
Case of Patient with AML with Complex Karyotype including Ultra-Rare t(4;8)(q32;q13), t(4;11)(q21;p15) and Familial Aggregation of Myeloid Malignancies

 , , , ,
, , , ,
Abstract
:1. Introduction
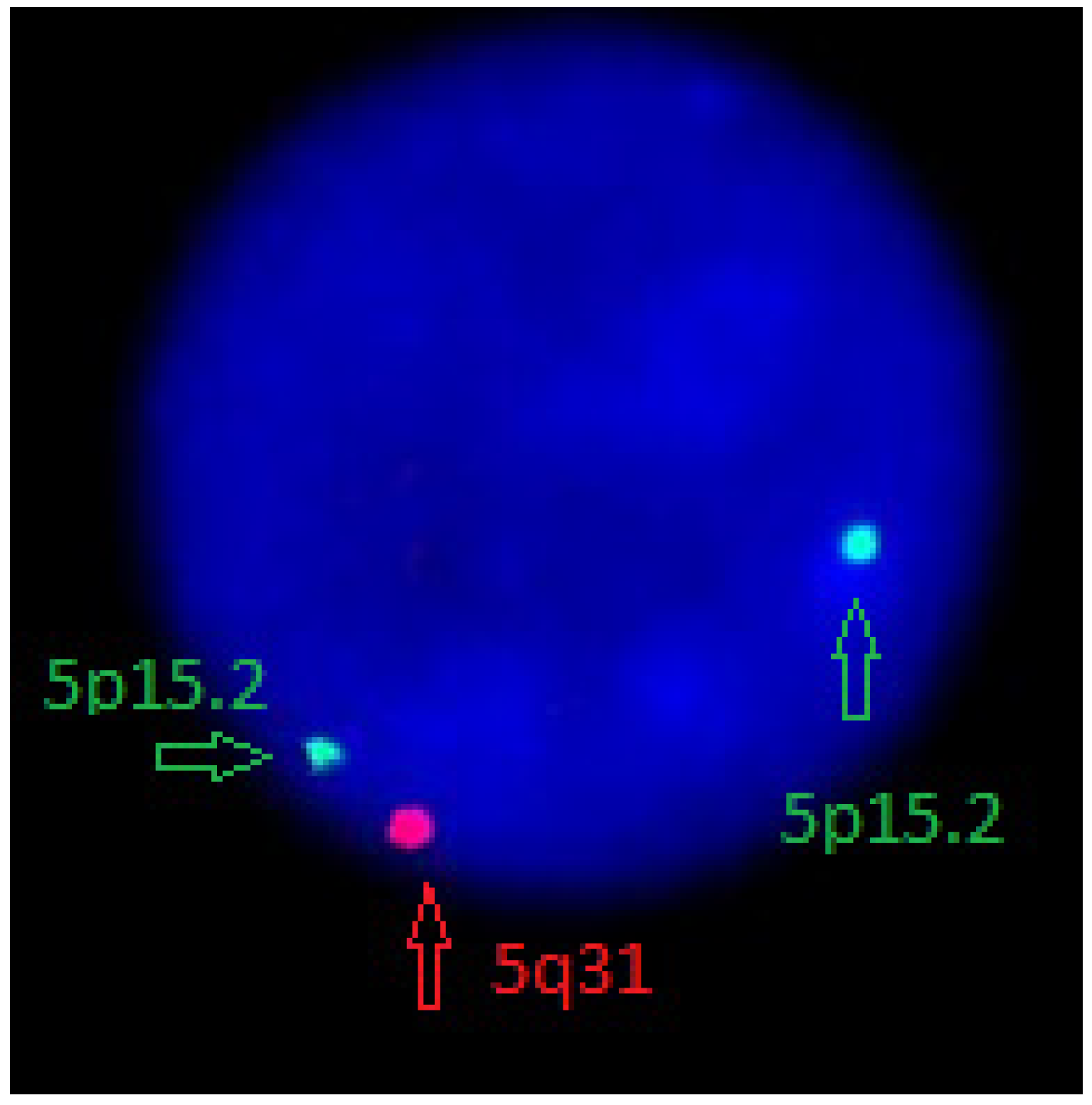
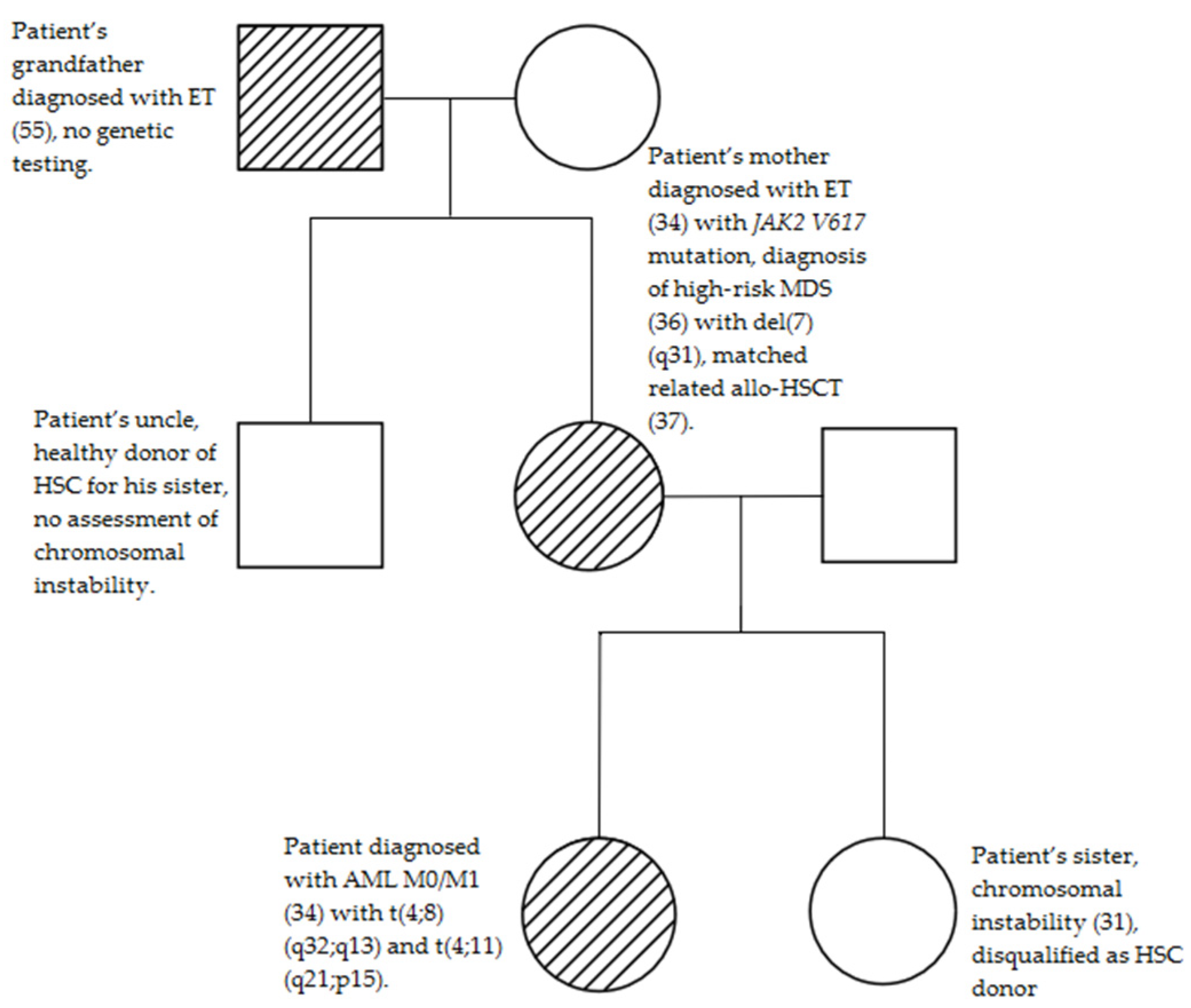
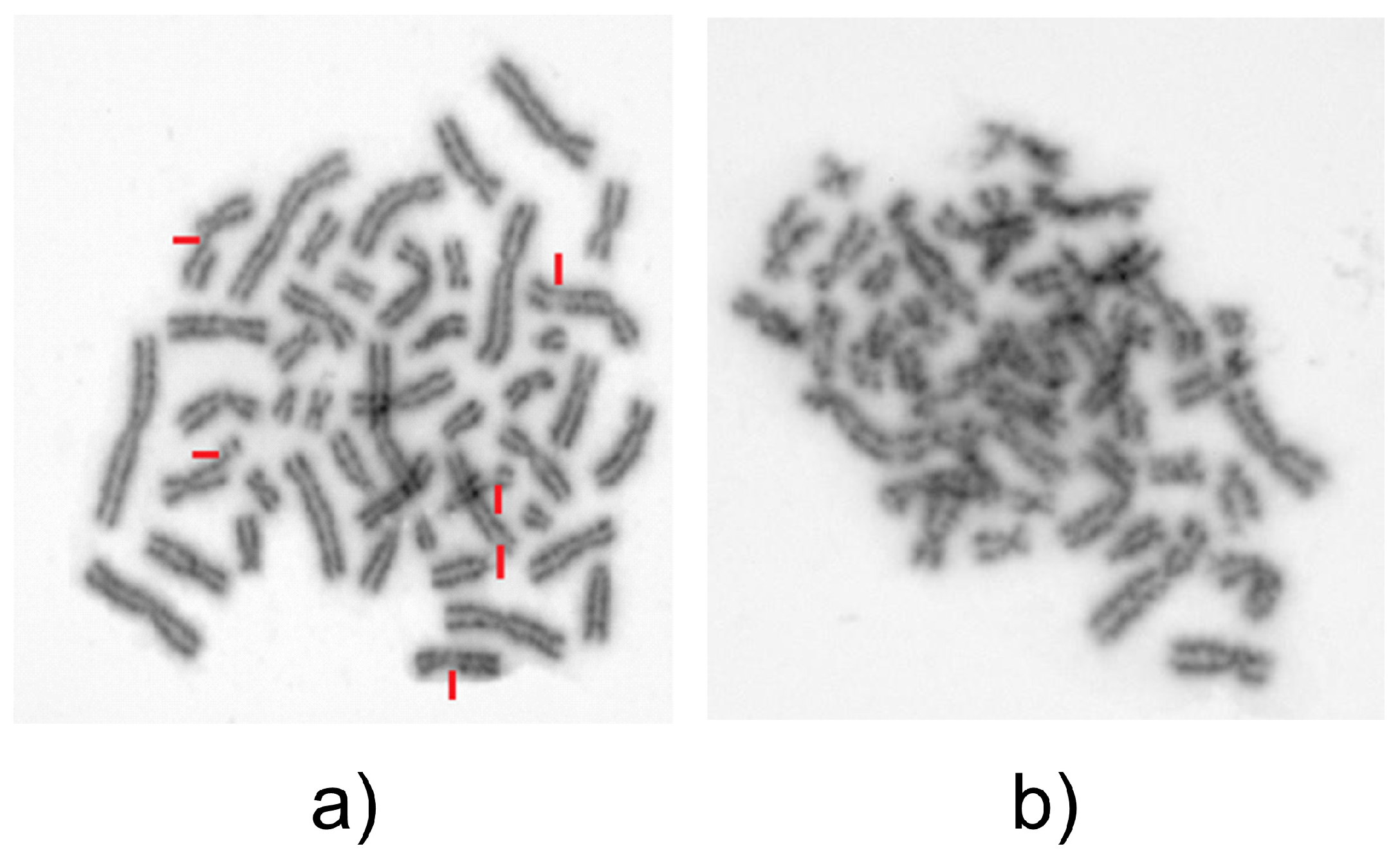
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sud, A.; Chattopadhyay, S.; Thomsen, H.; Sundquist, K.; Sundquist, J.; Houlston, R.S.; Hemminki, K. Familial risks of acute myeloid leukemia, myelodysplastic syndromes, and myeloproliferative neoplasms. Blood 2018, 132, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Landgren, O.; Goldin, L.R.; Kristinsson, S.Y.; Helgadottir, E.A.; Samuelsson, J.; Björkholm, M. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood 2008, 112, 2199–2204. [Google Scholar] [CrossRef] [PubMed]
- Mannelli, F. Acute Myeloid Leukemia Evolving from Myeloproliferative Neoplasms: Many Sides of a Challenging Disease. J. Clin. Med. 2021, 10, 436. [Google Scholar] [CrossRef] [PubMed]
- Björkholm, M.; Kristinsson, S.Y.; Landgren, O.; Goldin, L.R. No familial aggregation in chronic myeloid leukemia. Blood 2013, 122, 460–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barquinero, J.F.; Barrios, L.; Ribas, M.; Egozcue, J.; Caballín, M.R. Cytogenetic sensitivity of three Fanconi anemia heterozygotes to bleomycin and ionizing radiation. Cancer Genet. Cytogenet. 2001, 124, 80–83. [Google Scholar] [CrossRef]
