

Non-Transfusion-Dependent Thalassemia: A Panoramic Review


Abstract
1. Introduction
2. Genetic Heterogeneity
2.1. Primary Modifiers
2.2. Secondary Modifiers
2.3. Tertiary Modifiers
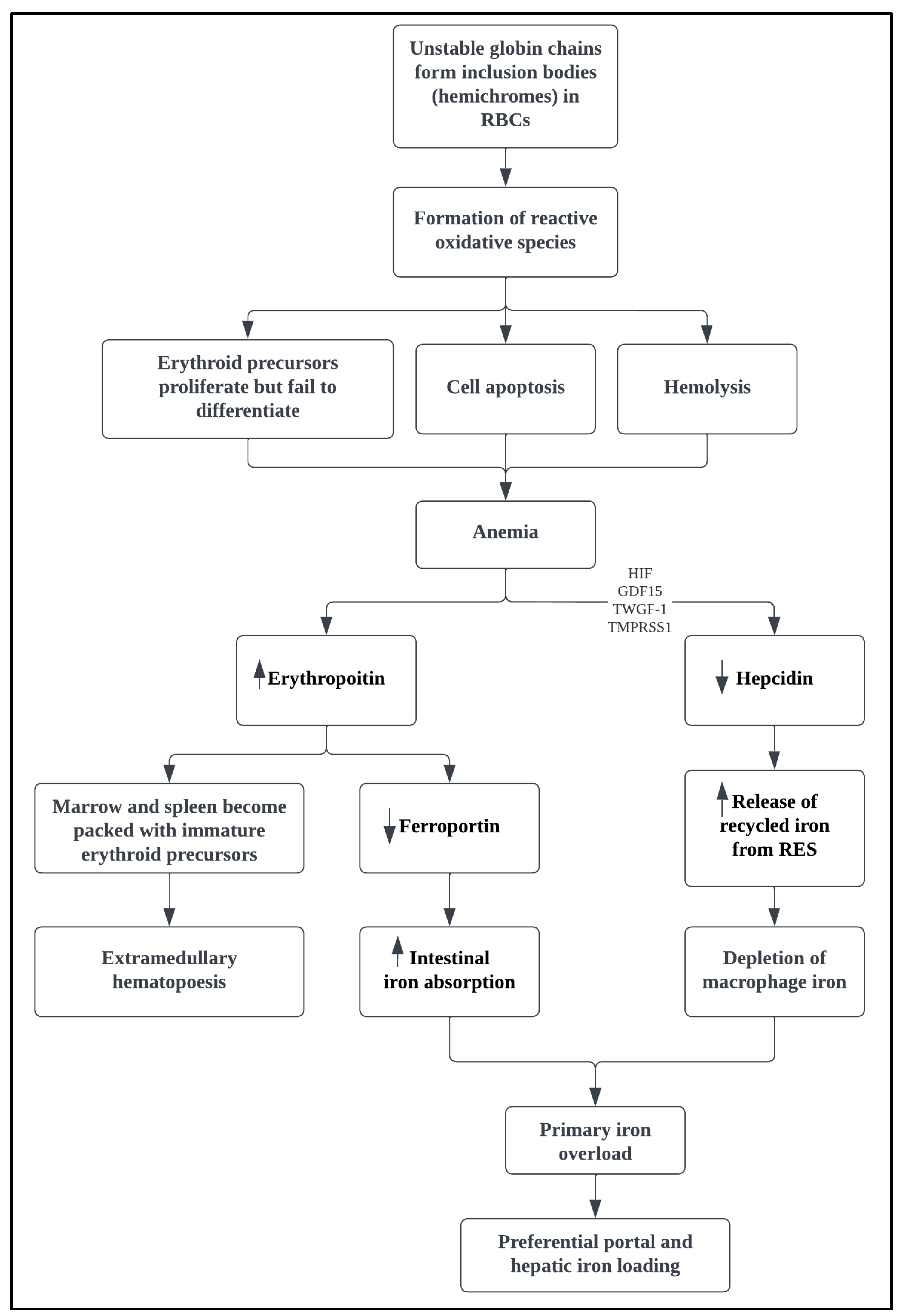
3. Pathophysiology
4. Clinical Presentation
5. Complications
6. Management
6.1. Conventional Therapy
6.1.1. Transfusion Therapy
6.1.2. Splenectomy
6.1.3. Iron Chelation
6.1.4. Hemoglobin F-Stimulating Agents
6.2. Management of Specific Complications
6.3. Novel Therapies
6.3.1. Correcting Globin Chain Imbalance
Hematopoietic Stem Cell Transplantation
Gene Therapy and Editing
6.3.2. Improving IEs
Activin Receptor-II Ligand Traps
Pyruvate Kinase Activators
Janus Kinase 2 Inhibitors
6.3.3. Improving Iron Dysregulation
7. Clinical Practice Points
8. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Musallam, K.M.; Cappellini, M.D.; Viprakasit, V.; Kattamis, A.; Rivella, S.; Taher, A.T. Revisiting the non-transfusion-dependent (NTDT) vs. transfusion-dependent (TDT) thalassemia classification 10 years later. Am. J. Hematol. 2021, 96, E54–E56. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Rivella, S.; Vichinsky, E.; Rachmilewitz, E.A. Non-transfusion-dependent thalassemias. Haematologica 2013, 98, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E. Non-transfusion-dependent thalassemia and thalassemia intermedia: Epidemiology, complications, and management. Curr. Med. Res. Opin. 2016, 32, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Asadov, C.; Alimirzoeva, Z.; Mammadova, T.; Aliyeva, G.; Gafarova, S.; Mammadov, J. beta-Thalassemia intermedia: A comprehensive overview and novel approaches. Int. J. Hematol. 2018, 108, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Radwan, A.; Viprakasit, V. When to consider transfusion therapy for patients with non-transfusion-dependent thalassaemia. Vox Sang. 2015, 108, 1–10. [Google Scholar] [CrossRef]
- Adly, A.A.; Ismail, E.A. Management of Children with beta-Thalassemia Intermedia: Overview, Recent Advances, and Treatment Challenges. J. Pediatr. Hematol. Oncol. 2018, 40, 253–268. [Google Scholar] [CrossRef]
- Jaing, T.H.; Chang, T.Y.; Chen, S.H.; Lin, C.W.; Wen, Y.C.; Chiu, C.C. Molecular genetics of beta-thalassemia: A narrative review. Medicine 2021, 100, e27522. [Google Scholar] [CrossRef]
- Danjou, F.; Anni, F.; Galanello, R. Beta-thalassemia: From genotype to phenotype. Haematologica 2011, 96, 1573–1575. [Google Scholar] [CrossRef]
- Bashir, S.; Mahmood, S.; Mohsin, S.; Tabassum, I.; Ghafoor, M.; Sajjad, O. Modulatory effect of single nucleotide polymorphism in Xmn1, BCL11A and HBS1L-MYB loci on foetal haemoglobin levels in beta-thalassemia major and Intermedia patients. J. Pak. Med. Assoc. 2021, 71, 1394–1398. [Google Scholar] [CrossRef]
- Bauer, D.E.; Kamran, S.C.; Lessard, S.; Xu, J.; Fujiwara, Y.; Lin, C.; Shao, Z.; Canver, M.C.; Smith, E.C.; Pinello, L.; et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science 2013, 342, 253–257. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Marsella, M.; Zanforlin, N. The natural history of thalassemia intermedia. Ann. N. Y. Acad. Sci. 2010, 1202, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D. beta-Thalassemias. N. Engl. J. Med. 2021, 384, 727–743. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, J.; Tarhini, A.; Bou-Fakhredin, R.; Saliba, A.N.; Cappellini, M.D.; Taher, A.T. Non-Transfusion-Dependent Thalassemia: An Update on Complications and Management. Int. J. Mol. Sci. 2018, 19, 182. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.A.; Arlet, J.B.; Dussiot, M.; Moura, I.C.; Courtois, G.; Hermine, O. Ineffective erythropoiesis in beta -thalassemia. Sci. World J. 2013, 2013, 394295. [Google Scholar] [CrossRef]
