
The Impact of Epithelial–Mesenchymal Transition and Metformin on Pancreatic Cancer Chemoresistance: A Pathway towards Individualized Therapy

 ,
, 
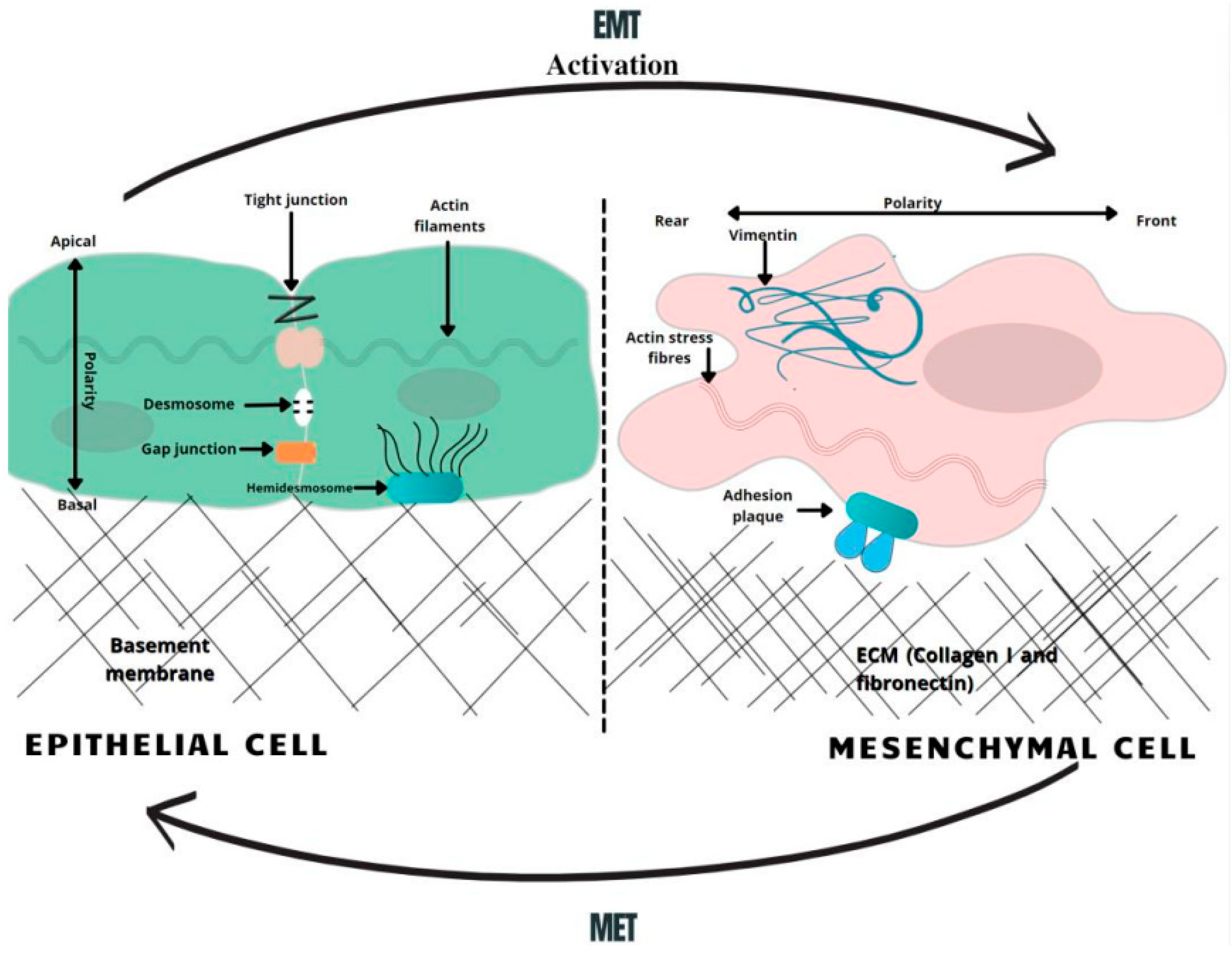
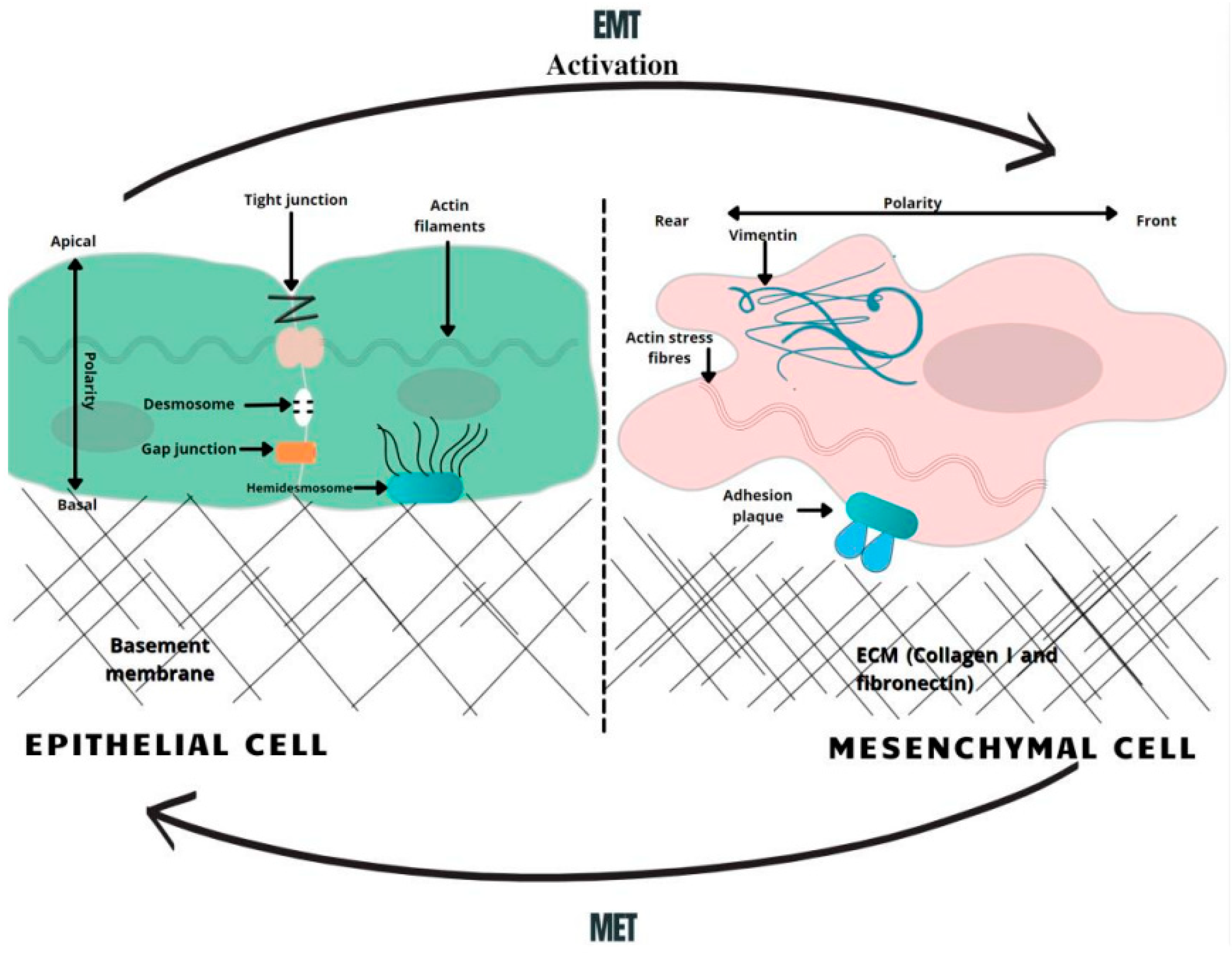{kind=link}
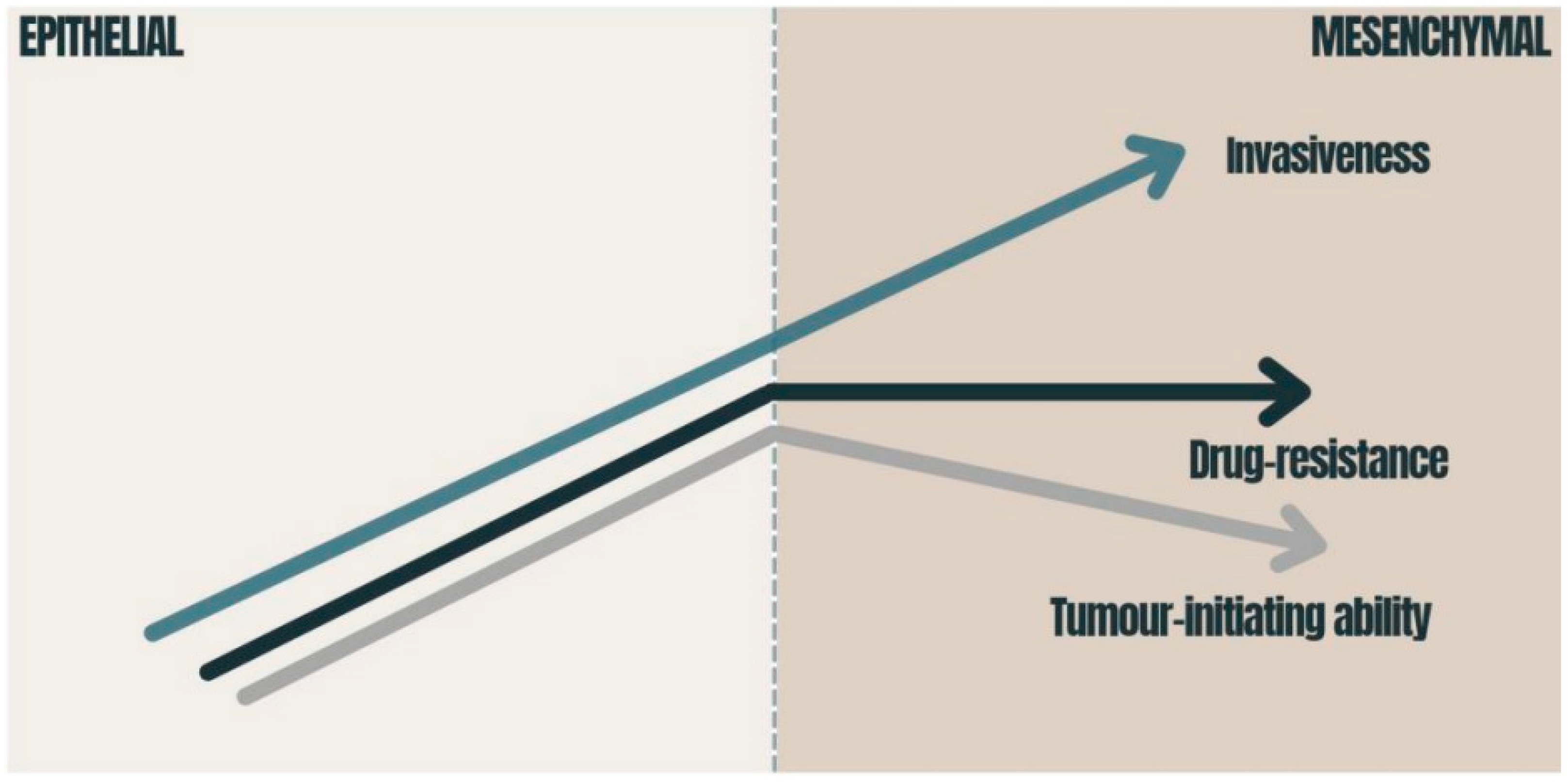
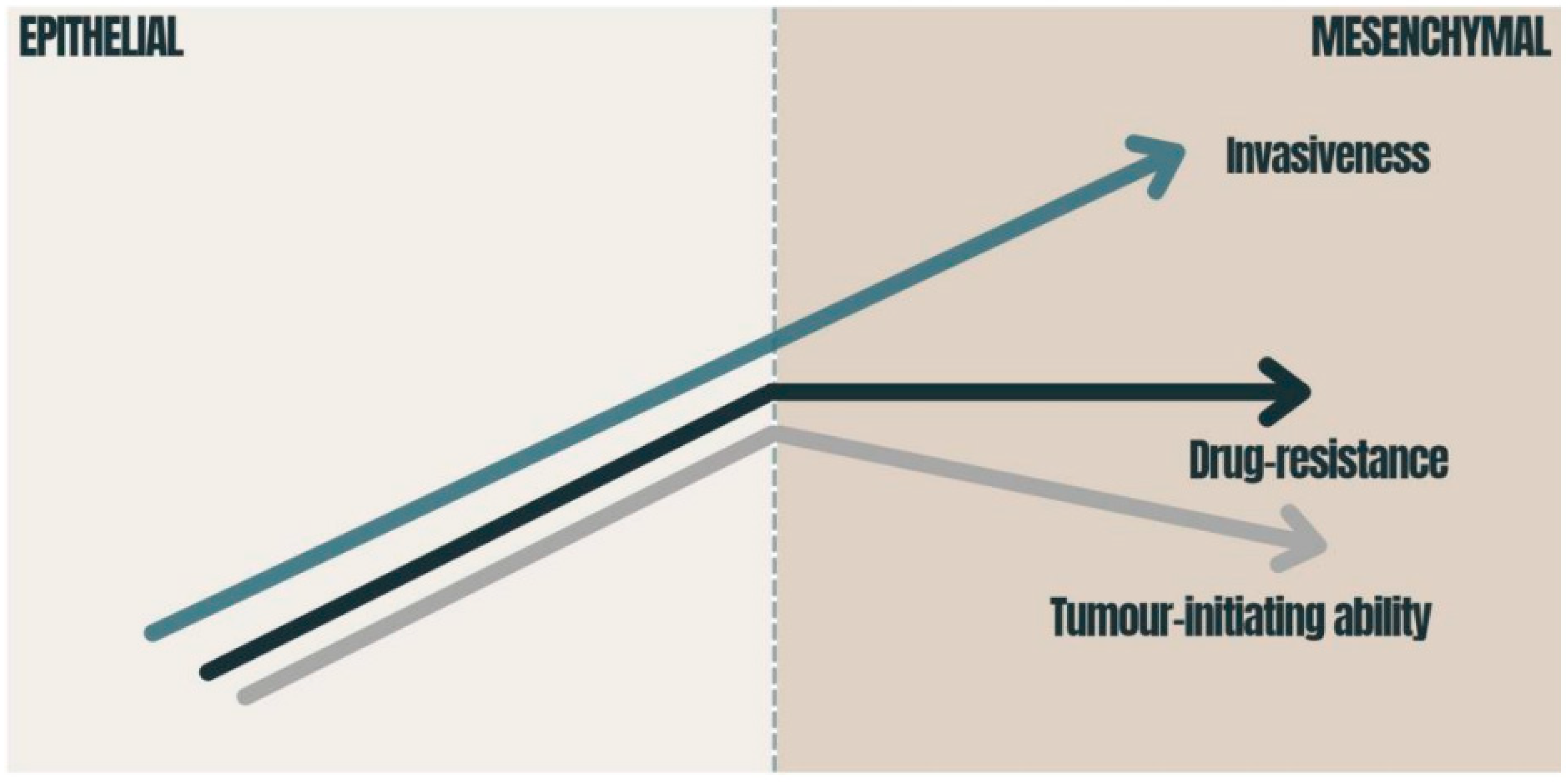{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Epithelial–Mesenchymal Transition
3.1. Epithelial–Mesenchymal Transition and Cancer Stem Cells
3.2. Chemoresistance and EMT Impact on Oncotherapy
4. Metformin and PDAC
4.1. Role of mTOR Mediation and CSC in an AMPK-Dependent Manner
4.2. Metformin and miRNAs
4.3. CSCs’ Resistance to Metabolic Deprivation: A Potential Target in PDAC Treatment?
4.4. Clinical Trials
5. Conclusions
6. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vincent, A.; Herman, J.; Schulick, R.; Hruban, R.H.; Goggins, M. Pancreatic cancer. Lancet 2011, 378, 607–620. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Chandana, S.R.; Babiker, H.M.; Mahadevan, D. Therapeutic trends in pancreatic ductal adenocarcinoma (PDAC). Expert Opin. Investig. Drugs 2019, 28, 161–177. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hessmann, E.; Buchholz, S.M.; Demir, I.E.; Singh, S.K.; Gress, T.M.; Ellenrieder, V.; Neesse, A. Microenvironmental determinants of pancreatic cancer. Physiol. Rev. 2020, 100, 1707–1751. [Google Scholar] [CrossRef]
- Candido, S.; Abrams, S.L.; Steelman, L.; Lertpiriyapong, K.; Martelli, A.M.; Cocco, L.; Ratti, S.; Follo, M.Y.; Murata, R.M.; Rosalen, P.L.; et al. Metformin influences drug sensitivity in pancreatic cancer cells. Adv. Biol. Regul. 2018, 68, 13–30. [Google Scholar] [CrossRef]
- Chen, K.; Qian, W.; Jiang, Z.; Cheng, L.; Li, J.; Sun, L.; Zhou, C.C.; Gao, L.P.; Lei, M.; Yan, B.; et al. Metformin suppresses cancer initiation and progression in genetic mouse models of pancreatic cancer. Mol. Cancer 2017, 16, 131. [Google Scholar] [CrossRef]
- Del Barco, S.; Vazquez-Martin, A.; Cufí, S.; Oliveras-Ferraros, C.; Bosch-Barrera, J.; Joven, J.; Martin-Castillo, B.; Menendez, J.A. Metformin: Multi-faceted protection against cancer. Oncotarget 2011, 2, 896–917. [Google Scholar] [CrossRef]
- Zhou, P.T.; Li, B.; Liu, F.R.; Zhang, M.C.; Wang, Q.; Liu, Y.H.; Yao, Y.; Li, D. The epithelial to mesenchymal transition (EMT) and cancer stem cells: Implication for treatment resistance in pancreatic cancer. Mol. Cancer 2017, 16, 52. [Google Scholar] [CrossRef] [PubMed]