- Ludwig, L.B.; Valiati, V.H.; Palazzo, R.P.; Jardim, L.B.; da Rosa, D.P.; Bona, S.; Rodrigues, G.; Marroni, N.P.; Prá, D.; Maluf, S.W. Chromosome instability and oxidative stress markers in patients with ataxia telangiectasia and their parents. BioMed Res. Int. 2013, 2013, 762048. [Google Scholar] [PubMed] [Green Version]
- Hsu, T.C. Differential mutagen susceptibility in cultured lymphocytes of normal individuals and cancer patients. Cancer Genet. Cytogenet. 1985, 17, 307–313. [Google Scholar] [CrossRef]
- Hsu, T.C. Sensitivity to genotoxic effects of bleomycin in humans: Possible relationship to environmental carcinogenesis. Int. J. Cancer 1989, 43, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Hussey, D.J.; Nicola, M.; Moore, S.; Peters, G.B.; Dobrovic, A. The (4;11)(q21;p15) translocation fuses the NUP98 and RAP1GDS1 genes and is recurrent in T-cell acute lymphocytic leukemia. Blood 1999, 94, 2072–2079. [Google Scholar] [CrossRef]
- van Zutven, L.J.C.M.; Önen, E.; Velthuizen, S.C.J.M.; van Drunen, E.; von Bergh, A.R.M.; van den Heuvel-Eibrink, M.M.; Veronese, A.; Mecucci, C.; Negrini, M.; de Greef, G.E.; et al. Identification of NUP98 abnormalities in acute leukemia: JARID1A (12p13) as a new partner gene. Genes Chromosomes Cancer 2006, 45, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Thangavelu, M.; Huang, B.; Lemieux, M.; Tom, W.; Richkind, K.E. A t(4;11)(q21;p15) in a case of T-cell lymphoma and a case of acute myelogenous leukemia. Cancer Genet. Cytogenet. 