- Rivella, S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012, 26, S12–S15. [Google Scholar] [CrossRef]
- Bou-Fakhredin, R.; Bazarbachi, A.H.; Chaya, B.; Sleiman, J.; Cappellini, M.D.; Taher, A.T. Iron Overload and Chelation Therapy in Non-Transfusion Dependent Thalassemia. Int. J. Mol. Sci. 2017, 18, 2778. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef]
- Wallace, D.F. The Regulation of Iron Absorption and Homeostasis. Clin. Biochem. Rev. 2016, 37, 51–62. [Google Scholar]
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T.; et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009, 114, 181–186. [Google Scholar] [CrossRef]
- Musallam, K.M.; Taher, A.T.; Duca, L.; Cesaretti, C.; Halawi, R.; Cappellini, M.D. Levels of growth differentiation factor-15 are high and correlate with clinical severity in transfusion-independent patients with beta thalassemia intermedia. Blood Cells Mol. Dis. 2011, 47, 232–234. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 2016, 172, 512–523. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Motta, I.; Musallam, K.M.; Taher, A.T. Redefining thalassemia as a hypercoagulable state. Ann. N. Y. Acad. Sci. 2010, 1202, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Eldor, A.; Rachmilewitz, E.A. The hypercoagulable state in thalassemia. Blood 2002, 99, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Sirachainan, N. Thalassemia and the hypercoagulable state. Thromb. Res. 2013, 132, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Eldor, A.; Lellouche, F.; Goldfarb, A.; Rachmilewitz, E.A.; Maclouf, J. In vivo platelet activation in beta-thalassemia major reflected by increased platelet-thromboxane urinary metabolites. Blood 1991, 77, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Del Principe, D.; Menichelli, A.; di Giulio, S.; de Matteis, W.; Cianciulli, P.; Papa, G. PADGEM/GMP-140 expression on platelet membranes from homozygous beta thalassaemic patients. Br. J. Haematol. 1993, 84, 111–117. [Google Scholar] [CrossRef]
- Eldor, A.; Durst, R.; Hy-Am, E.; Goldfarb, A.; Gillis, S.; Rachmilewitz, E.A.; Abramov, A.; MacLouf, J.; Godefray, Y.C.; de Raucourt, E.; et al. A chronic hypercoagulable state in patients with beta-thalassaemia major is already present in childhood. Br. J. Haematol. 1999, 107, 739–746. [Google Scholar] [CrossRef]
- Shirahata, A.; Funahara, Y.; Opartkiattikul, N.; Fucharoen, S.; Laosombat, V.; Yamada, K. Protein C and protein S deficiency in thalassemic patients. Southeast Asian J. Trop. Med. Public Health 1992, 23, 65–73. [Google Scholar]
- Visudhiphan, S.; Ketsa-Ard, K.; Tumliang, S.; Piankijagum, A. Significance of blood coagulation and platelet profiles in relation to pulmonary thrombosis in beta-thalassemia/Hb E. Southeast Asian J. Trop. Med. Public Health 1994, 25, 449–456. [Google Scholar]
- Viprakasit, V.; Tyan, P.; Rodmai, S.; Taher, A.T. Identification and key management of non-transfusion-dependent thalassaemia patients: Not a rare but potentially under-recognised condition. Orphanet J. Rare Dis. 2014, 9, 131. [Google Scholar] [CrossRef]
- Viprakasit, V.; Ekwattanakit, S. Clinical Classification, Screening and Diagnosis for Thalassemia. Hematol. Oncol. Clin. N. Am. 2018, 32, 193–211. [Google Scholar] [CrossRef]
- Taher, A.T.; Musallam, K.M.; El-Beshlawy, A.; Karimi, M.; Daar, S.; Belhoul, K.; Saned, M.S.; Graziadei, G.; Cappellini, M.D. Age-related complications in treatment-naive patients with thalassaemia intermedia. Br. J. Haematol. 2010, 150, 486–489. [Google Scholar] [CrossRef]
- Munkongdee, T.; Chen, P.; Winichagoon, P.; Fucharoen, S.; Paiboonsukwong, K. Update in Laboratory Diagnosis of Thalassemia. Front. Mol. Biosci. 2020, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Cappellini, M.D. How I manage medical complications of beta-thalassemia in adults. Blood 2018, 132, 1781–1791. [Google Scholar] [CrossRef]