- Beuran, M.; Negoi, I.; Paun, S.; Ion, A.D.; Bleotu, C.; Negoi, R.I.; Hostiuc, S. The epithelial to mesenchymal transition in pancreatic cancer: A systematic review. Pancreatology 2015, 15, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Lehúede, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res. 2016, 76, 5201–5208. [Google Scholar] [CrossRef] [PubMed]
- Gzil, A.; Zarębska, I.; Bursiewicz, W.; Antosik, P.; Grzanka, D.; Szylberg, Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol. Biol. Rep. 2019, 46, 6629–6645. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-I.; Jang, S.I.; Hong, J.; Kim, C.H.; Kwon, S.S.; Park, J.S.; Lim, J.-B. Cancer-initiating cells in human pancreatic cancer organoids are maintained by interactions with endothelial cells. Cancer Lett. 2021, 498, 42–53. [Google Scholar] [CrossRef]
- Preca, B.-T.; Bajdak, K.; Mock, K.; Sundararajan, V.; Pfannstiel, J.; Maurer, J.; Wellner, U.; Hopt, U.T.; Brummer, T.; Brabletz, S.; et al. A self-enforcing CD44s/ZEB1 feedback loop maintains EMT and stemness properties in cancer cells. Int. J. Cancer 2015, 137, 2566–2577. [Google Scholar] [CrossRef]
- Li, C.; Wu, J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; di Magliano, M.P.; Simeone, D.M. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, 2218–2227.e5. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Xia, M.F.; Jin, K.; Wang, S.F.; Wei, H.; Fan, C.M.; Wu, Y.F.; Li, X.L.; Li, X.Y.; Li, G.Y.; et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 2018, 17, 20. [Google Scholar] [CrossRef]
- Neuzillet, C.; Couvelard, A.; Tijeras-Raballand, A.; de Mestier, L.; de Gramont, A.; Bédossa, P.; Paradis, V.; Sauvanet, A.; Bachet, J.-B.; Ruszniewski, P.; et al. High c-Met expression in stage I–II pancreatic adenocarcinoma: Proposal for an immunostaining scoring method and correlation with poor prognosis. Histopathology 2015, 67, 664–676. [Google Scholar] [CrossRef]
- Lux, A.; Kahlert, C.; Grützmann, R.; Pilarsky, C. c-Met and PD-l1 on circulating exosomes as diagnostic and prognostic markers for pancreatic cancer. Int. J. Mol. Sci. 2019, 20, 3305. [Google Scholar] [CrossRef]
- Jiang, J.H.; Liu, C.; Cheng, H.; Lu, Y.; Qin, Y.; Xu, Y.F.; Xu, J.; Long, J.; Liu, L.; Ni, Q.X.; et al. Epithelial-mesenchymal transition in pancreatic cancer: Is it a clinically significant factor? Biochim. Biophys. Acta-Rev. Cancer 2015, 1855, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Schober, M.; Jesenofsky, R.; Faissner, R.; Weidenauer, C.; Hagmann, W.; Michl, P.; Heuchel, R.L.; Haas, S.L.; Löhr, J.-M. Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014, 6, 2137–2154. [Google Scholar] [CrossRef]
