

Perinatal Diagnosis and Management of a Case with Interrupted Aortic Arch, Pulmonary Valve Dysplasia and 22q11.2 Deletion: A Case Report

 ,
, 
Abstract
:1. Introduction
2. Case Report
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Volpe, P.; Tuo, G.; De Robertis, V.; Campobasso, G.; Marasini, M.; Tempesta, A.; Gentile, M.; Rembouskos, G. Fetal interrupted aortic arch: 2D-4D echocardiography, associations and outcome. Ultrasound Obstet. Gynecol. 2010, 35, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Riggs, K.W.; Anderson, R.H.; Spicer, D.; Morales, D.L. Coarctation and interrupted aortic arch. In Anderson′s Pediatric Cardiology; Wernovsky, G., Anderson, R.H., Kumar, K., Mussatto, K.A., Redington, A.N., Tweddell, J.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 843–864. [Google Scholar]
- Bassett, A.S.; McDonald-McGinn, D.M.; Devriendt, K.; Digilio, M.C.; Goldenberg, P.; Habel, A.; Marino, B.; Oskarsdottir, S.; Philip, N.; Sullivan, K.; et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J. Pediatr. 2011, 159, 332–339.e1. [Google Scholar] [CrossRef] [PubMed]
- Oskarsdottir, S.; Vujic, M.; Fasth, A. Incidence and prevalence of the 22q11.2 deletion syndrome: A population-based study in Western Sweden. Arch. Dis. Child. 2004, 89, 148–151. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers 2015, 1, 15071. [Google Scholar] [CrossRef]
- Volpe, P.; Marasini, M.; Caruso, G.; Marzullo, A.; Buonadonna, A.L.; Arciprete, P.; Di Paolo, S.; Volpe, G.; Gentile, M. 22q11 deletions in fetuses with malformations of the outflow tracts or interruption of the aortic arch: Impact of additional ultrasound signs. Prenat. Diagn. 2003, 23, 752–757. [Google Scholar] [CrossRef]
