades. J. Mol. Cell Biol. 2016, 8, 93–100. [Google Scholar] [CrossRef]
- Hudkins, K.L.; Pichaiwong, W.; Wietecha, T.; Kowalewska, J.; Banas, M.C.; Spencer, M.W.; Mühlfeld, A.; Koelling, M.; Pippin, J.W.; Shankland, S.J.; et al. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 1533–1542. [Google Scholar] [CrossRef]
- Hayden, M.; Banks, W. Deficient Leptin Cellular Signaling Plays a Key Role in Brain Ultrastructural Remodeling in Obesity and Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2021, 22, 5427. [Google Scholar] [CrossRef]
- Grundy, S.M. Metabolic syndrome: A multiplex cardiovascular risk factor. J. Clin. Endocrinol. 2007, 92, 399–404. [Google Scholar] [CrossRef]
- Alberti, K.G.M.M.; Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z.; Cleeman, J.I.; Donato, K.A.; Fruchart, J.C.; James, W.P.T.; Loria, C.M.; Smith, S.C., Jr.; et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009, 120, 1640–1645. [Google Scholar] [CrossRef]
- Panchal, S.K.; Brown, L. Rodent models for metabolic syndrome research. J. Biomed. Biotechnol. 2011, 2011, 351982. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An Inflammatory Disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Wilson, P.W.F.; Grundy, S.M. The Metabolic Syndrome. A Practical Guide to Origins and Treatment: Part II. Circulation 2003, 108, 1537–1540. [Google Scholar] [CrossRef]
- de Roos, B.; Mavrommatis, Y.; Brouwer, I.A. Long-chain n-3 polyunsaturated fatty acids: New insights into mechanisms relating to inflammation and coronary heart disease. Br. J. Pharmacol. 2009, 158, 413–428. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Tokgözoğlu, L.; Libby, P. The dawn of a new era of targeted lipid-lowering therapies. Eur. Heart J. 2022, 43, 3198–3208. [Google Scholar] [CrossRef]
- Rubins, H.B.; Robins, S.J.; Collins, D.; Fye, C.L.; Anderson, J.W.; Elam, M.B.; Faas, F.H.; Linares, E.; Schaefer, E.J.; Schectman, G.; et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N. Engl. J. Med. 1999, 341, 410–418. [Google Scholar] [CrossRef]
- Hayden, M.R.; Joginpally, T.; Salam, M.; Sowers, J.R. Childhood and adolescent obesity in cardiorenal metabolic syndrome and Type 2 diabetes: A clinical vignette and ultrastructure study. Diabetes Manag. 2011, 1, 601–614. [Google Scholar] [CrossRef]
- Paz-Filho, G.; Mastronardi, C.; Wong, M.-L.; Licinio, J. Leptin therapy, insulin sensitivity, and glucose homeostasis. Indian J. Endocrinol. Metab. 2012, 16, S549–S555. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, M.; Zoico, E.; Fantin, F.; Panourgia, M.P.; Di Francesco, V.; Tosoni, P.; Solerte, B.; Vettor, R.; Bosello, O. Relation Between Leptin and the Metabolic Syndrome in Elderly Women. J. Gerontol. Ser. A 2004, 59, M396–M400. [Google Scholar] [CrossRef] [PubMed]
- Van Gaal, L.F.; Wauters, M.A.; Mertens, I.L.; Considine, R.V.; De Leeuw, I.H. Clinical endocrinology of human leptin. Int. J. Obes. 1999, 23 (Suppl. S1), M29–M36. [Google Scholar] [CrossRef] [PubMed]
- Margetic, S.; Gazzola, C.; Pegg, G.G.; Hill, R.A. Leptin: A review of its peripheral actions and interactions. Int. J. Obes. 2002, 26, 1407–1433. [Google Scholar] [CrossRef] [PubMed]
- Dong, M. What fans the fire: Insights into mechanisms of leptin in metabolic syndrome-associated heart diseases. Curr. Pharm. Des. 2014, 20, 652–658. [Google Scholar] [CrossRef]
- Patel, S.B.; Reams, G.P.; Spear, R.M.; Freeman, R.H.; Villarreal, D. Leptin: Linking obesity, the metabolic syndrome, and cardiovascular disease. Curr. Hypertens. Rep. 2008, 10, 131–137. [Google Scholar] [CrossRef]
- Donahue, R.P.; Prineas, R.J.; Zimmet, P.; Bean, J.A.; De Courten, M.; Collier, G.; Goldberg, R.B.; Skyler, J.S.; Schneiderman, N. Is fasting leptin associated with insulin resistance among nondiabetic individuals? Diabetes Care 1999, 22, 1092–1096. [Google Scholar] [CrossRef]
- Moore, J.X.; Chaudhary, N.; Akinyemiju, T. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988–2012. Prev. Chronic Dis. 2017, 14, E24. [Google Scholar] [CrossRef]
- Ford, E.S.; Giles, W.H.; Dietz, W.H. Prevalence of the metabolic syndrome among US adults: Findings from the third National Health and Nutrition Examination Survey. JAMA 2002, 287, 356–359. [Google Scholar] [CrossRef]
- Ford, E.S.; Giles, W.H.; Mokdad, A.H. Increasing prevalence of the metabolic syndrome among U.S. Adults. Diabetes Care 2004, 27, 2444–2449. [Google Scholar] [CrossRef]
- Reaven, G. Metabolic syndrome: Pathophysiology and implications for management of cardiovascular disease. Circulation 2002, 106, 286–288. [Google Scholar] [CrossRef]
- Hayden, M.R. An Immediate and Long-Term Complication of COVID-19 May Be Type 2 Diabetes Mellitus: The Central Role of β-Cell Dysfunction, Apoptosis and Exploration of Possible Mechanisms. Cells 2020, 9, 2475. [Google Scholar] [CrossRef]
- Jaikaran, E.T.; Clark, A. Islet amyloid and type 2 diabetes: From molecular misfolding to islet pathophysiology. Biochim. Biophys. Acta 2001, 1537, 179–203. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C.; Kerklo, M.M.; Nicolls, M.R. Type 2 diabetes mellitus as a conformational disease. JOP 2005, 6, 287–302. [Google Scholar]
- Westwell-Roper, C.Y.; Chehroudi, C.A.; Denroche, H.; Courtade, J.A.; Ehses, J.; Verchere, C.B. IL-1 mediates amyloid-associated islet dysfunction and inflammation in human islet amyloid polypeptide transgenic mice. Diabetologia 2015, 58, 575–585. [Google Scholar] [CrossRef]