- Gradiz, R.; Silva, H.C.; Carvalho, L.; Botelho, M.F.; Mota-Pinto, A. MIA PaCa-2 and PANC-1—pancreas ductal adenocarcinoma cell lines with neuroendocrine differentiation and somatostatin receptors. Sci. Rep. 2016, 6, 21648. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 170. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Sancho, P.; Alcala, S.; Usachov, V.; Hermann, P.C.; Sainz, B. The ever-changing landscape of pancreatic cancer stem cells. Pancreatology 2016, 16, 489–496. [Google Scholar] [CrossRef]
- Lyle, S.; Moore, N. Quiescent, slow-cycling stem cell populations in cancer: A review of the evidence and discussion of significance. J. Oncol. 2010, 2011, 396076. [Google Scholar] [CrossRef]
- Bhagwandin, V.J.; Bishop, J.M.; Wright, W.E.; Shay, J.W. The Metastatic Potential and Chemoresistance of Human Pancreatic Cancer Stem Cells. PLoS ONE 2016, 11, e0148807. [Google Scholar] [CrossRef]
- Hu, X.; Chen, W. Role of epithelial-mesenchymal transition in chemoresistance in pancreatic ductal adenocarcinoma. World J. Clin. Cases 2021, 9, 4998. [Google Scholar] [CrossRef] [PubMed]
- Celià-Terrassa, T.; Jolly, M.K. Cancer stem cells and epithelial-to-mesenchymal transition in cancer metastasis. Cold Spring Harb. Perspect. Med. 2020, 10, a036905. [Google Scholar] [CrossRef] [PubMed]
- Cannon, A.; Thompson, C.; Hall, B.R.; Jain, M.; Kumar, S.; Batra, S.K. Desmoplasia in pancreatic ductal adenocarcinoma: Insight into pathological function and therapeutic potential. Genes Cancer 2018, 9, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Kong, D.; Ahmad, A.; Bao, B.; Sarkar, F.H. Pancreatic cancer stem cells: Emerging target for designing novel therapy. Cancer Lett. 2013, 338, 94–100. [Google Scholar] [CrossRef]
- Adamska, A.; Falasca, M. ATP-binding cassette transporters in progression and clinical outcome of pancreatic cancer: What is the way forward? World J. Gastroenterol. 2018, 24, 3222–3236. [Google Scholar] [CrossRef]
- Awaji, M.; Singh, R.K. Cancer-associated fibroblasts’ functional heterogeneity in pancreatic ductal adenocarcinoma. Cancers 2019, 11, 290. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell 2018, 45, 681–695.e4. [Google Scholar] [CrossRef]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Donaher, J.L.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-β4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef]