- Volpe, P.; Marasini, M.; Caruso, G.; Gentile, M. Prenatal diagnosis of interruption of the aortic arch and its association with deletion of chromosome 22q11. Ultrasound Obstet. Gynecol. 2002, 20, 327–331. [Google Scholar] [CrossRef]
- Blagowidow, N.; Nowakowska, B.; Schindewolf, E.; Grati, F.R.; Putotto, C.; Breckpot, J.; Swillen, A.; Crowley, T.B.; Loo, J.C.Y.; Lairson, L.A.; et al. Prenatal screening and diagnostic considerations for 22q11.2 microdeletions. Genes 2023, 14, 160. [Google Scholar] [CrossRef]
- Palmer, L.D.; Butcher, N.J.; Boot, E.; Hodgkinson, K.A.; Heung, T.; Chow, E.W.C.; Guna, A.; Crowley, T.B.; Zackai, E.; McDonald-McGinn, D.M.; et al. Elucidating the Diagnostic Odyssey of 22q11.2 Deletion Syndrome. Am. J. Med. Genet. A 2018, 176, 936–944. [Google Scholar] [CrossRef]
- Szczawińska-Popłonyk, A.; Schwartzmann, E.; Chmara, Z.; Głukowska, A.; Krysa, T.; Majchrzycki, M.; Olejnicki, M.; Ostrowska, P.; Babik, J. Chromosome 22q11.2 deletion syndrome: A comprehensive review of molecular genetics in the context of multidisciplinary clinical approach. Int. J. Mol. Sci. 2023, 24, 8317. [Google Scholar] [CrossRef]
- Dar, P.; Jacobsson, B.; Clifton, R.; Egbert, M.; Malone, F.; Wapner, R.J.; Roman, A.S.; Khalil, A.; Faro, R.; Madankumar, R.; et al. Cell-free DNA screening for prenatal detection of 22q11.2 deletion syndrome. Am. J. Obstet. Gynecol. 2022, 227, 79.e1–79.e11. [Google Scholar] [CrossRef]
- Bevilacqua, E.; Jani, J.C.; Chaoui, R.; Suk, E.A.; Palma-Dias, R.; Ko, T.; Warsof, S.; Stokowski, R.; Jones, K.J.; Grati, F.R.; et al. Performance of a targeted cell-free DNA prenatal test for 22q11.2 deletion is a large clinical cohort. Ultrasound Obstet. Gynecol. 