- Taher, A.; Isma’eel, H.; Cappellini, M.D. Thalassemia intermedia: Revisited. Blood Cells Mol. Dis. 2006, 37, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Haddad, A.; Tyan, P.; Radwan, A.; Mallat, N.; Taher, A. beta-Thalassemia Intermedia: A Bird’s-Eye View. Turk. J. Haematol. 2014, 31, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Wood, J.C.; Motta, I.; Graziadei, G.; Tamim, H.; Taher, A.T. Elevated liver iron concentration is a marker of increased morbidity in patients with beta thalassemia intermedia. Haematologica 2011, 96, 1605–1612. [Google Scholar] [CrossRef]
- Musallam, K.M.; Cappellini, M.D.; Taher, A.T. Evaluation of the 5 mg/g liver iron concentration threshold and its association with morbidity in patients with beta-thalassemia intermedia. Blood Cells Mol. Dis. 2013, 51, 35–38. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, G.; Wang, M.; Wei, X.; Pan, L.; Liu, J.; Lei, Y.; Peng, P.; Long, L.; Lai, Y. Iron overload status in patients with non-transfusion-dependent thalassemia in China. Ther. Adv. Hematol. 2022, 13, 20406207221084639. [Google Scholar] [CrossRef]
- Musallam, K.M.; Cappellini, M.D.; Wood, J.C.; Taher, A.T. Iron overload in non-transfusion-dependent thalassemia: A clinical perspective. Blood Rev. 2012, 26, S16–S19. [Google Scholar] [CrossRef]
- Taher, A.; Musallam, K.M.; Rassi, F.E.; Duca, L.; Inati, A.; Koussa, S.; Cappellini, M.D. Levels of non-transferrin-bound iron as an index of iron overload in patients with thalassaemia intermedia. Br. J. Haematol. 2009, 146, 569–572. [Google Scholar] [CrossRef]
- Taher, A.T.; Musallam, K.M.; Karimi, M.; El-Beshlawy, A.; Belhoul, K.; Daar, S.; Saned, M.S.; El-Chafic, A.H.; Fasulo, M.R.; Cappellini, M.D. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: The OPTIMAL CARE study. Blood 2010, 115, 1886–1892. [Google Scholar] [CrossRef]
- Taher, A.; Isma’eel, H.; Mehio, G.; Bignamini, D.; Kattamis, A.; Rachmilewitz, E.A.; Cappellini, M.D. Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thromb. Haemost. 2006, 96, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Karimi, M.; El-Beshlawy, A.; Belhoul, K.; Daar, S.; Saned, M.; Cesaretti, C.; Cappellini, M.D. Splenectomy and thrombosis: The case of thalassemia intermedia. J. Thromb. Haemost. 2010, 8, 2152–2158. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Sankaran, V.G.; Cappellini, M.D.; Duca, L.; Nathan, D.G.; Taher, A.T. Fetal hemoglobin levels and morbidity in untransfused patients with beta-thalassemia intermedia. Blood 2012, 119, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Haidar, R.; Mhaidli, H.; Taher, A.T. Paraspinal extramedullary hematopoiesis in patients with thalassemia intermedia. Eur. Spine J. 2010, 19, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Fraidenburg, D.R.; Machado, R.F. Pulmonary hypertension associated with thalassemia syndromes. Ann. N. Y. Acad. Sci. 2016, 1368, 127–139. [Google Scholar] [CrossRef]
- Aessopos, A.; Farmakis, D. Pulmonary hypertension in beta-thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 342–349. [Google Scholar] [CrossRef]
- Aessopos, A.; Farmakis, D.; Karagiorga, M.; Voskaridou, E.; Loutradi, A.; Hatziliami, A.; Joussef, J.; Rombos, J.; Loukopoulos, D. Cardiac involvement in thalassemia intermedia: A multicenter study. Blood 2001, 97, 3411–3416. [Google Scholar] [CrossRef]
- Derchi, G.; Galanello, R.; Bina, P.; Cappellini, M.D.; Piga, A.; Lai, M.E.; Quarta, A.; Casu, G.; Perrotta, S.; Pinto, V.; et al. Prevalence and risk factors for pulmonary arterial hypertension in a large group of beta-thalassemia patients using right heart catheterization: A Webthal study. Circulation 2014, 129, 338–345. [Google Scholar] [CrossRef]