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Vignjevic, D.M. Modes of cancer cell invasion and the role of the microenvironment. Curr. Opin. Cell Biol. 2015, 36, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Schernthaner, G.; Schernthaner, G.H. The right place for metformin today. Diabetes Res. Clin. Pract. 2020, 159, 107946. [Google Scholar] [CrossRef]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin pathways: Pharmacokinetics and pharmacodynamics. Pharm. Genom. 2012, 22, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Hsu, C.-C.; Wahlqvist, M.L.; Tsai, H.-N.; Chang, Y.-H.; Huang, Y.-C. Type 2 diabetes increases and metformin reduces total, colorectal, liver and pancreatic cancer incidences in Taiwanese: A representative population prospective cohort study of 800,000 individuals. BMC Cancer 2011, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.; Cafferty, F.H.; Vale, C.; Langley, R.E. Metformin as an adjuvant treatment for cancer: A systematic review and meta-analysis. Ann. Oncol. 2016, 27, 2184–2195. [Google Scholar] [CrossRef]
- Wan, G.; Sun, X.; Li, F.; Wang, X.; Li, C.; Li, H.; Yu, X.; Cao, F. Cellular Physiology and Biochemistry Cellular Physiology and Biochemistry Survival Benefit of Metformin Adjuvant Treatment For Pancreatic Cancer Patients: A Systematic Review and Meta-Analysis. Cell Physiol. Biochem. 2018, 49, 837–847. [Google Scholar] [CrossRef]
- Sadeghi, N.; Abbruzzese, J.L.; Yeung, S.C.J.; Hassan, M.; Li, D. Metformin use is associated with better survival of diabetic patients with pancreatic cancer. Clin. Cancer Res. 2012, 18, 2905–2912. [Google Scholar] [CrossRef]
- Duan, W.X.; Chen, K.; Jiang, Z.D.; Chen, X.; Sun, L.K.; Li, J.H.; Lei, J.J.; Xu, Q.H.; Ma, J.G.; Li, X.Q.; et al. Desmoplasia suppression by metformin-mediated AMPK activation inhibits pancreatic cancer progression. Cancer Lett. 2017, 385, 225–233. [Google Scholar] [CrossRef]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGf-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. Development 2012, 139, 1259–1273. [Google Scholar] [CrossRef]
- Khezri, M.R.; Melekinejad, H.; Majidi-Zolbanin, N.; Ghasemnejad-Berenji, M. Anticancer potential of metformin: Focusing on gastrointestinal cancers. Cancer Chemother. Pharmacol. 2021, 87, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Yi, B.; Riker, A.I.; Xi, Y. Metformin and cancer immunity. Acta Pharmacol. Sin. 2020, 41, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, M.; Venkatesan, N.; Ogeyingbo, O.D.; Bhandari, R.; Botleroo, R.A.; Kareem, R.; Ahmed, R.; Elshaikh, A.O. Magic of a Common Sugar Pill in Cancer: Can Metformin Raise Survival in Pancreatic Cancer Patients? Cureus 2021, 13, e16916. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.; Khawaja, K.I.; Masud, F.; Saif, M.W. Metformin and pancreatic cancer: Is there a role? Cancer Chemother. Pharmacol. 2016, 77, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.X.; Qian, W.K.; Zhou, C.C.; Cao, J.Y.; Qin, T.; Xiao, Y.; Cheng, L.; Li, J.; Chen, K.; Li, X.Q.; et al. Metformin suppresses the invasive ability of pancreatic cancer cells by blocking autocrine TGF-ß1 signaling. Oncol. Rep. 2018, 40, 1495–1502. [Google Scholar] [CrossRef]
- Wu, Q.; Miele, L. The Role of EMT in Pancreatic Cancer Progression. Pancreat. Disord. Ther. 2012, 2, 2012–2014. [Google Scholar] [CrossRef]
- Incio, J.; Suboj, P.; Chin, S.M.; Vardam-Kaur, T.; Liu, H.; Hato, T.; Babykutty, S.; Chen, I.; Deshpande, V.; Jain, R.K.; et al. Metformin reduces desmoplasia in pancreatic cancer by reprogramming stellate cells and tumor-associated macrophages. PLoS ONE 2015, 10, e0141392. [Google Scholar] [CrossRef]
- Zi, F.; Zi, H.; Li, Y.; He, J.; Shi, Q.; Cai, Z. Metformin and cancer: An existing drug for cancer prevention and therapy (review). Oncol. Lett. 2018, 15, 683–690. [Google Scholar] [CrossRef]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Morales, D.R.; Morris, A.D. Metformin in cancer treatment and prevention. Annu. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- New, M.; Van Acker, T.; Long, J.S.; Sakamaki, J.I.; Ryan, K.M.; Tooze, S.A. Molecular pathways controlling autophagy in pancreatic cancer. Front. Oncol. 2017, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res. 2011, 71, 3196–3201. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Mao, W.; Zhai, Y.; Tong, C.; Liu, M.; Ma, L.; Yu, X.L.; Li, S.S. Anti-tumor activity of metformin: From metabolic and epigenetic perspectives. Oncotarget 2017, 8, 5619–5628. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Fang, L.; Fang, Q.; Zheng, X.; Huang, P. Effects of metformin on survival outcomes of pancreatic cancer patients with diabetes: A meta-analysis. Mol. Clin. Oncol. 2017, 8, 483–488. [Google Scholar] [CrossRef]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2018, 50, 133–143. [Google Scholar] [CrossRef]
- Sahra, I.B.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef]