- D’Alessio, D.A.; Verchere, C.B.; Kahn, S.E.; Hoagland, V.; Baskin, D.G.; Palmiter, R.D.; Ensinck, J.W. Pancreatic Expression and Secretion of Human Islet Amyloid Polypeptide in a Transgenic Mouse. Diabetes 1994, 43, 1457–1461. [Google Scholar] [CrossRef]
- Haataja, L.; Gurlo, T.; Huang, C.J.; Butler, P.C. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr. Rev. 2008, 29, 303–316. [Google Scholar] [CrossRef]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar] [CrossRef]
- Cooper, M.E.; McNally, P.G.; Phillips, P.A.; Johnston, C.I. Amylin stimulates plasma renin concentrations in humans. Hypertension 1995, 26, 460–464. [Google Scholar] [CrossRef]
- Hull, R.L.; Westermark, G.T.; Westermark, P.; Kahn, S.E. Islet amyloid: A critical entity in the pathogenesis of type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 3629–3643. [Google Scholar] [CrossRef]
- Kahn, S.E.; Andrikopoulos, S.; Verchere, C.B. Islet amyloid: A long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes 1999, 48, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Biessels, G.J.; Despa, F. Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nat. Rev. Endocrinol. 2018, 14, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Despa, F.; Goldstein, L.B. Amylin Dyshomeostasis Hypothesis: Small Vessel-Type Ischemic Stroke in the Setting of Type-2 Diabetes. Stroke 2021, 52, e244–e249. [Google Scholar] [CrossRef] [PubMed]
- Ly, H.; Despa, F. Hyperamylinemia as a risk factor for accelerated cognitive decline in diabetes. Expert Rev. Proteom. 2015, 12, 575–577. [Google Scholar] [CrossRef]
- Ly, H.; Despa, F. Diabetes-related Amylin Dyshomeostasis: A Contributing Factor to Cerebrovascular Pathology and Dementia. J. Lipid Atheroscler. 2019, 8, 144–151. [Google Scholar] [CrossRef]
- Despa, F.; Decarli, C. Amylin: What might be its role in Alzheimer’s disease and how could this affect therapy? Expert Rev. Proteom. 2013, 10, 403–405. [Google Scholar] [CrossRef]
- Opie, E.L. Pathological changes affecting the islands of langerhans of the pancreas. J. Boston Soc. Med. Sci. 1900, 4, 251–260. [Google Scholar]
- Opie, E.L. The relation oe diabetes mellitus to lesions of the pancreas. Hyaline degeneration of the islands oe langerhans. J. Exp. Med. 1901, 5, 527–540. [Google Scholar] [CrossRef]
- Ma, Z.A.; Zhao, Z.; Turk, J. Mitochondrial Dysfunction andβ-Cell Failure in Type 2 Diabetes Mellitus. Exp. Diabetes Res. 2012, 2012, 703538. [Google Scholar] [CrossRef]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289. [Google Scholar] [CrossRef]
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165838. [Google Scholar] [CrossRef]
- Weksler-Zangen, S. Is Type 2 Diabetes a Primary Mitochondrial Disorder? Cells 2022, 11, 1617. [Google Scholar] [CrossRef]
- Guzzardi, M.A.; Guiducci, L.; Campani, D.; La Rosa, F.; Insilla, A.C.; Bartoli, A.; Cabiati, M.; De Sena, V.; Del Ry, S.; Burchielli, S.; et al. Leptin resistance before and after obesity: Evidence that tissue glucose uptake underlies adipocyte enlargement and liver steatosis/steatohepatitis in Zucker rats from early-life stages. Int. J. Obes. 2022, 46, 50–58. [Google Scholar] [CrossRef]
- Ghadge, A.A.; Khaire, A.A. Leptin as a predictive marker for metabolic syndrome. Cytokine 2019, 121, 154735. [Google Scholar] [CrossRef]
- Kelly, A.S.; Metzig, A.M.; Schwarzenberg, S.J.; Norris, A.L.; Fox, C.K.; Steinberger, J. Hyperleptinemia and Hypoadiponectinemia in Extreme Pediatric Obesity. Metab. Syndr. Relat. Disord. 2012, 10, 123–127. [Google Scholar] [CrossRef]
- Frühbeck, G.; Catalán, V.; Rodríguez, A.; Gómez-Ambrosi, J. Adiponectin-leptin ratio: A promising index to estimate adipose tissue dysfunction. Relation with obesity-associated cardiometabolic risk. Adipocyte 2018, 7, 57–62. [Google Scholar] [CrossRef]
- Fisman, E.Z.; Tenenbaum, A. Adiponectin: A manifold therapeutic target for metabolic syndrome, diabetes, and coronary disease? Cardiovasc. Diabetol. 2014, 13, 103. [Google Scholar] [CrossRef]
- Ghadge, A.A.; Khaire, A.A.; Kuvalekar, A.A. Adiponectin: A potential therapeutic target for metabolic syndrome. Cytokine Growth Factor Rev. 2018, 39, 151–158. [Google Scholar] [CrossRef]
- Menon, B.; Krishnan, R. Role of Leptin in Acute Ischemic Stroke. J. Neurosci. Rural Pract. 2018, 09, 376–380. [Google Scholar] [CrossRef]
- Söderberg, S.; Stegmayr, B.; Ahlbeck-Glader, C.; Slunga-Birgander, L.; Ahrén, B.; Olsson, T. High leptin levels are associated with stroke. Cerebrovasc. Dis. 2003, 15, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Beltowski, J. Leptin and atherosclerosis. Atherosclerosis 2006, 189, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.; Polotsky, V.Y. Leptin and Leptin Resistance in the Pathogenesis of Obstructive Sleep Apnea: A Possible Link to Oxidative Stress and Cardiovascular Complications. Oxidative Med. Cell. Longev. 2018, 2018, 5137947. [Google Scholar] [CrossRef] [PubMed]
- United Nations Department of Economics and Social Affairs Populations Division Population Ageing and Sustainable Development. Available online: https://www.un.org/development/desa/pd/sites/www.un.org.development.desa.pd/files/wpp2022_summary_of_results.pdf (accessed on 30 January 2023).