- Phrommintikul, A.; Sukonthasarn, A.; Kanjanavanit, R.; Nawarawong, W. Splenectomy: A strong risk factor for pulmonary hypertension in patients with thalassaemia. Heart 2006, 92, 1467–1472. [Google Scholar] [CrossRef]
- Morris, C.R.; Kim, H.Y.; Trachtenberg, F.; Wood, J.; Quinn, C.T.; Sweeters, N.; Kwiatkowski, J.L.; Thompson, A.A.; Giardina, P.J.; Boudreaux, J.; et al. Risk factors and mortality associated with an elevated tricuspid regurgitant jet velocity measured by Doppler-echocardiography in thalassemia: A Thalassemia Clinical Research Network report. Blood 2011, 118, 3794–3802. [Google Scholar] [CrossRef]
- Karimi, M.; Musallam, K.M.; Cappellini, M.D.; Daar, S.; El-Beshlawy, A.; Belhoul, K.; Saned, M.S.; Temraz, S.; Koussa, S.; Taher, A.T. Risk factors for pulmonary hypertension in patients with beta thalassemia intermedia. Eur. J. Intern. Med. 2011, 22, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Inati, A.; Noureldine, M.A.; Mansour, A.; Abbas, H.A. Endocrine and bone complications in β-thalassemia intermedia: Current understanding and treatment. Biomed. Res. Int. 2015, 2015, 813098. [Google Scholar] [CrossRef]
- Dede, A.D.; Trovas, G.; Chronopoulos, E.; Triantafyllopoulos, I.K.; Dontas, I.; Papaioannou, N.; Tournis, S. Thalassemia-associated osteoporosis: A systematic review on treatment and brief overview of the disease. Osteoporos. Int. 2016, 27, 3409–3425. [Google Scholar] [CrossRef]
- Vogiatzi, M.G.; Macklin, E.A.; Fung, E.B.; Cheung, A.M.; Vichinsky, E.; Olivieri, N.; Kirby, M.; Kwiatkowski, J.L.; Cunningham, M.; Holm, I.A.; et al. Bone disease in thalassemia: A frequent and still unresolved problem. J. Bone Miner. Res. 2009, 24, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Hashemieh, M.; Azarkeivan, A.; Radfar, M.; Saneifard, H.; Hosseini-Zijoud, S.M.; Noghabaei, G.; Yaseri, M. Prevalence of Osteoporosis among Thalassemia Patients from Zafar Adult Thalassemia Clinic, Iran. Iran. J. Blood Cancer 2014, 6, 143–148. [Google Scholar]
- Charoenngam, N.; Rittiphairoj, T.; Ponvilawan, B. Fracture prevalence in thalassemia: A systematic review and meta-analysis. Arch. Osteoporos. 2021, 16, 171. [Google Scholar] [CrossRef]
- Mehta, V.; Kirubarajan, A.; Sabouhanian, A.; Jayawardena, S.M.; Chandrakumaran, P.; Thangavelu, N.; Cader, R.; Mettananda, S.; Bandara, D.; Khan, S.; et al. Leg Ulcers: A Report in Patients with Hemoglobin E Beta Thalassemia and Review of the Literature in Severe Beta Thalassemia. Acta Haematol. 2022, 145, 334–343. [Google Scholar] [CrossRef]
- Matta, B.N.; Abbas, O.; Maakaron, J.E.; Koussa, S.; Daderian, R.H.; Taher, A.T. Leg ulcers in patients with beta-thalassaemia intermedia: A single centre’s experience. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1245–1250. [Google Scholar] [CrossRef]
- Gimmon, Z.; Wexler, M.R.; Rachmilewitz, E.A. Juvenile leg ulceration in beta-thalassemia major and intermedia. Plast. Reconstr. Surg. 1982, 69, 320–325. [Google Scholar] [CrossRef]
- Spanos, T.; Karageorga, M.; Ladis, V.; Peristeri, J.; Hatziliami, A.; Kattamis, C. Red cell alloantibodies in patients with thalassemia. Vox Sang. 1990, 58, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Al-Riyami, A.Z.; Al-Mahrooqi, S.; Al-Hinai, S.; Al-Hosni, S.; Al-Madhani, A.; Daar, S. Transfusion therapy and alloimmunization in Thalassemia Intermedia: A 10 year experience at a tertiary care university hospital. Transfus. Apher. Sci. 2014, 51, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Ang, A.L.; Lim, C.Y.; Ng, W.Y.; Lam, J.C.M. Non-transfusion dependent thalassemia is independently associated with higher alloimmunization risk than transfusion dependent thalassemia and would benefit the most from extended red cell antigen-matching. Transfusion 2021, 61, 2566–2577. [Google Scholar] [CrossRef]