- Hassan, Z.; Schneeweis, C.; Wirth, M.; Veltkamp, C.; Dantes, Z.; Feuerecker, B.; Ceyhan, G.O.; Knauer, S.K.; Weichert, C.; Schmid, R.M.; et al. MTOR inhibitor-based combination therapies for pancreatic cancer. Br. J. Cancer 2018, 118, 366–377. [Google Scholar] [CrossRef]
- Papaconstantinou, I.G.; Manta, A.; Gazouli, M.; Lyberopoulou, A.; Lykoudis, P.M.; Polymeneas, G.; Voros, D. Expression of MicroRNAs in Patients With Pancreatic Cancer and Its Prognostic Significance. Pancreas 2013, 42, 67–71. [Google Scholar] [CrossRef]
- Funamizu, N.; Ray Lacy, C.; Kamada, M.; Yanaga, K.; Manome, Y. MicroRNA-200b and -301 are associated with gemcitabine response as biomarkers in pancreatic carcinoma cells. Int. J. Oncol. 2019, 54, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.A.; Al-Qahtani, M.H.; Shay, J.W. Role of miRNAs in human cancer metastasis: Implications for therapeutic intervention. Semin. Cancer Biol. 2017, 44, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.L.; Zhang, J.H.; Wu, X.Z.; Yan, T.; Lv, W. MiR-15b promotes epithelial-mesenchymal transition by inhibiting SMURF2 in pancreatic cancer. Int. J. Oncol. 2015, 47, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Sun, J.; Wang, X.; Wang, H.; Pei, H.; Zhang, Z. MicroRNA-153 is a prognostic marker and inhibits cell migration and invasion by targeting SNAI1 in human pancreatic ductal adenocarcinoma. Oncol. Rep. 2015, 34, 595–602. [Google Scholar] [CrossRef]
- Liu, G.; Ji, L.; Ke, M.; Ou, Z.; Tang, N.; Li, Y. miR-125a-3p is responsible for chemosensitivity in PDAC by inhibiting epithelial-mesenchymal transition via Fyn. Biomed. Pharmacother. 2018, 106, 523–531. [Google Scholar] [CrossRef]
- Hiramoto, H.; Muramatsu, T.; Ichikawa, D.; Tanimoto, K.; Yasukawa, S.; Otsuji, E.; Inazawa, J. MiR-509-5p and miR-1243 increase the sensitivity to gemcitabine by inhibiting epithelial-mesenchymal transition in pancreatic cancer. Sci. Rep. 2017, 7, 4002. [Google Scholar] [CrossRef]
- Pan, G.; Liu, Y.; Shang, L.; Zhou, F.; Yang, S. EMT-associated microRNAs and their roles in cancer stemness and drug resistance. Cancer Commun. 2021, 41, 199–217. [Google Scholar] [CrossRef]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Quirantes, R.; Segura-Carretero, A.; Micol, V.; Joven, J.; Bosch-Barrera, J.; Del Barco, S.; Martin-Castillo, B.; et al. Metformin lowers the threshold for stress-induced senescence: A role for the microRNA-200 family and miR-205. Cell Cycle 2012, 11, 1235–1246. [Google Scholar] [CrossRef]
- Bao, B.; Wang, Z.; Ali, S.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Banerjee, S.; Kong, D.; Li, Y.W.; Thakur, S.; et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev. Res. 2012, 5, 355–364. [Google Scholar] [CrossRef]