- Ma, X.H.; Muzumdar, R.; Yang, X.M.; Gabriely, I.; Berger, R.; Barzilai, N. Aging Is Associated With Resistance to Effects of Leptin on Fat Distribution and Insulin Action. J. Gerontol. A Biol. Sci. Med. Sci. 2002, 57, B225–B231. [Google Scholar] [CrossRef] [PubMed]
- Gabriely, I.; Ma, X.H.; Yang, X.M.; Rossetti, L.; Barzilai, N. Leptin resistance during aging is independent of fat mass. Diabetes 2002, 51, 1016–1021. [Google Scholar] [CrossRef]
- Stern, J.H.; Rutkowski, J.M.; Scherer, P.E. Adiponectin, Leptin, and Fatty Acids in the Maintenance of Metabolic Homeostasis through Adipose Tissue Crosstalk. Cell Metab. 2016, 23, 770–784. [Google Scholar] [CrossRef]
- Messerli, F.H.; Williams, B.; Ritz, E. Essential hypertension. Lancet 2007, 370, 591–603. [Google Scholar] [CrossRef]
- Sowers, J.R. Insulin resistance and hypertension. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1597–H1602. [Google Scholar] [CrossRef]
- Addison, S.; Stas, S.; Hayden, M.R.; Sowers, J.R. Insulin Resistance and Blood Pressure. Curr. Hypertens. Rep. 2008, 10, 319–325. [Google Scholar] [CrossRef]
- Correia, M.L.G.; Rahmouni, K. Role of leptin in the cardiovascular and endocrine complications of metabolic syndrome. Diabetes, Obes. Metab. 2006, 8, 603–610. [Google Scholar] [CrossRef]
- Haynes, W.G. Role of leptin in obesity-related hypertension. Exp. Physiol. 2005, 90, 683–688. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Ungvari, Z.; Itani, H.; Richmond, B.; Vergeade, A.; Rahman, S.M.J.; Boutaud, O.; Blackwell, T.; Massion, P.P.; Harrison, D.G.; et al. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1417–H1427. [Google Scholar] [CrossRef]
- Kim, J.; Wei, Y.; Sowers, J.R. Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef]
- Nicolson, G.L. Metabolic Syndrome and Mitochondrial Function: Molecular Replacement and Antioxidant Supplements to Prevent Membrane Peroxidation and Restore Mitochondrial Function. J. Cell. Biochem. 2007, 100, 1352–1369. [Google Scholar] [CrossRef]
- Van Guldener, C.; Nanayakkara, P.W.B.; Stehouwer, C.D.A. Homocysteine and blood pressure. Curr. Hypertens. Rep. 2003, 5, 26–31. [Google Scholar] [CrossRef]
- Lopes-Vicente, W.R.P.; Rodrigues, S.; Cepeda, F.X.; Jordão, C.P.; Costa-Hong, V.; Dutra-Marques, A.C.B.; Carvalho, J.C.; Alves, M.J.N.N.; Bortolotto, L.A.; Trombetta, I.C. Arterial stiffness and its association with clustering of metabolic syndrome risk factors. Diabetol. Metab. Syndr. 2017, 9, 87. [Google Scholar] [CrossRef]
- Schillaci, G.; Pirro, M.; Vaudo, G.; Mannarino, M.R.; Savarese, G.; Pucci, G.; Franklin, S.S.; Mannarino, E. Metabolic Syndrome Is Associated with Aortic Stiffness in Untreated Essential Hypertension. Hypertension 2005, 45, 1078–1082. [Google Scholar] [CrossRef]
- Mitchell, G.F.; Hwang, S.-J.; Vasan, R.S.; Larson, M.; Pencina, M.J.; Hamburg, N.; Vita, J.; Levy, D.; Benjamin, E. Arterial stiffness and cardiovascular events: The Framingham Heart Study. Circulation 2010, 121, 505–511. [Google Scholar] [CrossRef]
- Howes, L.G.; Brillante, D.G.; O’Sullivan, A.J. Arterial stiffness in insulin resistance: The role of nitric oxide and angiotensin II receptors. Vasc. Health Risk Manag. 2009, 5, 73–78. [Google Scholar] [CrossRef]
- Jia, G.; Aroor, A.R.; Demarco, V.G.; Martinez-Lemus, L.A.; Meininger, G.A.; Sowers, J.R. Vascular stiffness in insulin resistance and obesity. Front. Physiol. 2015, 6, 231. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, J.; Chaudhry, M.A.; Nie, Y.; Sun, S.; Carmon, J.; Shah, P.T.; Bai, F.; Pratt, R.; Brickman, C.; et al. Metabolic Syndrome and Salt-Sensitive Hypertension in Polygenic Obese TALLYHO/JngJ Mice: Role of Na/K-ATPase Signaling. Int. J. Mol. Sci. 2019, 20, 3495. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T. Aldosterone in salt-sensitive hypertension and metabolic syndrome. J. Mol. Med. 2008, 86, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Alexander, C.M.; Landsman, P.B.; Grundy, S.M. Metabolic Syndrome and Hyperglycemia: Congruence and Divergence. Am. J. Cardiol. 2006, 98, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Oguntibeju, O.O. Type 2 diabetes mellitus, oxidative stress and inflammation: Examining the links. Int. J. Physiol. Pathophysiol. Pharmacol. 2019, 11, 45–63. [Google Scholar]
- Yang, Y.; Hayden, M.R.; Sowers, S.; Bagree, S.V.; Sowers, J.R. Retinal redox stress and remodeling in cardiometabolic syndrome and diabetes. Oxidative Med. Cell. Longev. 2010, 3, 392–403. [Google Scholar] [CrossRef]