- Taher, A.; Musallam, K.; Cappellini, M.D. (Eds.) Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT), 2nd ed.; Thalassaemia International Federation: Nicosia, Cyprus, 2017. [Google Scholar]
- Phadke, S.R.; Agarwal, S. Phenotype score to grade the severity of thalassemia intermedia. Indian J. Pediatr. 2003, 70, 477–481. [Google Scholar] [CrossRef]
- Sripichai, O.; Makarasara, W.; Munkongdee, T.; Kumkhaek, C.; Nuchprayoon, I.; Chuansumrit, A.; Chuncharunee, S.; Chantrakoon, N.; Boonmongkol, P.; Winichagoon, P.; et al. A scoring system for the classification of beta-thalassemia/Hb E disease severity. Am. J. Hematol. 2008, 83, 482–484. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Neumayr, L.; Trimble, S.; Giardina, P.J.; Cohen, A.R.; Coates, T.; Boudreaux, J.; Neufeld, E.J.; Kenney, K.; Grant, A.; et al. Transfusion complications in thalassemia patients: A report from the Centers for Disease Control and Prevention (CME). Transfusion 2014, 54, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Al-Salem, A.H.; Al-Dabbous, I.; Bhamidibati, P. The role of partial splenectomy in children with thalassemia. Eur. J. Pediatr. Surg. 1998, 8, 334–338. [Google Scholar] [CrossRef]
- Sheikha, A.K.; Salih, Z.T.; Kasnazan, K.H.; Khoshnaw, M.K.; Al-Maliki, T.; Al-Azraqi, T.A.; Zafer, M.H. Prevention of overwhelming postsplenectomy infection in thalassemia patients by partial rather than total splenectomy. Can. J. Surg. 2007, 50, 382–386. [Google Scholar] [PubMed]
- Taher, A.; Rassi, F.E.; Isma’eel, H.; Koussa, S.; Inati, A.; Cappellini, M.D. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica 2008, 93, 1584–1586. [Google Scholar] [CrossRef]
- Taher, A.T.; Cappellini, M.D.; Aydinok, Y.; Porter, J.B.; Karakas, Z.; Viprakasit, V.; Siritanaratkul, N.; Kattamis, A.; Wang, C.; Zhu, Z.; et al. Optimising iron chelation therapy with deferasirox for non-transfusion-dependent thalassaemia patients: 1-year results from the THETIS study. Blood Cells Mol. Dis. 2016, 57, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Porter, J.B.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Galanello, R.; Karakas, Z.; Lawniczek, T.; et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann. Hematol. 2013, 92, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghe, C.N.; Kontoghiorghes, G.J. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with non-transfusion-dependent thalassemia syndromes. Drug Des. Dev. Ther. 2016, 10, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T.; Cappellini, M.D.; Sankaran, V.G. Clinical experience with fetal hemoglobin induction therapy in patients with beta-thalassemia. Blood 2013, 121, 2199–2212. [Google Scholar] [CrossRef] [PubMed]
- Mabaera, R.; West, R.J.; Conine, S.J.; Macari, E.R.; Boyd, C.D.; Engman, C.A.; Lowrey, C.H. A cell stress signaling model of fetal hemoglobin induction: What doesn’t kill red blood cells may make them stronger. Exp. Hematol. 2008, 36, 1057–1072. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.T.; Huang, S.Z.; Ren, Z.R.; Lu, Z.H.; Zeng, F.Y.; Schechter, A.N.; Rodgers, G.P. Hydroxyurea therapy in beta-thalassaemia intermedia: Improvement in haematological parameters due to enhanced beta-globin synthesis. Br. J. Haematol. 1995, 90, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Zohaib, M.; Ansari, S.H.; Shamsi, T.S.; Zubarev, R.A.; Zarina, S. Pharmacoproteomics Profiling of Plasma from beta-Thalassemia Patients in Response to Hydroxyurea Treatment. J. Clin. Pharmacol. 2019, 59, 98–106. [Google Scholar] [CrossRef]