- Ma, M.L.; Ma, C.F.; Li, P.P.; Ma, C.X.; Ping, F.; Li, W.; Xu, L.L.; Zhang, H.B.; Sun, Q.; Li, Y.X. Low glucose enhanced metformin’s inhibitory effect on pancreatic cancer cells by suppressing glycolysis and inducing energy stress via up-regulation of miR-210-5p. Cell Cycle 2020, 19, 2168–2181. [Google Scholar] [CrossRef]
- Nimmakayala, R.K.; Leon, F.; Rachagani, S.; Rauth, S.; Nallasamy, P.; Marimuthu, S.; Shailendra, G.K.; Chhonker, Y.S.; Chugh, S.; Chirravuri, R.; et al. Metabolic programming of distinct cancer stem cells promotes metastasis of pancreatic ductal adenocarcinoma. Oncogene 2021, 40, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Andriani, F.; Bertolini, G.; Facchinetti, F.; Baldoli, E.; Moro, M.; Casalini, P.; Caserini, R.; Milione, M.; Leone, G.; Pelosi, G.; et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol. Oncol. 2016, 10, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Wicha, M.S. Metabolic plasticity of cancer stem cells. Oncotarget 2015, 6, 35141–35142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Guo, X.L. Combinational strategies of metformin and chemotherapy in cancers. Cancer Chemother. Pharmacol. 2016, 78, 13–26. [Google Scholar] [CrossRef]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Crusz, S.; Heeschen, C. Studying Pancreatic Cancer Stem Cell Characteristics for Developing New Treatment Strategies. J. Vis. Exp. 2015, 100, 52801. [Google Scholar] [CrossRef]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef]
- Suzuki, K.; Takeuchi, O.; Suzuki, Y.; Kitagawa, Y. Mechanisms of metformin’s anti-tumor activity against gemcitabine-resistant pancreatic adenocarcinoma. Int. J. Oncol. 2019, 54, 764–772. [Google Scholar] [CrossRef]
- Kordes, S.; Pollak, M.N.; Zwinderman, A.H.; Mathôt, R.A.; Weterman, M.J.; Beeker, A.; Punt, C.J.; Richel, D.J.; Wilmink, J.W. Metformin in patients with advanced pancreatic cancer: A double-blind, randomised, placebo-controlled phase 2 trial. Lancet Oncol. 2015, 16, 839–847. [Google Scholar] [CrossRef]
- Broadhurst, P.J.; Hart, A.R. Metformin as an Adjunctive Therapy for Pancreatic Cancer: A Review of the Literature on Its Potential Therapeutic Use. Dig. Dis. Sci. 2018, 63, 2840–2852. [Google Scholar] [CrossRef]
