
Radiological and Physiological Predictors of IPF Mortality

 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Study Population and Collection Data
2.2. Physiological Data
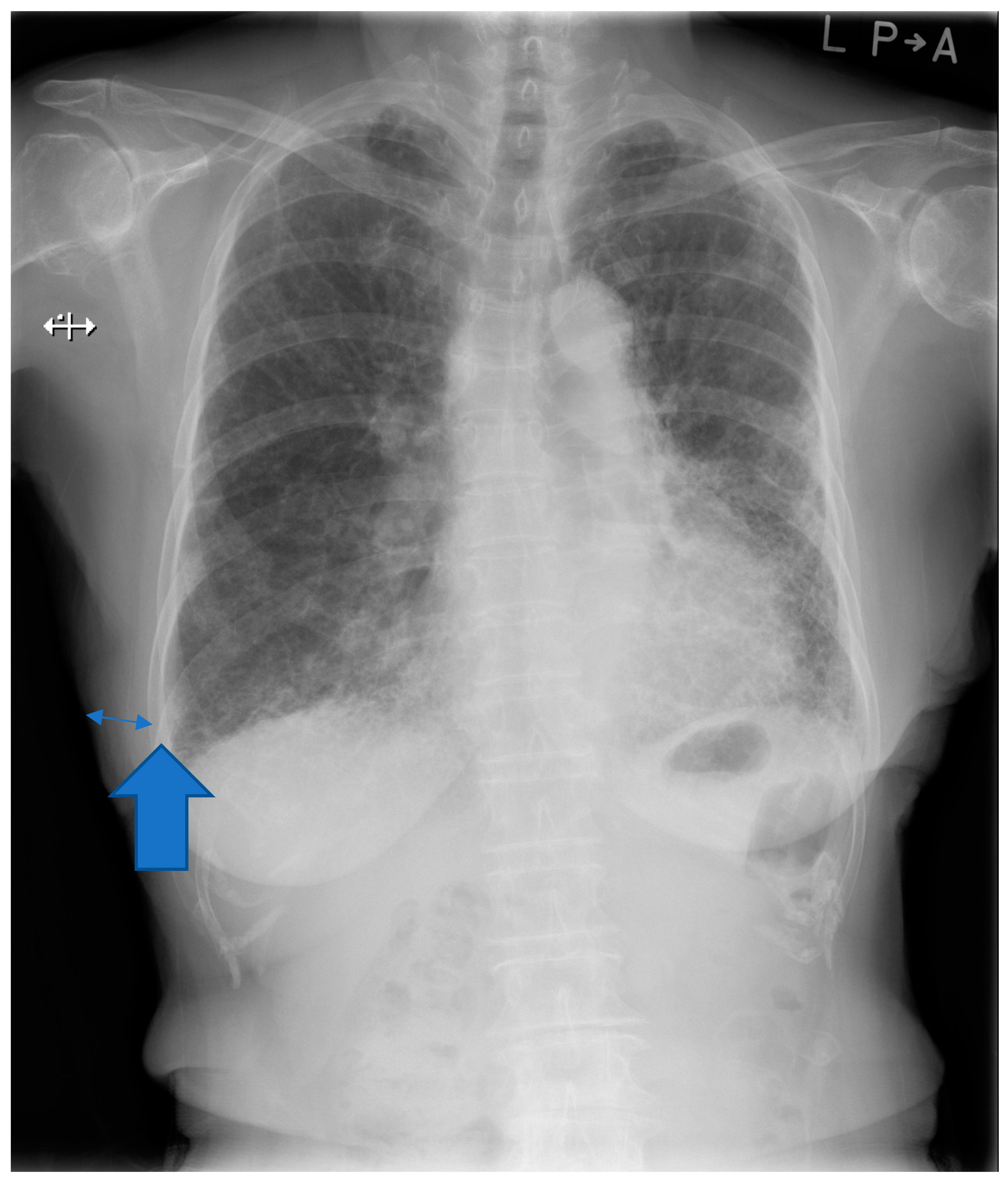