- Hayden, M.R.; Salam, M.; Sowers, J.R. Reactive Oxygen Species and Diabetic Peripheral Neuropathy—A Closer Look; Lahey, I., Ed.; Systems Biology of Reactive Oxygen Species and Antioxidants; Springer: Berlin/Heidelberg, Germany, 2014; Chapter 149; pp. 3375–3400. [Google Scholar]
- Hayden, M.R.; Whaley-Connell, A.; Sowers, J.R. Renal redox stress and remodeling in metabolic syndrome, type 2 diabetes mellitus, and diabetic nephropathy: Paying homage to the podocyte. Am. J. Nephrol. 2005, 25, 553–569. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R. Isletopathy in Type 2 diabetes mellitus: Implications of islet RAS, islet fibrosis, islet amyloid, remodeling, and oxidative stress. Antioxid. Redox Signal. 2007, 9, 891–910. [Google Scholar] [CrossRef]
- Jia, G.; Hill, M.A.; Sowers, J.R. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ. Res. 2018, 122, 624–638. [Google Scholar] [CrossRef]
- Climie, R.E.; van Sloten, T.T.; Bruno, R.-M.; Taddei, S.; Empana, J.-P.; Stehouwer, C.D.; Sharman, J.E.; Boutouyrie, P.; Laurent, S. Macrovasculature and Microvasculature at the Crossroads between Type 2 Diabetes Mellitus and Hypertension. Hypertension 2019, 73, 1138–1149. [Google Scholar] [CrossRef]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; DeMarco, V.G. Ultrastructural Remodeling of The Neurovascular Unit in the Female Diabetic db/db Model—Part I: Astrocyte. Neuroglia 2018, 1, 220–244. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; Demarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model–Part II: Microglia and Mitochondria. Neuroglia 2018, 1, 311–326. [Google Scholar] [CrossRef]
- Hayden, M.R.; Grant, D.G.; Aroor, A.R.; Demarco, V.G. Ultrastructural Remodeling of the Neurovascular Unit in the Female Diabetic db/db Model—Part III: Oligodendrocyte and Myelin. Neuroglia 2018, 1, 351–367. [Google Scholar] [CrossRef]
- Hayden, M.R.; Chowdhury, N.A.; Cooper, S.A.; Whaley-Connell, A.; Habibi, J.; Witte, L.; Wiedmeyer, C.; Manrique, C.M.; Lastra, G.; Ferrario, C.; et al. Proximal tubule microvilli remodeling and albuminuria in the Ren2 transgenic rat. Am. J. Physiol. Renal. Physiol. 2007, 292, F861–F867. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R. Childhood-Adolescent Obesity in the Cardiorenal Syndrome: Lessons from Animal Models. Cardiorenal Med. 2011, 1, 75–86. [Google Scholar] [CrossRef]
- Habibi, J.; Aroor, A.R.; Sowers, J.R.; Jia, G.; Hayden, M.R.; Garro, M.; Barron, B.; Mayoux, E.; Rector, R.S.; Whaley-Connell, A.; et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc. Diabetol. 2017, 16, 9. [Google Scholar] [CrossRef]
- Hayden, M.R.; Chowdhury, N.; Govindarajan, G.; Karuparthi, P.R.; Habibi, J.; Sowers, J.R. Myocardial myocyte remodeling and fibrosis in the cardiometabolic syndromee. J. CardioMetabolic Syndr. 2006, 1, 326–333. [Google Scholar] [CrossRef]
- Hayden, M.R. Empagliflozin Ameliorates Tunica Adiposa Expansion and Vascular Stiffening of the Descending Aorta in Female db/db Mice. Adipobiology 2020, 10, 41–54. [Google Scholar] [CrossRef]
- Salameh, T.S.; Shah, G.N.; Price, T.O.; Hayden, M.R.; Banks, W.A. Blood-Brain Barrier Disruption and Neurovascular Unit Dysfunction in Diabetic Mice: Protection with the Mitochondrial Carbonic Anhydrase Inhibitor Topiramate. J. Pharmacol. Exp. Ther. 2016, 359, 452–459. [Google Scholar] [CrossRef]
- Stover, P.J. One-Carbon Metabolism–Genome Interactions in Folate-Associated Pathologies. J. Nutr. 2009, 139, 2402–2405. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C. Homocysteine and reactive oxygen species in metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: The pleiotropic effects of folate supplementation. Nutr. J. 2004, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Finer, S.; Saravanan, P.; Hitman, G.; Yajnik, C. The role of the one-carbon cycle in the developmental origins of Type 2 diabetes and obesity. Diabet. Med. 2014, 31, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.B.; Baipadithaya, G.; Balakrishnan, A.; Hegde, M.; Vohra, M.; Ahamed, R.; Nagri, S.K.; Ramachandra, L.; Satyamoorthy, K. Elevated homocysteine levels in type 2 diabetes induce constitutive neutrophil extracellular traps. Sci. Rep. 2016, 6, 36362. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Ren, J.; Huang, J.; Li, D. Association of homocysteine with type 2 diabetes: A meta-analysis implementing Mendelian randomization approach. BMC Genom. 2013, 14, 867. [Google Scholar] [CrossRef]
- Piazzolla, G.; Candigliota, M.; Fanelli, M.; Castrovilli, A.; Berardi, E.; Antonica, G.; Battaglia, S.; Solfrizzi, V.; Sabbà, C.; Tortorella, C. Hyperhomocysteinemia is an independent risk factor of atherosclerosis in patients with metabolic syndrome. Diabetol. Metab. Syndr. 2019, 11, 87–89. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.-O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef]