2021, 58, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.; Wang, E.; Bogard, P.E.; Bevilacqua, E.; Hacker, C.; Wang, S.; Doshi, J.; White, K.; Kaplan, J.; Sparks, A.; et al. Prenatal screening for 22q11.2 deleletion using a targeted microarray-based cell-free DNA test. Fetal Diagn. Ther. 2018, 44, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Schindewolf, E.; Khalek, N.; Johnson, M.P.; Gebb, J.; Coleman, B.; Crowley, T.B.; Zackai, E.H.; McDonald-McGinn, D.M.; Moldenhauer, J.S. Expanding the fetal phenotype: Prenatal sonographic findings and perinatal outcomes in a cohort of patients with confirmed 22q11.2 deletion syndrome. Am. J. Med. Genet. A 2018, 176, 1735–1741. [Google Scholar] [CrossRef]
- Karl, K.; Heling, K.S.; Lopez, A.S.; Thiel, G.; Chaoui, R. Thymic-thoracic ratio in fetuses with trisomy 21, 18 or 13. Ultrasound Obstet. Gynecol. 2012, 40, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Noël, A.-C.; Pelluard, F.; Delezoide, A.-L.; Devisme, L.; Loeuillet, L.; Leroy, B.; Martin, A.; Bouvier, R.; Laquerriere, A.; Jeanne-Pasquier, C.; et al. Fetal phenotype associated with the 22q11 deletion. Am. J. Med. Genet. 2014, 164, 2724–2731. [Google Scholar] [CrossRef]
- Rauch, R.; Rauch, A.; Koch, A.; Zink, S.; Kaulitz, R.; Girisch, M.; Singer, H.; Hofbeck, M. Laterality of the aortic arch and anomalies of the subclavian artery-reliable indicators for 22q11.2 deletion syndromes? Eur. J. Pediatr. 2004, 163, 642–645. [Google Scholar] [CrossRef]
- Perolo, A.; De Robertis, V.; Cataneo, I.; Volpe, N.; Campobasso, G.; Frusca, T.; Ghi, T.; Prandstraller, D.; Pilu, G.; Volpe, P. Risk of 22q11.2 deletion in fetuses with right aortic arch and without intracardiac anomalies. Ultrasound Obstet. Gynecol. 2016, 48, 200–203. [Google Scholar] [CrossRef]