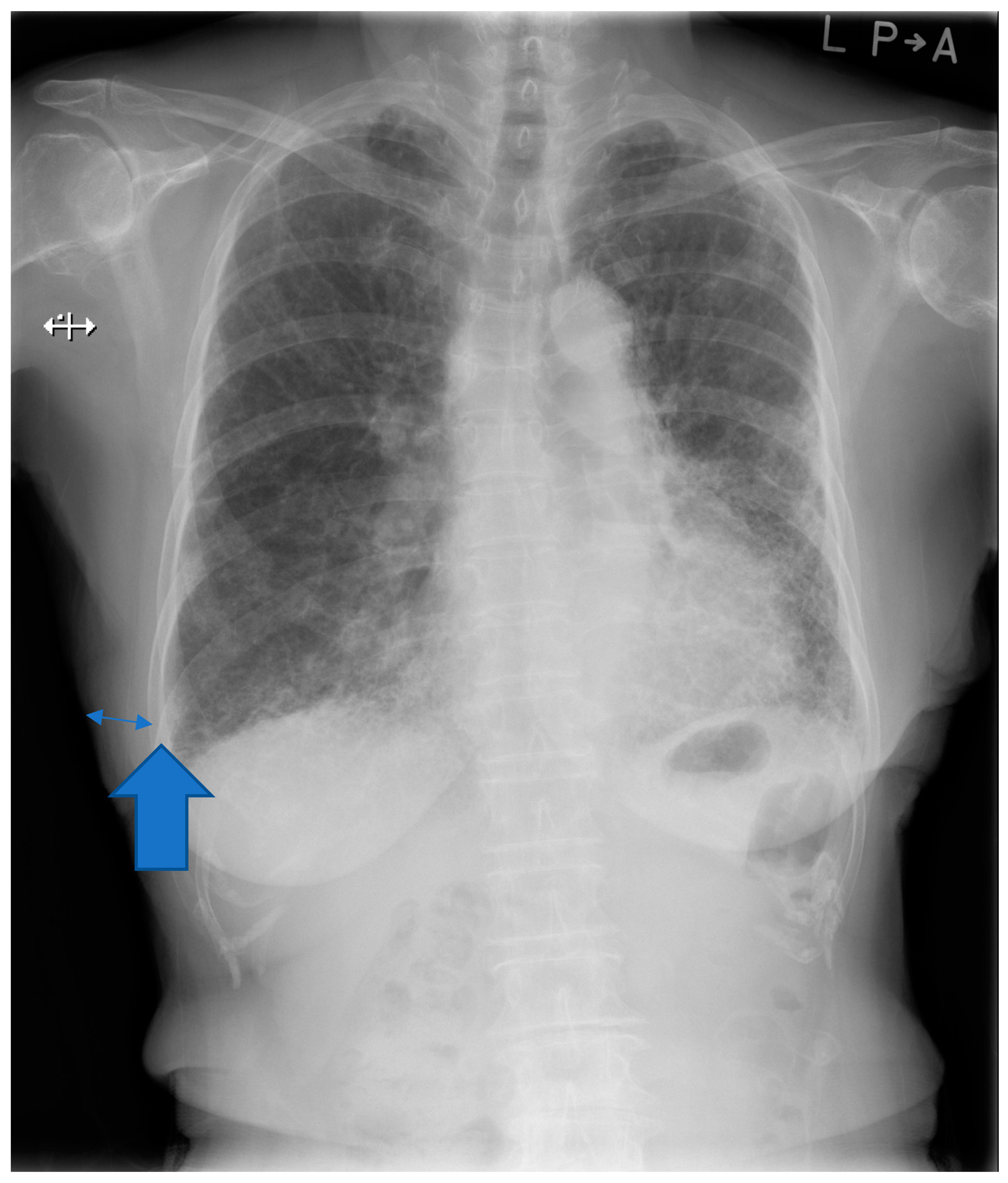
2.3. Chest Imaging Information
2.4. Survival Period
2.5. Data Analysis