- Burgess, K.; Bennett, C.; Mosnier, H.; Kwatra, N.; Bethel, F.; Jadavji, N.M. The Antioxidant Role of One-Carbon Metabolism on Stroke. Antioxidants 2020, 9, 1141. [Google Scholar] [CrossRef]
- Welch, G.N.; Loscalzo, J. Homocysteine and atherothrombosis. N. Engl. J. Med. 1998, 338, 1042–1050. [Google Scholar] [CrossRef]
- Arruda, V.R.; von Zuben, P.M.; Chiaparini, L.C.; Annichino-Bizzacchi, J.M.; Costa, F.F. The mutation Ala677 → Val in the methylene tetrahydrofolate reductase gene: A risk factor for arterial disease and venous thrombosis. Thromb. Haemost. 1997, 77, 818–821. [Google Scholar] [CrossRef]
- Feix, A.; Fritsche-Polanz, R.; Kletzmayr, J.; Vychytil, A.; Hörl, W.H.; Sunder-Plassmann, G.; Födinger, M. Increased prevalence of combined MTR and MTHFR genotypes among individuals with severely elevated total homocysteine plasma levels. Am. J. Kidney Dis. 2001, 38, 956–964. [Google Scholar] [CrossRef]
- Woo, K.S.; Chook, P.; Lolin, Y.I.; Sanderson, J.E.; Metreweli, C.; Celermajer, D.S. Folic acid improves arterial endothelial function in adults with hyperhomocystinemia. J. Am. Coll. Cardiol. 1999, 34, 2002–2006. [Google Scholar] [CrossRef]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Investig. 2013, 123, 540–541. [Google Scholar] [CrossRef]
- Hayden, M.R. Endothelial activation and dysfunction in metabolic syndrome, type 2 diabetes and coronavirus disease 2019. J. Int. Med. Res. 2020, 48, 300060520939746. [Google Scholar] [CrossRef]
- Christ, M.; Bauersachs, J.; Liebetrau, C.; Heck, M.; Günther, A.; Wehling, M. Glucose Increases Endothelial-Dependent Superoxide Formation in Coronary Arteries by NAD(P)H Oxidase Activation: Attenuation by the 3-Hydroxy-3-Methylglutaryl Coenzyme A. Diabetes 2002, 51, 2648–2652. [Google Scholar] [CrossRef]
- Padilla, J.; Ramirez-Perez, F.; Habibi, J.; Bostick, B.; Aroor, A.R.; Hayden, M.R.; Jia, G.; Garro, M.; DeMarco, V.; Manrique, C.; et al. Regular exercise reduces endothelial cortical stiffness in Western diet-fed female mice. Hypertension 2016, 68, 1236–1244. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; Aroor, A.R.; Martinez-Lemus, L.A.; DeMarco, V.G.; Ramirez-Perez, F.I.; Sun, Z.; Hayden, M.R.; Meininger, G.A.; Mueller, K.B.; et al. Endothelial Mineralocorticoid Receptor Mediates Diet Induced Aortic Stiffness in Females. Circ. Res. 2016, 118, 935–943. [Google Scholar] [CrossRef]
- Sowers, J.R.; Habibi, J.; Jia, G.; Bostick, B.; Manrique-Acevedo, C.; Lastra, G.; Yang, Y.; Chen, D.; Sun, Z.; Domeier, T.L.; et al. Endothelial sodium channel activation promotes cardiac stiffness and diastolic dysfunction in Western diet fed female mice. Metabolism 2020, 109, 154223. [Google Scholar] [CrossRef]
- Stepp, D.W.; Ou, J.; Ackerman, A.W.; Welak, S.; Klick, D.; Pritchard, K.A. Native LDL and minimally oxidized LDL differentially regulate superoxide anion in vascular endothelium in situ. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H750–H759. [Google Scholar] [CrossRef]
- Zou, M.-H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Verma, S.; Wang, C.-H.; Li, S.-H.; Dumont, A.S.; Fedak, P.; Badiwala, M.V.; Dhillon, B.; Weisel, R.D.; Li, R.-K.; Mickle, D.A.; et al. A self fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation 2002, 106, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Mussa, S.; Gastaldi, D.; Sadowski, J.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Mechanisms of increased vascular superoxide production in human diabetes mellitus: Role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 2002, 105, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Aroor, A.R.; Habibi, J.; Kandikattu, H.K.; Garro-Kacher, M.; Barron, B.; Chen, D.; Hayden, M.R.; Whaley-Connell, A.; Bender, S.B.; Klein, T.; et al. Dipeptidyl peptidase-4 (DPP-4) inhibition with linagliptin reduces western diet-induced myocardial TRAF3IP2 expression, inflammation and fibrosis in female mice. Cardiovasc. Diabetol. 2017, 16, 61. [Google Scholar] [CrossRef] [PubMed]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 2010, 88, 993–1001. [Google Scholar] [CrossRef]
- Vásquez-Vivar, J.; Kalyanaraman, B.; Martásek, P.; Hogg, N.; Masters, B.S.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef]
- Elitvinova, L.; Atochin, D.N.; Fattakhov, N.; Evasilenko, M.; Ezatolokin, P.; Kirienkova, E. Nitric oxide and mitochondria in metabolic syndrome. Front. Physiol. 2015, 6, 20. [Google Scholar] [CrossRef]