- Friedman, K. Preoperative physiology, imaging, and management of interrupted aortic arch. Semin. Cardiothorac. Vasc. Anesth. 2018, 22, 265–269. [Google Scholar] [CrossRef]
- Putotto, C.; Pugnaloni, F.; Unolt, M.; Maiolo, S.; Trezzi, M.; Digilio, M.C.; Cirillo, A.; Limongelli, G.; Marino, B.; Calcagni, G.; et al. 22q11.2 deletion syndrome: Impact of genetics in the treatment of conotruncal heart defects. Children 2022, 9, 772. [Google Scholar] [CrossRef]
- Yeoh, T.Y.; Scavonetto, F.; Hamlin, R.J.; Burkhart, H.M.; Sprung, J.; Weingarten, T.N. Perioperative management of patients with DiGeorge syndrome undergoing cardiac surgery. J. Cardiothorac. Vasc. Anesth. 2014, 28, 983–989. [Google Scholar] [CrossRef]
- Atallah, J.; Joffe, A.R.; Robertson, C.M.; Leonard, N.; Blakley, P.M.; Nettel-Aguirre, A.; Sauve, R.S.; Ross, D.B.; Rebeyka, I.M. Two-year general and neurodevelopmental outcome after neonatal complex cardiac surgery in patients with deletion 22q11.2: A comparative study. J. Thorac. Cardiovasc. Surg. 2007, 134, 772–779. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, C.; Jiang, X.; Chen, S. Is surgery necessary for adults with isolated interrupted aortic arch?: Case series with literature review. J. Card. Surg. 2021, 36, 2467–2475. [Google Scholar] [CrossRef]
- Gan, Y.; Zhang, P.; Liao, R.; Nie, Y.; Fu, Y. Novel interrupted aortic arch: A case report. J. Card. Surg. 2022, 37, 5639–5642. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Fan, G.; Zhang, W.; Liang, H. Interrupted aortic arch in adulthood: A rare case report. Asian J. Surg. 2023, 46, 2976–2977. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-W.; Li, H.; Pu, H. Isolated interrupted of aortic arch diagnosed using CT angiography: A case report and literature review. Medicine 2018, 97, e10569. [Google Scholar] [CrossRef] [PubMed]