- Algiraigri, A.H.; Wright, N.A.M.; Paolucci, E.O.; Kassam, A. Hydroxyurea for nontransfusion-dependent beta-thalassemia: A systematic review and meta-analysis. Hematol. Oncol. Stem Cell Ther. 2017, 10, 116–125. [Google Scholar] [CrossRef]
- Koren, A.; Levin, C.; Dgany, O.; Kransnov, T.; Elhasid, R.; Zalman, L.; Palmor, H.; Tamary, H. Response to hydroxyurea therapy in beta-thalassemia. Am. J. Hematol. 2008, 83, 366–370. [Google Scholar] [CrossRef]
- Karimi, M.; Zarei, T.; Bahmanimehr, A.; Aramesh, A.; Daryanoush, S.; Haghpanah, S. Long-term safety and efficacy of hydroxyurea in patients with non-transfusion-dependent beta-thalassemia: A comprehensive single-center experience. Ann. Hematol. 2021, 100, 2901–2907. [Google Scholar] [CrossRef]
- Myssina, S.; Huber, S.M.; Birka, C.; Lang, P.A.; Lang, K.S.; Friedrich, B.; Risler, T.; Wieder, T.; Lang, F. Inhibition of erythrocyte cation channels by erythropoietin. J. Am. Soc. Nephrol. 2003, 14, 2750–2757. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.T.; Vichinsky, E.P.; Sweeters, N.; Rachmilewitz, E. Darbepoetin alfa for the treatment of anaemia in alpha- or beta- thalassaemia intermedia syndromes. Br. J. Haematol. 2011, 154, 281–284. [Google Scholar] [CrossRef]
- Elalfy, M.S.; Adly, A.A.; Ismail, E.A.; Elhenawy, Y.I.; Elghamry, I.R. Therapeutic superiority and safety of combined hydroxyurea with recombinant human erythropoietin over hydroxyurea in young beta-thalassemia intermedia patients. Eur. J. Haematol. 2013, 91, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Rivella, S.; Taher, A.T. Management of non-transfusion-dependent beta-thalassemia (NTDT): The next 5 years. Am. J. Hematol. 2021, 96, E57–E59. [Google Scholar] [CrossRef] [PubMed]
- Makis, A.; Voskaridou, E.; Papassotiriou, I.; Hatzimichael, E. Novel Therapeutic Advances in beta-Thalassemia. Biology 2021, 10, 546. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Porter, J.B.; Viprakasit, V.; Taher, A.T. A paradigm shift on beta-thalassaemia treatment: How will we manage this old disease with new therapies? Blood Rev. 2018, 32, 300–311. [Google Scholar] [CrossRef]
- Canver, M.C.; Smith, E.C.; Sher, F.; Pinello, L.; Sanjana, N.E.; Shalem, O.; Chen, D.D.; Schupp, P.G.; Vinjamur, D.S.; Garcia, S.P.; et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 2015, 527, 192–197. [Google Scholar] [CrossRef]
- Masuda, T.; Wang, X.; Maeda, M.; Canver, M.C.; Sher, F.; Funnell, A.P.; Fisher, C.; Suciu, M.; Martyn, G.E.; Norton, L.J.; et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science 2016, 351, 285–289. [Google Scholar] [CrossRef]
- Wienert, B.; Funnell, A.P.; Norton, L.J.; Pearson, R.C.; Wilkinson-White, L.E.; Lester, K.; Vadolas, J.; Porteus, M.H.; Matthews, J.M.; Quinlan, K.G.; et al. Editing the genome to introduce a beneficial naturally occurring mutation associated with increased fetal globin. Nat. Commun. 2015, 6, 7085. [Google Scholar] [CrossRef]
- Goodman, M.A.; Malik, P. The potential of gene therapy approaches for the treatment of hemoglobinopathies: Achievements and challenges. Ther. Adv. Hematol. 2016, 7, 302–315. [Google Scholar] [CrossRef]
- Smith, A.R.; Schiller, G.J.; Vercellotti, G.M.; Kwiatkowski, J.L.; Krishnamurti, L.; Esrick, E.B.; Williams, D.A.; Miller, W.P.; Woolfson, A.; Walters, M.C. Preliminary results of a phase 1/2 clinical study of zinc finger nuclease-mediated editing of BCL11A in autologous hematopoietic stem cells for transfusion-dependent beta thalassemia. Blood 2019, 134, 3544. [Google Scholar] [CrossRef]