3. Results
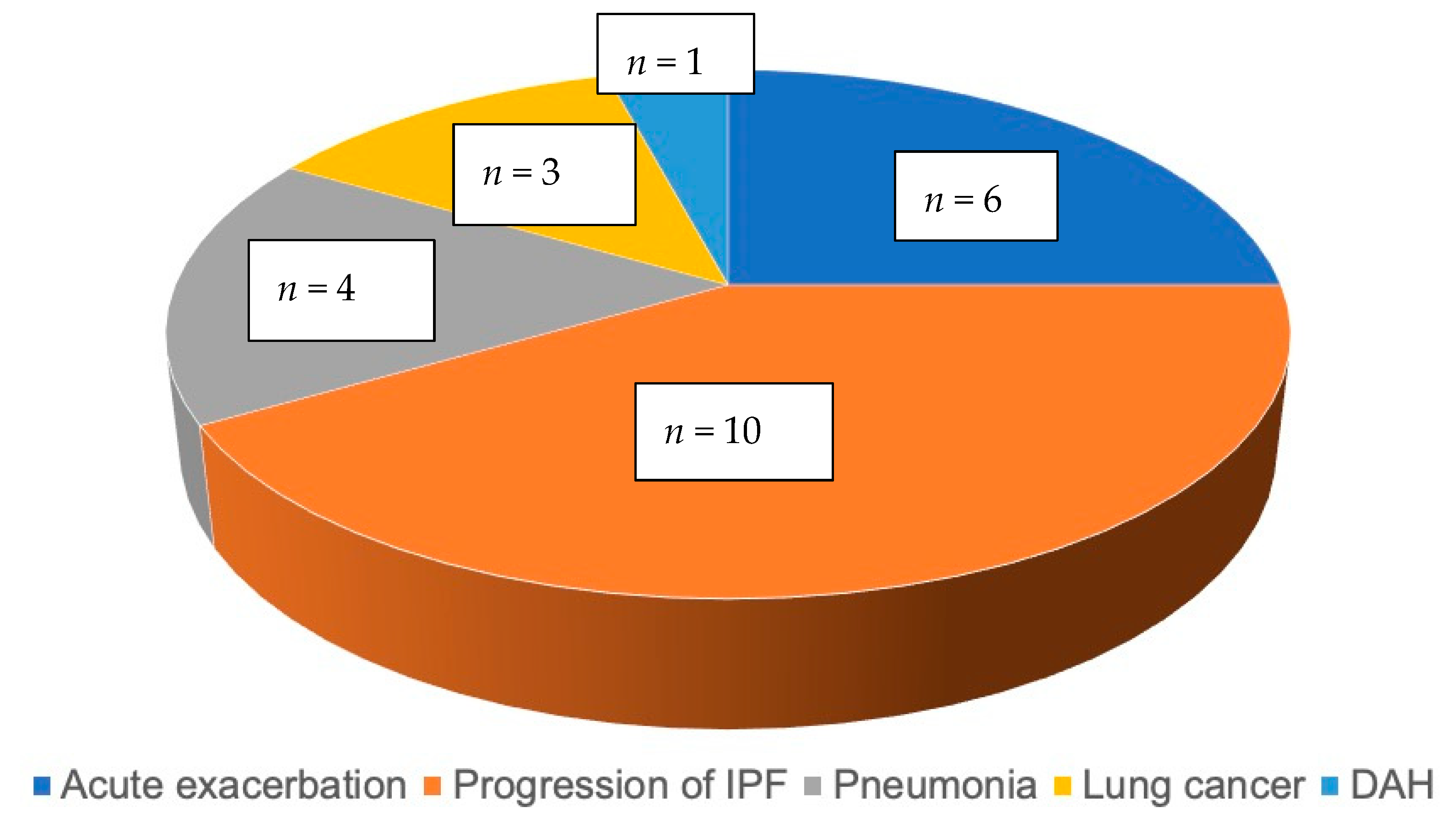
3.1. Baseline Description
3.2. Pulmonary Function Test