- Martin, S.D.; McGee, S.L. The role of mitochondria in the aetiology of insulin resistance and type 2 diabetes. Biochim. Biophys. Acta 2014, 1840, 1303–1312. [Google Scholar] [CrossRef]
- du Preez, H.N.; Aldous, C.; Hayden, M.R.; Kruger, H.G.; Lin, J. Pathogenesis of COVID-19 described through the lens of an undersulfated and degraded epithelial and endothelial glycocalyx. FASEB J. 2022, 36, e22052. [Google Scholar] [CrossRef]
- Ando, Y.; Okada, H.; Takemura, G.; Suzuki, K.; Takada, C.; Tomita, H.; Zaikokuji, R.; Hotta, Y.; Miyazaki, N.; Yano, H.; et al. Brain-Specific Ultrastructure of Capillary Endothelial Glycocalyx and Its Possible Contribution for Blood Brain Barrier. Sci. Rep. 2018, 8, 17523. [Google Scholar] [CrossRef]
- Sampei, S.; Okada, H.; Tomita, H.; Takada, C.; Suzuki, K.; Kinoshita, T.; Kobayashi, R.; Fukuda, H.; Kawasaki, Y.; Nishio, A.; et al. Endothelial Glycocalyx Disorders May Be Associated with Extended Inflammation during Endotoxemia in a Diabetic Mouse Model. Front. Cell Dev. Biol. 2021, 9, 623582. [Google Scholar] [CrossRef]
- Okada, H.; Takemura, G.; Suzuki, K.; Oda, K.; Takada, C.; Hotta, Y.; Miyazaki, N.; Tsujimoto, A.; Muraki, I.; Ando, Y.; et al. Three-dimensional ultrastructure of capillary endothelial glycocalyx under normal and experimental endotoxemic conditions. Crit. Care 2017, 21, 261. [Google Scholar] [CrossRef]
- Kutuzov, N.; Flyvbjerg, H.; Lauritzen, M. Contributions of the glycocalyx, endothelium, and extravascular compartment to the blood–brain barrier. Proc. Natl. Acad. Sci. USA 2018, 115, E9429–E9438. [Google Scholar] [CrossRef]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.M.J.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflug. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Hayden, M.R.; Sowers, J.R.; Tyagi, S.C. The central role of vascular extracellular matrix and basement membrane remodeling in metabolic syndrome and type 2 diabetes: The matrix preloaded. Cardiovasc. Diabetol. 2005, 4, 9. [Google Scholar] [CrossRef]
- Ericsson, A.; Tonelius, P.; Lal, M.; Sabirsh, A.; Böttcher, G.; William-Olsson, L.; Strömstedt, M.; Johansson, C.; Hyberg, G.; Tapani, S.; et al. The effects of dual PPARα/γ agonism compared with ACE inhibition in the BTBRob/ob mouse model of diabetes and diabetic nephropathy. Physiol. Rep. 2017, 5, e13186. [Google Scholar] [CrossRef]
- Kolářová, H.; Ambrůzová, B.; Šindlerová, L.; Klinke, A.; Kubala, L. Modulation of endothelial glycocalyx structure under inflammatory conditions. Mediat. Inflamm. 2014, 2014, 694312. [Google Scholar] [CrossRef]
- Dogné, S.; Flamion, B.; Caron, N. Endothelial Glycocalyx as a Shield Against Diabetic Vascular Complications: Involvement of Hyaluronan and Hyaluronidases. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1427–1439. [Google Scholar] [CrossRef]
- Stackhouse, T.L.; Mishra, A. Neurovascular Coupling in Development and Disease: Focus on Astrocytes. Front. Cell Dev. Biol. 2021, 9, 702832. [Google Scholar] [CrossRef]
- Alves, N.G.; Trujillo, A.N.; Breslin, J.W.; Yuan, S.Y. Sphingosine-1-Phosphate (S1P) Reduces Hemorrhagic Shock and Resuscitation-Induced Microvascular Leakage by Protecting Endothelial Mitochondrial Integrity. Shock 2019, 52, 423–433. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Xu, H.-X.; Li, J.-K.; Zhang, D.; Ma, X.-H.; Huang, L.-N.; Lü, J.-H.; Wang, X.-Z. Neferine Protects Endothelial Glycocalyx via Mitochondrial ROS in Lipopolysaccharide-Induced Acute Respiratory Distress Syndrome. Front. Physiol. 2018, 9, 102. [Google Scholar] [CrossRef] [PubMed]
- Alhayaza, R.; Haque, E.; Karbasiafshar, C.; Sellke, F.W.; Abid, M.R. The Relationship between Reactive Oxygen Species and Endothelial Cell Metabolism. Front. Chem. 2020, 8, 592688. [Google Scholar] [CrossRef] [PubMed]
- Koziel, A.; Sobieraj, I.; Jarmuszkiewicz, W. Increased activity of mitochondrial uncoupling protein 2 improves stress resistance in cultured endothelial cells exposed in vitro to high glucose levels. Am. J. Physiol. Circ. Physiol. 2015, 309, H147–H156. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Yen, W.; Cai, B.; Yang, J.; Zhang, L.; Zeng, M.; Tarbell, J.M.; Fu, B.M. Endothelial surface glycocalyx can regulate flow-induced nitric oxide production in microvessels in vivo. PLoS ONE 2015, 10, e0117133. [Google Scholar] [CrossRef]
- Valenti, D.; Vacca, R.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef]
- Broekhuizen, L.N.; Lemkes, B.A.; Mooij, H.L.; Meuwese, M.C.; Verberne, H.; Holleman, F.; Schlingemann, R.O.; Nieuwdorp, M.; Stroes, E.S.G.; Vink, H. Effect of sulodexide on endothelial glycocalyx and vascular permeability in patients with type 2 diabetes mellitus. Diabetologia 2010, 53, 2646–2655. [Google Scholar] [CrossRef]