- Onalan, M.A.; Temur, B.; Aydın, S.; Suzan, D.; Demir, I.H.; Odemis, E.; Erek, E. Management of interrupted aortic arch with associated anomalies: A single-center experience. World J. Pediatr. Congenit. Heart Surg. 2021, 12, 706–714. [Google Scholar] [CrossRef]
- Chen, P.C.; Cubberley, A.T.; Reyes, K.; Zurakowski, D.; Baird, C.W.; Pigula, F.A.; Geva, T.; Emani, S.M. Predictors of reintervention after repair of interrupted aortic arch with ventricular septal defect. Ann. Thorac. Surg. 2013, 96, 621–628. [Google Scholar] [CrossRef]
- Hidalgo, J.; Dickerson, J.C.; Burnsed, B.; Luqman, A.; Shiflett, J.M. Middle cerebral artery aneurysm rupture in a neonate with interrupted aortic arch: Case report. Childs Nerv. Syst. 2017, 33, 999–1003. [Google Scholar] [CrossRef]
- Ghosh, A.; Liu, A.; Mora, B.; Agarwala, B. Giant aortic arch aneurysm after interrupted aortic arch repair. Pediatr. Cardiol. 2010, 31, 1104–1106. [Google Scholar] [CrossRef]
- Takahashi, K.; Kuwahara, T.; Nagatsu, M. Interruption of the aortic arch at the isthmus with DiGeorge syndrome and 22q11.2 deletion. Cardiol. Young 1999, 9, 516–518. [Google Scholar] [CrossRef]
- Lewin, M.B.; Lindsay, E.A.; Jurecic, V.; Goytia, B.; Towbin, J.; Baldini, A. A genetic aetiology for interruption of the aortic arch type B. Am. J. Cardiol. 1997, 80, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Hofbeck, M.; Leipold, G.; Klinge, J.; Trautmann, U.; Kirsch, M.; Singer, H.; Pfeiffer, R.A. Incidence and significance of 22q11.2 hemizygosity in patients with interrupted aortic arch. Am. J. Med. Genet. 1998, 78, 322–331. [Google Scholar] [CrossRef]