- Reni, M.; Dugnani, E.; Cereda, S.; Belli, C.; Balzano, G.; Nicoletti, R.; Liberati, D.; Pasquale, V.; Scavini, M.; Maggiora, P.; et al. (Ir)relevance of Metformin Treatment in Patients with Metastatic Pancreatic Cancer: An Open-Label, Randomized Phase II Trial. Clin. Cancer Res. 2016, 22, 1076–1085. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Elgogary, A.; Xu, Q.; Poore, B.; Alt, J.; Zimmermann, S.C.; Zhao, L.; Fu, J.; Chen, B.W.; Xia, S.Y.; Liu, Y.F.; et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E5328–E5336. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Liu, Y.; Bi, Y.; Zhang, Z.-J. Metformin and pancreatic cancer survival: Real effect or immortal time bias? Int. J. Cancer 2019, 145, 1822–1828. [Google Scholar] [CrossRef] [PubMed]
- Frouws, M.A.; Mulder, B.G.S.; Bastiaannet, E.; Zanders, M.M.J.; Van Herk-Sukel, M.P.P.; De Leede, E.M.; Bonsing, B.A.; Mieog, J.S.; Van de Velde, C.J.H.; Liefers, G.-J.; et al. No association between metformin use and survival in patients with pancreatic cancer. Medicine 2017, 96, e6229. [Google Scholar] [CrossRef]
- Dong, Y.-W.; Shi, Y.-Q.; He, L.-W.; Cui, X.-Y.; Su, P.-Z. Effects of metformin on survival outcomes of pancreatic cancer: A meta-analysis. Oncotarget 2017, 8, 55478–55488. [Google Scholar] [CrossRef]
- Singh, R.R.; O’reilly, E.M. New Treatment Strategies for Metastatic Pancreatic Ductal Adenocarcinoma. Drugs 2020, 80, 647–669. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gulla, A.; Andriusaityte, U.; Zdanys, G.T.; Babonaite, E.; Strupas, K.; Kelly, H. The Impact of Epithelial–Mesenchymal Transition and Metformin on Pancreatic Cancer Chemoresistance: A Pathway towards Individualized Therapy. Medicina 2022, 58, 467. https://doi.org/10.3390/medicina58040467
Gulla A, Andriusaityte U, Zdanys GT, Babonaite E, Strupas K, Kelly H. The Impact of Epithelial–Mesenchymal Transition and Metformin on Pancreatic Cancer Chemoresistance: A Pathway towards Individualized Therapy. Medicina. 2022; 58(4):467. https://doi.org/10.3390/medicina58040467
Chicago/Turabian StyleGulla, Aiste, Urte Andriusaityte, Gabrielius Tomas Zdanys, Elena Babonaite, Kestutis Strupas, and Helena Kelly. 2022. "The Impact of Epithelial–Mesenchymal Transition and Metformin on Pancreatic Cancer Chemoresistance: A Pathway towards Individualized Therapy" Medicina 58, no. 4: 467. https://doi.org/10.3390/medicina58040467
APA StyleGulla, A., Andriusaityte, U., Zdanys, G. T., Babonaite, E., Strupas, K., & Kelly, H. (2022). The Impact of Epithelial–Mesenchymal Transition and Metformin on Pancreatic Cancer Chemoresistance: A Pathway towards Individualized Therapy. Medicina, 58(4), 467. https://doi.org/10.3390/medicina58040467