2002, 132, 109–115. [Google Scholar] [CrossRef]
- Lam, D.H.; Aplan, P.D. NUP98 gene fusions in hematologic malignancies. Leukemia 2001, 15, 1689–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Oliveira Lisboa, M.; Brofman, P.R.S.; Schmid-Braz, A.T.; Rangel-Pozzo, A.; Mai, S. Chromosomal Instability in Acute Myeloid Leukemia. Cancers 2021, 13, 2655. [Google Scholar] [CrossRef] [PubMed]
- Berger, G.; van den Berg, E.; Sikkema-Raddatz, B.; Abbott, K.M.; Sinke, R.J.; Bungener, L.B.; Mulder, A.B.; Vellenga, E. Re-emergence of acute myeloid leukemia in donor cells following allogeneic transplantation in a family with a germline DDX41 mutation. Leukemia 2017, 31, 520–522. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Test Sample without Bleomycin (n = 100 Cells) | Test Sample with Bleomycin (n = 100 Cells) | Control Sample without Bleomycin (n = 50 Cells) | Control Sample without Bleomycin (n = 50 Cells) | |
|---|---|---|---|---|
| Number of chromatid breaks | 0 | 127 | 0 | 0 |
| Breaks/cell ratio | 0 | 1.27 | 0 | 0 |