- Frangoul, H.; Bobruff, Y.; Cappellini, M.D.; Corbacioglu, S.; Fernandez, C.M.; de la Fuente, J.; Grupp, S.A.; Handgretinger, R.; Ho, T.W.; Imren, S. Safety and efficacy of CTX001 in patients with transfusion-dependent β-thalassemia and sickle cell disease: Early results from the climb THAL-111 and climb SCD-121 studies of autologous CRISPR-CAS9-modified CD34+ hematopoietic stem and progenitor cells. Blood 2020, 136, 3–4. [Google Scholar] [CrossRef]
- Walters, M.C.; Smith, A.R.; Schiller, G.J.; Esrick, E.B.; Williams, D.A.; Gogoleva, T.; Rouy, D.; Cockroft, B.M.; Vercellotti, G.M. Updated Results of a Phase 1/2 Clinical Study of Zinc Finger Nuclease-Mediated Editing of BCL11A in Autologous Hematopoietic Stem Cells for Transfusion-Dependent Beta Thalassemia. Blood 2021, 138, 3974. [Google Scholar] [CrossRef]
- Soderberg, S.S.; Karlsson, G.; Karlsson, S. Complex and context dependent regulation of hematopoiesis by TGF-beta superfamily signaling. Ann. N. Y. Acad. Sci. 2009, 1176, 55–69. [Google Scholar] [CrossRef]
- Vaidya, A.; Kale, V.P. TGF-beta signaling and its role in the regulation of hematopoietic stem cells. Syst. Synth. Biol. 2015, 9, 1–10. [Google Scholar] [CrossRef]
- Taher, A.T.; Cappellini, M.D. Luspatercept for beta-thalassemia: Beyond red blood cell transfusions. Expert Opin. Biol. Ther. 2021, 21, 1363–1371. [Google Scholar] [CrossRef]
- Suragani, R.N.; Cawley, S.M.; Li, R.; Wallner, S.; Alexander, M.J.; Mulivor, A.W.; Gardenghi, S.; Rivella, S.; Grinberg, A.V.; Pearsall, R.S.; et al. Modified activin receptor IIB ligand trap mitigates ineffective erythropoiesis and disease complications in murine beta-thalassemia. Blood 2014, 123, 3864–3872. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T.; Georgiev, P.; Kuo, K.H.M.; Coates, T.; Voskaridou, E.; Liew, H.K.; Pazgal-Kobrowski, I.; Forni, G.L.; et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent beta-Thalassemia. N. Engl. J. Med. 2020, 382, 1219–1231. [Google Scholar] [CrossRef]
- Taher, A.T.; Viprakasit, V.; Hermine, O.; Porter, J.B.; Piga, A.; Kuo, K.H.; Coates, T.D.; Voskaridou, E.; Khelif, A.; Kattamis, A. Sustained reductions in red blood cell (RBC) transfusion burden and events in β-thalassemia with luspatercept: Longitudinal results of the BELIEVE trial. Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Taher, A.; Cappellini, M.; Kattamis, A. The BEYOND study: Results of a phase 2, double-blind, randomized, placebo-controlled multicenter study of luspatercept in adult patients with non-tranfusion dependent beta-thalassemia. In Proceedings of the 26th Congress of the European Hematology Association (EHA), Virtual Congress Platform, 11 June 2021. [Google Scholar]
- Musallam, K.M.; Bou-Fakhredin, R.; Cappellini, M.D.; Taher, A.T. 2021 update on clinical trials in beta-thalassemia. Am. J. Hematol. 2021, 96, 1518–1531. [Google Scholar] [CrossRef]
- Aizawa, S.; Harada, T.; Kanbe, E.; Tsuboi, I.; Aisaki, K.; Fujii, H.; Kanno, H. Ineffective erythropoiesis in mutant mice with deficient pyruvate kinase activity. Exp. Hematol. 2005, 33, 1292–1298. [Google Scholar] [CrossRef]
- Grace, R.F.; Rose, C.; Layton, D.M.; Galacteros, F.; Barcellini, W.; Morton, D.H.; van Beers, E.J.; Yaish, H.; Ravindranath, Y.; Kuo, K.H.M.; et al. Safety and Efficacy of Mitapivat in Pyruvate Kinase Deficiency. N. Engl. J. Med. 2019, 381, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Matte, A.; Federti, E.; Kung, C.; Kosinski, P.A.; Narayanaswamy, R.; Russo, R.; Federico, G.; Carlomagno, F.; Desbats, M.A.; Salviati, L. The pyruvate kinase activator mitapivat reduces hemolysis and improves anemia in a β-thalassemia mouse model. J. Clin. Investig. 2021, 131, e144206. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of neoplastic and inflammatory disorders. Pharmacol. Res. 2022, 183, 106362. [Google Scholar] [CrossRef]