- Lee, M.; McGeer, E.G.; McGeer, P.L. Sodium thiosulfate attenuates glial-mediated neuroinflammation in degenerative neurological diseases. J. Neuroinflamm. 2016, 13, 32. [Google Scholar] [CrossRef]
- du Preez, H.N.; Aldous, C.; Hayden, M.R.; Kruger, H.G.; Lin, J. N-acetylcysteine and other sulfur-donors as a preventative and adjunct therapy for COVID-19. Adv. Pharmacol. Pharm. Sci. J. 2022, 2022, 4555490. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Spiegelman, B.M. Tumor necrosis factor alpha: A key component of the obesity-diabetes link. Diabetes 1994, 43, 1271–1278. [Google Scholar] [CrossRef]
- Esposito, K.; Giugliano, D. The metabolic syndrome and inflammation: Association or causation? Nutr. Metab. Cardiovasc. Dis. 2004, 14, 228–232. [Google Scholar] [CrossRef]
- Mohammad, S.; Thiemermann, C. Role of Metabolic Endotoxemia in Systemic Inflammation and Potential Interventions. Front. Immunol. 2021, 11, 594150. [Google Scholar] [CrossRef]
- Singh, H.; Miyamoto, S.; Darshi, M.; Torralba, M.; Kwon, K.; Sharma, K.; Pieper, R. Gut Microbial Changes in Diabetic db/db Mice and Recovery of Microbial Diversity upon Pirfenidone Treatment. Microorganisms 2020, 8, 1347. [Google Scholar] [CrossRef]
- Di Tommaso, N.; Santopaolo, F.; Gasbarrini, A.; Ponziani, F.R. The Gut–Vascular Barrier as a New Protagonist in Intestinal and Extraintestinal Diseases. Int. J. Mol. Sci. 2023, 24, 1470. [Google Scholar] [CrossRef]
- Tilg, H.; Kaser, A. Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Investig. 2011, 121, 2126–2132. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Schmidt, M.I.; Duncan, B.B.; Sharrett, A.R.; Lindberg, G.; Savage, P.J.; Offenbacher, S.; Azambuja, M.I.; Tracy, R.P.; Heiss, G. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): A cohort study. Lancet 1999, 353, 1649–1652. [Google Scholar] [CrossRef]
- Duncan, B.B.; Schmidt, M.I.; Pankow, J.S.; Ballantyne, C.M.; Couper, D.; Vigo, A.; Hoogeveen, R.; Folsom, A.R.; Heiss, G. Low-grade systemic inflammation and the development of type 2 diabetes: The atherosclerosis risk in communities study. Atherosclerosis Risk in Communities Study. Diabetes 2003, 52, 1799–1805. [Google Scholar] [CrossRef]
- Scheithauer, T.P.M.; Rampanelli, E.; Nieuwdorp, M.; Vallance, B.A.; Verchere, C.B.; van Raalte, D.H.; Herrema, H. Gut Microbiota as a Trigger for Metabolic Inflammation in Obesity and Type 2 Diabetes. Front. Immunol. 2020, 11, 571731. [Google Scholar] [CrossRef]
- de Git, K.C.G.; Adan, R.A.H. Leptin resistance in diet-induced obesity: The role of hypothalamic inflammation. Obes. Rev. 2015, 16, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, A.; Sánchez-Jiménez, F.; Vilariño-García, T.; Sánchez-Margalet, V. Role of Leptin in Inflammation and Vice Versa. Int. J. Mol. Sci. 2020, 21, 5887. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, K.; MacIver, N.J. The Role of the Adipokine Leptin in Immune Cell Function in Health and Disease. Front. Immun. Front. Immunol. 2021, 11, 622468. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.; Lago, R.; Gomez, R.; Dieguez, C.; Lago, F.; Gómez-Reino, J.; Gualillo, O. Towards a pro-inflammatory and immunomodulatory emerging role of leptin. Rheumatology 2006, 45, 944–950. [Google Scholar] [CrossRef]
- Martin, S.S.; Qasim, A.; Reilly, M.P. Leptin resistance: A possible interface of inflammation and metabolism in obesity-related cardiovascular disease. J. Am. Coll. Cardiol. 2008, 52, 1201–1210. [Google Scholar] [CrossRef]
- Hayden, M.R. Overview of Neuroglia Activation, Chronic Neuroinflammation, Remodeling, and Impaired Cognition Due to Perivascular Adipose Tissue-Derived Extracellular Vesicle Exosomes in Obesity and Diabetes. Neuroglia 2022, 3, 112–138. [Google Scholar] [CrossRef]
- Lau, D.C.; Dhillon, B.; Yan, H.; Szmitko, P.E.; Verma, S. Adipokines: Molecular links between obesity and atheroslcerosis. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2031–H2041. [Google Scholar] [CrossRef]
- Molica, F.; Morel, S.; Kwak, B.R.; Rohner-Jeanrenaud, F.; Steffens, S. Adipokines at the crossroad between obesity and cardiovascular disease. Thromb. Haemost. 2015, 113, 553–566. [Google Scholar] [CrossRef]
- Mohammadi, M.H.; Gozashti, M.; Aghadavood, M.; Mehdizadeh, M.R.; Hayatbakhsh, M.M. Clinical Significance of Serum IL-6 and TNF-α Levels in Patients with Metabolic Syndrome. Rep. Biochem. Mol. Biol. 2017, 6, 74–79. [Google Scholar]
- Bing, C. Is interleukin-1β a culprit in macrophage-adipocyte crosstalk in obesity? Adipocyte 2015, 4, 149–152. [Google Scholar] [CrossRef]