- Hoeffding, L.K.; Trabjerg, B.B.; Olsen, L.; Mazin, W.; Sparsø, T.; Vangkilde, A.; Mortensen, P.B.; Pedersen, C.B.; Werge, T. Risk of psychiatric disorders among individuals with the 22q11.2 deletion or duplication: A Danish nationwide, register-based study. JAMA Psychiatry 2017, 74, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Vajdi, A.; Kushan-Wells, L.; Helleman, G.; Hansen, L.P.; Jonas, R.K.; Jalbrzikowski, M.; Kingsbury, L.; Raznahan, A.; Bearden, C.E. Reciprocal copy number variations at 22q11.2 produce distinct and convergent neurobehavioral impairments relevant for schizophrenia and autism spectrum disorder. Biol. Psychiatry 2020, 88, 260–272. [Google Scholar] [CrossRef]
- Chawner, S.J.; Doherty, J.L.; Anney, R.J.L.; Antshel, K.M.; Bearden, C.E.; Bernier, R.; Chung, W.K.; Clements, C.C.; Curran, S.R.; Cuturilo, G.; et al. A genetics- first approach to dissecting the heterogeneity of autism: Phenotypic comparison of autism risk copy number variants. Am. J. Pshychiatry 2021, 178, 77–86. [Google Scholar] [CrossRef]
- Kortanek, E.S.; McDonald, N.M.; Nosco, E.E.; MacNaughton, G.A.; Lin, A.; Jeste, S.S.; Bearden, C.E. Early developmental concerns in 22q11.2 deletion and duplication carriers. Res. Autism Spectr. Disord. 2022, 97, 102026. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case Highlights | Details |
|---|---|
| Maternal age | 34 years |
| Gestational age at cardiologic diagnosis | 22 weeks |
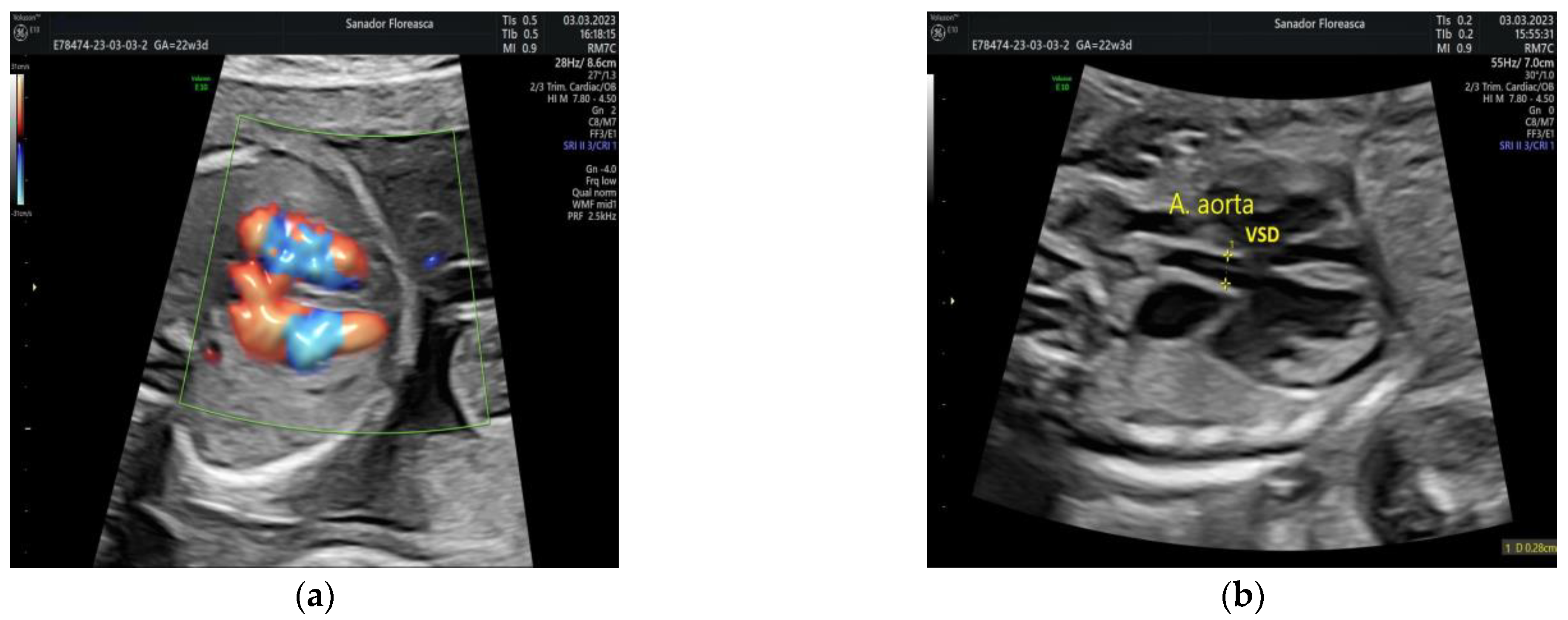
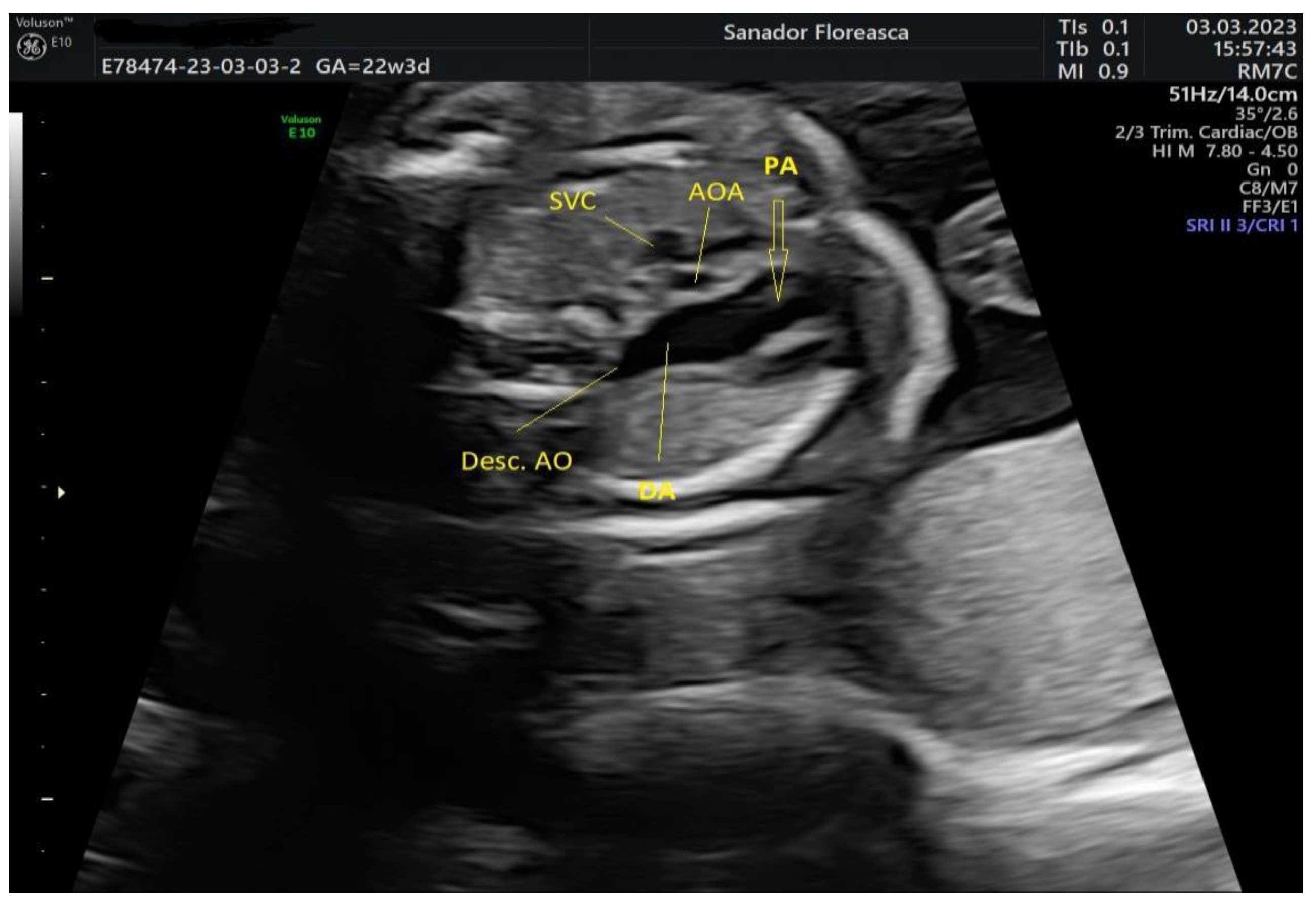
| Cardiologic diagnosis | IAA type B, malalignment-type VSD, pulmonary valve dyplasia, ARSA |
| Genetic diagnosis (amniocentesis with microarray) | 22q11.2 deletion |
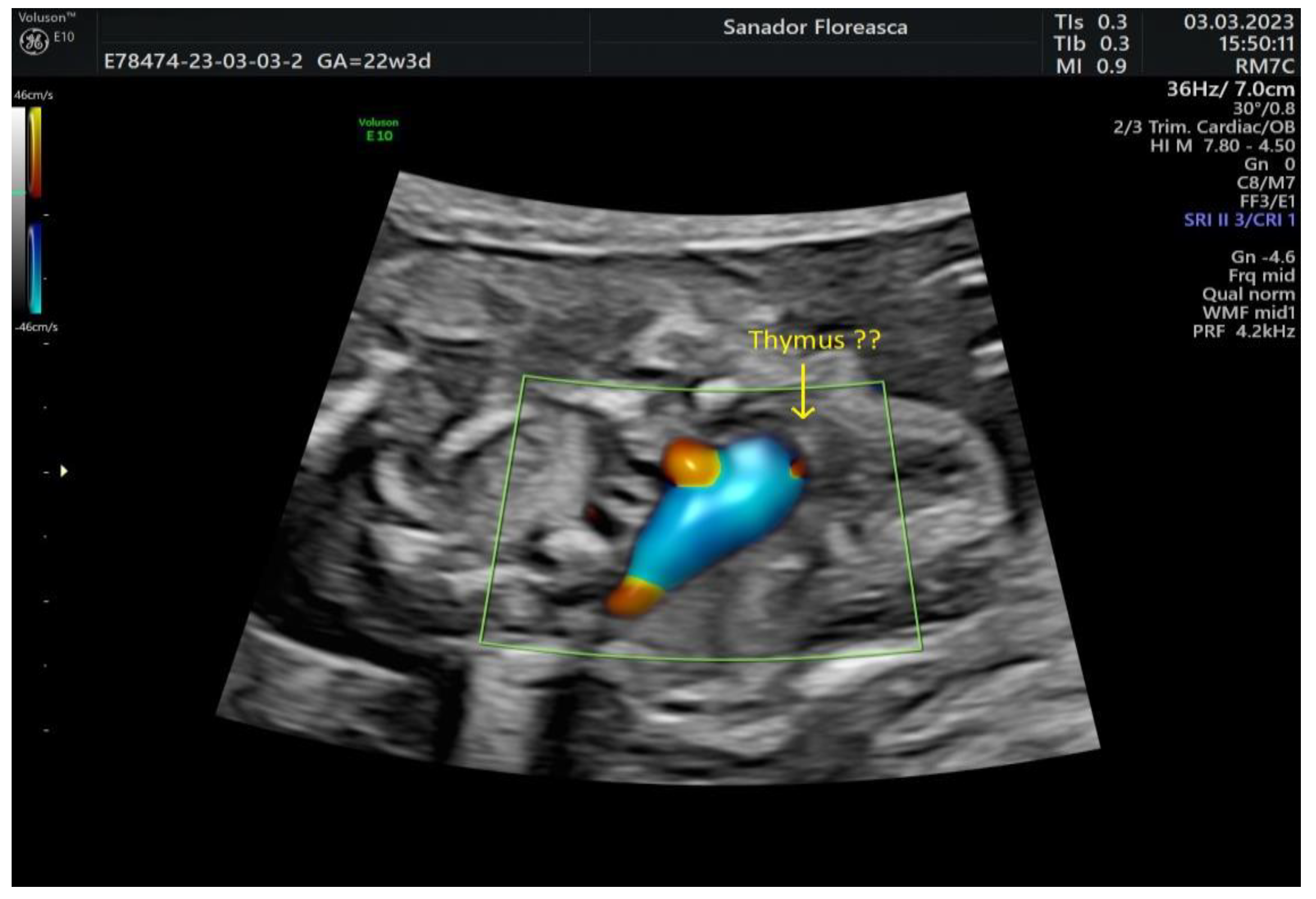
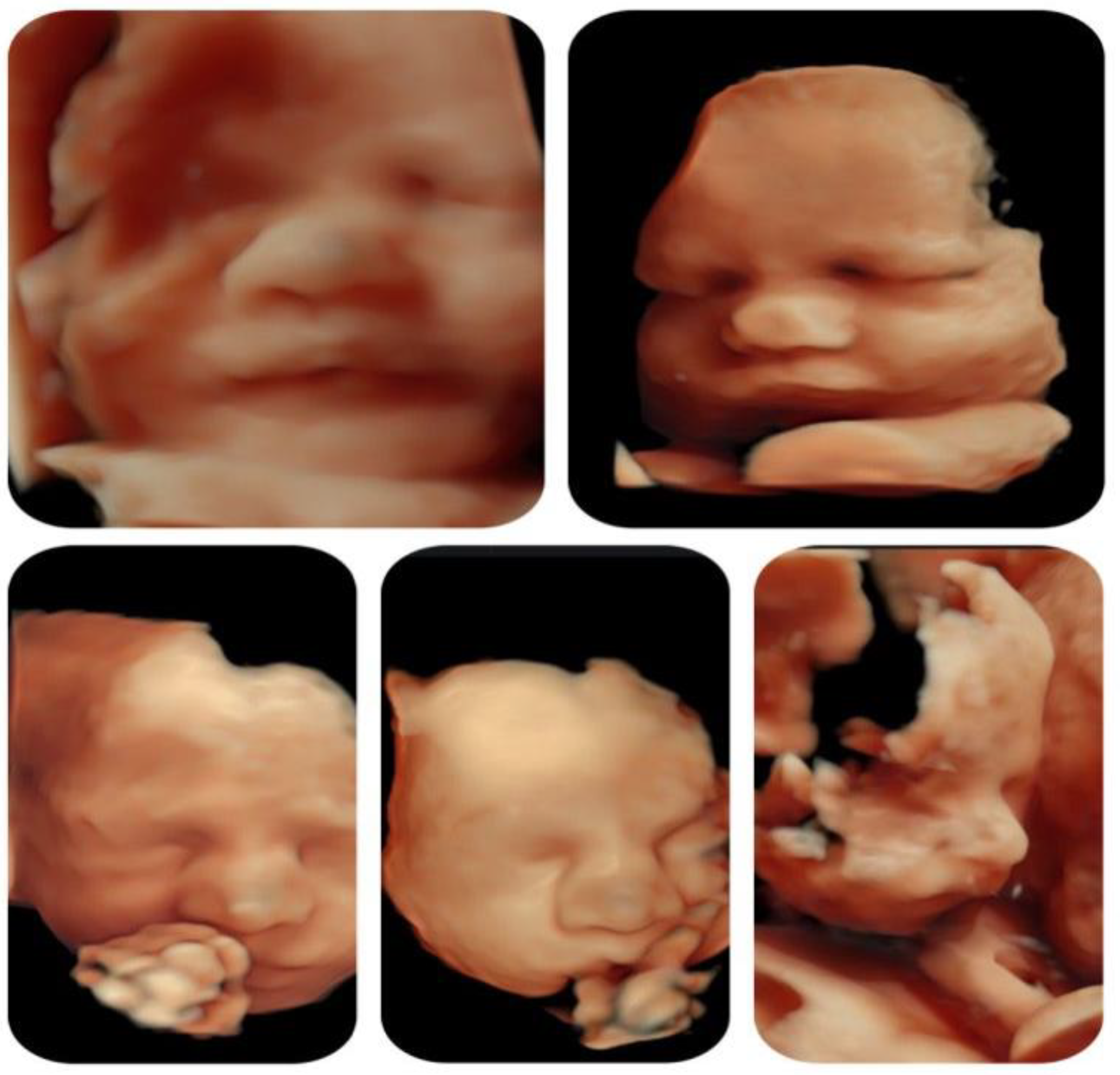
| Other ultrasonographic findings | Polyhydramnios Enlarged CSP Thymus hypoplasia Bulbous nose |