- Casu, C.; Presti, V.L.; Oikonomidou, P.R.; Melchiori, L.; Abdulmalik, O.; Ramos, P.; Rivella, S. Short-term administration of JAK2 inhibitors reduces splenomegaly in mouse models of beta-thalassemia intermedia and major. Haematologica 2018, 103, e46–e49. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Karakas, Z.; Cassinerio, E.; Siritanaratkul, N.; Kattamis, A.; Maggio, A.; Rivella, S.; Hollaender, N.; Mahuzier, B.; Gadbaw, B.; et al. Efficacy and safety of ruxolitinib in regularly transfused patients with thalassemia: Results from a phase 2a study. Blood 2018, 131, 263–265. [Google Scholar] [CrossRef]
- Taneja, K.; Verma, C.; Mahajan, A. Can ruxolitinib avert splenectomy in patients with thalassaemia: A short term case series. Br. J. Haematol. 2022, 196, 1111–1113. [Google Scholar] [CrossRef]
- Casu, C.; Chessa, R.; Liu, A.; Gupta, R.; Drakesmith, H.; Fleming, R.; Ginzburg, Y.Z.; MacDonald, B.; Rivella, S. Minihepcidins improve ineffective erythropoiesis and splenomegaly in a new mouse model of adult beta-thalassemia major. Haematologica 2020, 105, 1835–1844. [Google Scholar] [CrossRef]
- Nai, A.; Pagani, A.; Mandelli, G.; Lidonnici, M.R.; Silvestri, L.; Ferrari, G.; Camaschella, C. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood 2012, 119, 5021–5029. [Google Scholar] [CrossRef]
- Guo, S.; Casu, C.; Gardenghi, S.; Booten, S.; Aghajan, M.; Peralta, R.; Watt, A.; Freier, S.; Monia, B.P.; Rivella, S. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J. Clin. Investig. 2013, 123, 1531–1541. [Google Scholar] [CrossRef]
- Schmidt, P.J.; Toudjarska, I.; Sendamarai, A.K.; Racie, T.; Milstein, S.; Bettencourt, B.R.; Hettinger, J.; Bumcrot, D.; Fleming, M.D. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood 2013, 121, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Manolova, V.; Nyffenegger, N.; Flace, A.; Altermatt, P.; Varol, A.; Doucerain, C.; Sundstrom, H.; Durrenberger, F. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of beta-thalassemia. J. Clin. Investig. 2019, 130, 491–506. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical Criteria | TDT More Likely | NTDT More Likely |
|---|---|---|
| Age at presentation | ≤2 years | >2 years |
| Degree of anemia at presentation | Severe | Mild to severe |
| Clinical anemia affecting daily living | Yes | No |
| Splenomegaly at presentation | Severe | Mild to severe |
| Jaundice | No | Mild |
| Skeletal deformities | Yes | Negative to mild |
| Growth retardation | Moderate to Severe | Negative to moderate |
| Transfusion requirements | Lifelong, dependence for survival | None, occasional, or frequent but temporary |
| Hb levels | 6–7 gm/dL | 8–10 gm/dL |
| Nucleated RBCs | Numerous | Negative to few |
| Reticulocytes | >10% | <10% |
| Therapy | Indications | |
|---|---|---|
| Blood transfusion | Acute stress | |
|
| |
|
| |
Progressive changes from childhood $
| ||
Complications $
| ||
| Splenectomy |
| |
| Hydroxyurea |
| |
| Complication | Screening and Management |
|---|---|
| Thrombotic disease |
|
| Pulmonary hypertension |
|
| Extramedullary hematopoiesis |
|
| Leg ulcers |
|
| Endocrine and bone disease # |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shash, H. Non-Transfusion-Dependent Thalassemia: A Panoramic Review. Medicina 2022, 58, 1496. https://doi.org/10.3390/medicina58101496
Shash H. Non-Transfusion-Dependent Thalassemia: A Panoramic Review. Medicina. 2022; 58(10):1496. https://doi.org/10.3390/medicina58101496
Chicago/Turabian StyleShash, Hwazen. 2022. "Non-Transfusion-Dependent Thalassemia: A Panoramic Review" Medicina 58, no. 10: 1496. https://doi.org/10.3390/medicina58101496
APA StyleShash, H. (2022). Non-Transfusion-Dependent Thalassemia: A Panoramic Review. Medicina, 58(10), 1496. https://doi.org/10.3390/medicina58101496