- Henderson, G. Plasma Free Fatty Acid Concentration as a Modifiable Risk Factor for Metabolic Disease. Nutrients 2021, 13, 2590. [Google Scholar] [CrossRef]
- Bussler, S.; Penke, M.; Flemming, G.; Elhassan, Y.S.; Kratzsch, J.; Sergeyev, E.; Lipek, T.; Vogel, M.; Spielau, U.; Körner, A.; et al. Novel Insights in the Metabolic Syndrome in Childhood and Adolescence. Horm. Res. Paediatr. 2017, 88, 181–193. [Google Scholar] [CrossRef]
- Gao, X.; Salomon, C.; Freeman, D.J. Extracellular Vesicles from Adipose Tissue—A Potential Role in Obesity and Type 2 Diabetes? Front. Endocrinol. 2017, 8, 202. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, C.; Wei, M.; Yang, G.; Yuan, L. Multifaceted Roles of Adipose Tissue-Derived Exosomes in Physiological and Pathological Conditions. Front. Physiol. 2021, 12, 669429. [Google Scholar] [CrossRef]
- Deng, Z.-B.; Poliakov, A.; Hardy, R.W.; Clements, R.; Liu, C.; Liu, Y.; Wang, J.; Xiang, X.; Zhang, S.; Zhuang, X.; et al. Adipose tissue exosome-like vesicles mediate activation of macrophage-induced insulin resistance. Diabetes 2009, 58, 2498–2505. [Google Scholar] [CrossRef]
- Pan, Y.; Hui, X.; Hoo, R.L.C.; Ye, D.; Chan, C.Y.C.; Feng, T.; Wang, Y.; Lam, K.S.L.; Xu, A. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J. Clin. Investig. 2019, 129, 834–849. [Google Scholar] [CrossRef]
- Kwan, H.Y.; Chen, M.; Xu, K.; Chen, B. The impact of obesity on adipocyte-derived extracellular vesicles. Cell. Mol. Life Sci. 2021, 78, 7275–7288. [Google Scholar] [CrossRef]
- Żbikowski, A.; Błachnio-Zabielska, A.; Galli, M.; Zabielski, P. Adipose-Derived Exosomes as Possible Players in the Development of Insulin Resistance. Int. J. Mol. Sci. 2021, 22, 7427. [Google Scholar] [CrossRef]
- Guglielmi, V.; D’Adamo, M.; Menghini, R.; Marina, C.; Paolo, G.; Massimo, F.; Paolo, S. MicroRNA 21 is up-regulated in adipose tissue of obese diabetic subjects. Nutr. Health Aging 2017, 4, 141–145. [Google Scholar] [CrossRef]
- Tryggestad, J.B.; Teague, A.M.; Sparling, D.P.; Jiang, S.; Chernausek, S.D. Macrophage-Derived microRNA-155 Increases in Obesity and Influences Adipocyte Metabolism by Targeting Peroxisome Proliferator-Activated Receptor Gamma. Obesity 2019, 27, 1856–1864. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Morris, K.; Wood, M.J.A. The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130507. [Google Scholar] [CrossRef] [PubMed]
- Riva, P.; Ratti, A.; Venturin, M. The Long Non-Coding RNAs in Neurodegenerative Diseases: Novel Mechanisms of Pathogenesis. Curr. Alzheimer Res. 2016, 13, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Chadt, A.; Scherneck, S.; Joost, H.G.; Al-Hasanin, H. Molecular links between Obesity and Diabetes: “Diabesity”. In Endotext [Internet]; MDText.com, Inc.: South Dartmouth, MA, USA, 2000; Updated 2018. Available online: www.endotext.org. https://www.ncbi.nlm.nih.gov/books/NBK279051/ (accessed on 19 January 2023).
- Simons, L.A.; Simons, J.; Friedlander, Y.; McCallum, J. Is Prediction of Cardiovascular Disease and All-cause Mortality Genuinely Driven by the Metabolic Syndrome, and Independently from its Component Variables? The Dubbo Study. Heart Lung Circ. 2011, 20, 214–219. [Google Scholar] [CrossRef]
- Reilly, M.P.; Rader, D.J. The Metabolic Syndrome More than the Sum of Its Parts? Circulation 2003, 108, 1546–1551. [Google Scholar] [CrossRef]
- Reaven, G.M. The metabolic syndrome: Is this diagnosis necessary? Am. J. Clin. Nutr. 2006, 83, 1237–1247. [Google Scholar] [CrossRef]
- Kahn, R.; Buse, J.; Ferrannini, E.; Stern, M. American Diabetes Association; European Association for the Study of Diabetes. The metabolic syndrome: Time for a critical appraisal: Joint statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2005, 28, 2289–2304. [Google Scholar] [CrossRef]
- Ferrannini, E. Metabolic syndrome: A solution in search of a problem. J. Clin. Endocrinol. Metab. 2007, 92, 396–398. [Google Scholar] [CrossRef]
























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayden, M.R. Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease. Medicina 2023, 59, 561. https://doi.org/10.3390/medicina59030561
Hayden MR. Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease. Medicina. 2023; 59(3):561. https://doi.org/10.3390/medicina59030561
Chicago/Turabian StyleHayden, Melvin R. 2023. "Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease" Medicina 59, no. 3: 561. https://doi.org/10.3390/medicina59030561
APA StyleHayden, M. R. (2023). Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease. Medicina, 59(3), 561. https://doi.org/10.3390/medicina59030561