| Delivery route | Vaginal |
| Birthweight | 3420 g |
| Postnatal echocardiography | Prenatal diagnosis confirmed |
| Heart surgery | Performed at 7 days of life Median sternotomy Extended latero-terminal anastomosis of the aortic arch and closure of VSD with bovine pericardial patch, closure of the patent ductus arteriosus Prophylactic antibiotics+ anticoagulants for 10 days |
| Total lentgh of stay in the cardiology unit | 3 weeks |
| Recommendations at discharge | Sternal precautions for 10 weeks One year prophylactic therapy with ASA + endocarditis prophylaxis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vlădăreanu, R.; Maier, C.; Tocariu, R.; Șerban, M.; Brătilă, E. Perinatal Diagnosis and Management of a Case with Interrupted Aortic Arch, Pulmonary Valve Dysplasia and 22q11.2 Deletion: A Case Report. Medicina 2023, 59, 1838. https://doi.org/10.3390/medicina59101838
Vlădăreanu R, Maier C, Tocariu R, Șerban M, Brătilă E. Perinatal Diagnosis and Management of a Case with Interrupted Aortic Arch, Pulmonary Valve Dysplasia and 22q11.2 Deletion: A Case Report. Medicina. 2023; 59(10):1838. https://doi.org/10.3390/medicina59101838
Chicago/Turabian StyleVlădăreanu, Radu, Călina Maier, Raluca Tocariu, Marcela Șerban, and Elvira Brătilă. 2023. "Perinatal Diagnosis and Management of a Case with Interrupted Aortic Arch, Pulmonary Valve Dysplasia and 22q11.2 Deletion: A Case Report" Medicina 59, no. 10: 1838. https://doi.org/10.3390/medicina59101838
APA StyleVlădăreanu, R., Maier, C., Tocariu, R., Șerban, M., & Brătilă, E. (2023). Perinatal Diagnosis and Management of a Case with Interrupted Aortic Arch, Pulmonary Valve Dysplasia and 22q11.2 Deletion: A Case Report. Medicina, 59(10), 1838. https://doi.org/10.3390/medicina59101838