| % of damaged cells | 0 | 26 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milczarek, S.; Studniak, E.; Baumert, B.; Janowski, M.; Bonda, W.; Pietrzak, J.; Łanocha, A.; Paczkowska, E.; Zdziarska, B.; Machaliński, B. Case of Patient with AML with Complex Karyotype including Ultra-Rare t(4;8)(q32;q13), t(4;11)(q21;p15) and Familial Aggregation of Myeloid Malignancies. Medicina 2022, 58, 105. https://doi.org/10.3390/medicina58010105
Milczarek S, Studniak E, Baumert B, Janowski M, Bonda W, Pietrzak J, Łanocha A, Paczkowska E, Zdziarska B, Machaliński B. Case of Patient with AML with Complex Karyotype including Ultra-Rare t(4;8)(q32;q13), t(4;11)(q21;p15) and Familial Aggregation of Myeloid Malignancies. Medicina. 2022; 58(1):105. https://doi.org/10.3390/medicina58010105
Chicago/Turabian StyleMilczarek, Sławomir, Ewa Studniak, Bartłomiej Baumert, Michał Janowski, Wioleta Bonda, Joanna Pietrzak, Aleksandra Łanocha, Edyta Paczkowska, Barbara Zdziarska, and Bogusław Machaliński. 2022. "Case of Patient with AML with Complex Karyotype including Ultra-Rare t(4;8)(q32;q13), t(4;11)(q21;p15) and Familial Aggregation of Myeloid Malignancies" Medicina 58, no. 1: 105. https://doi.org/10.3390/medicina58010105
APA StyleMilczarek, S., Studniak, E., Baumert, B., Janowski, M., Bonda, W., Pietrzak, J., Łanocha, A., Paczkowska, E., Zdziarska, B., & Machaliński, B. (2022). Case of Patient with AML with Complex Karyotype including Ultra-Rare t(4;8)(q32;q13), t(4;11)(q21;p15) and Familial Aggregation of Myeloid Malignancies. Medicina, 58(1), 105. https://doi.org/10.3390/medicina58010105