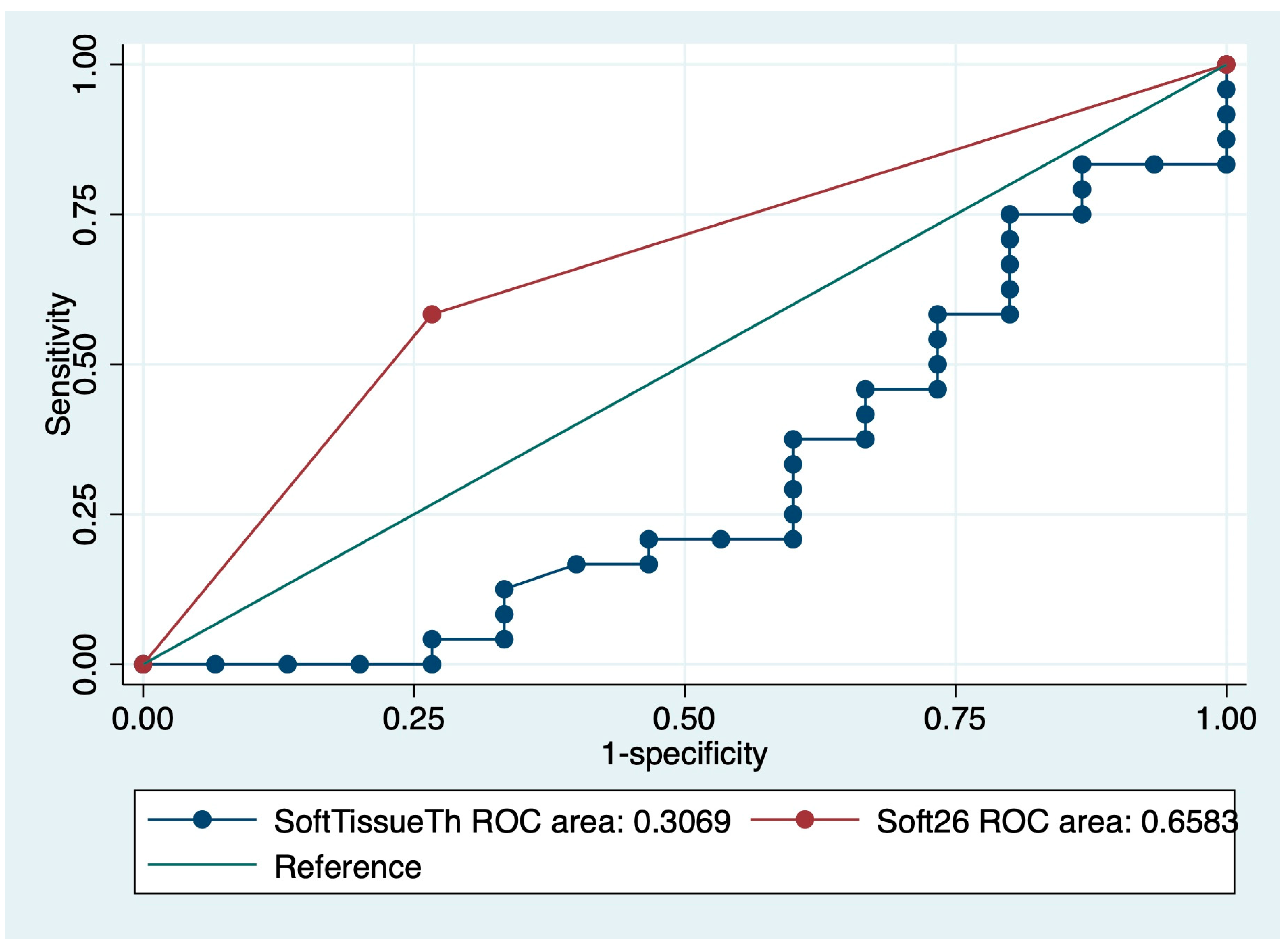
3.3. Hazard Analyses for IPF Mortality
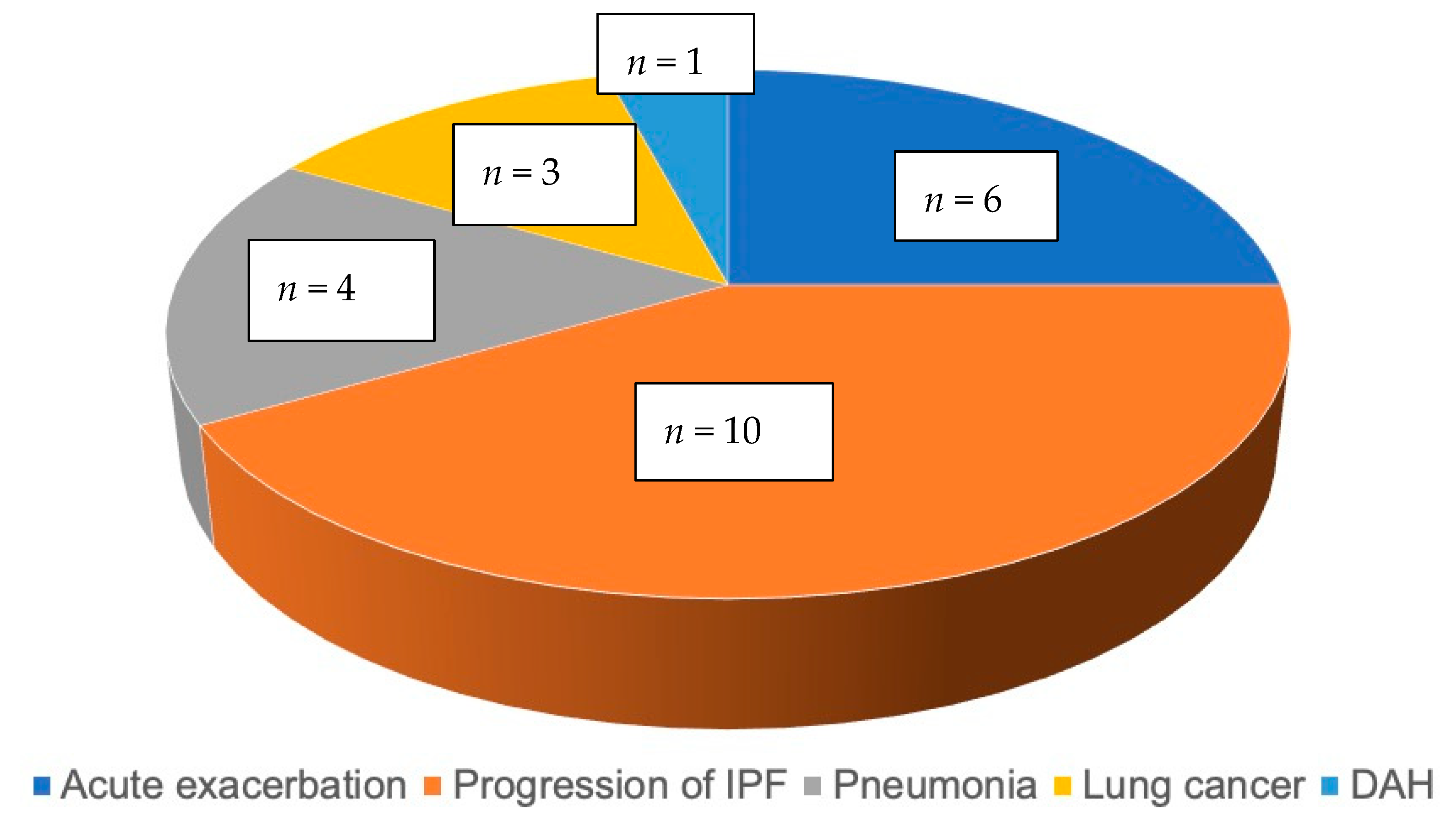
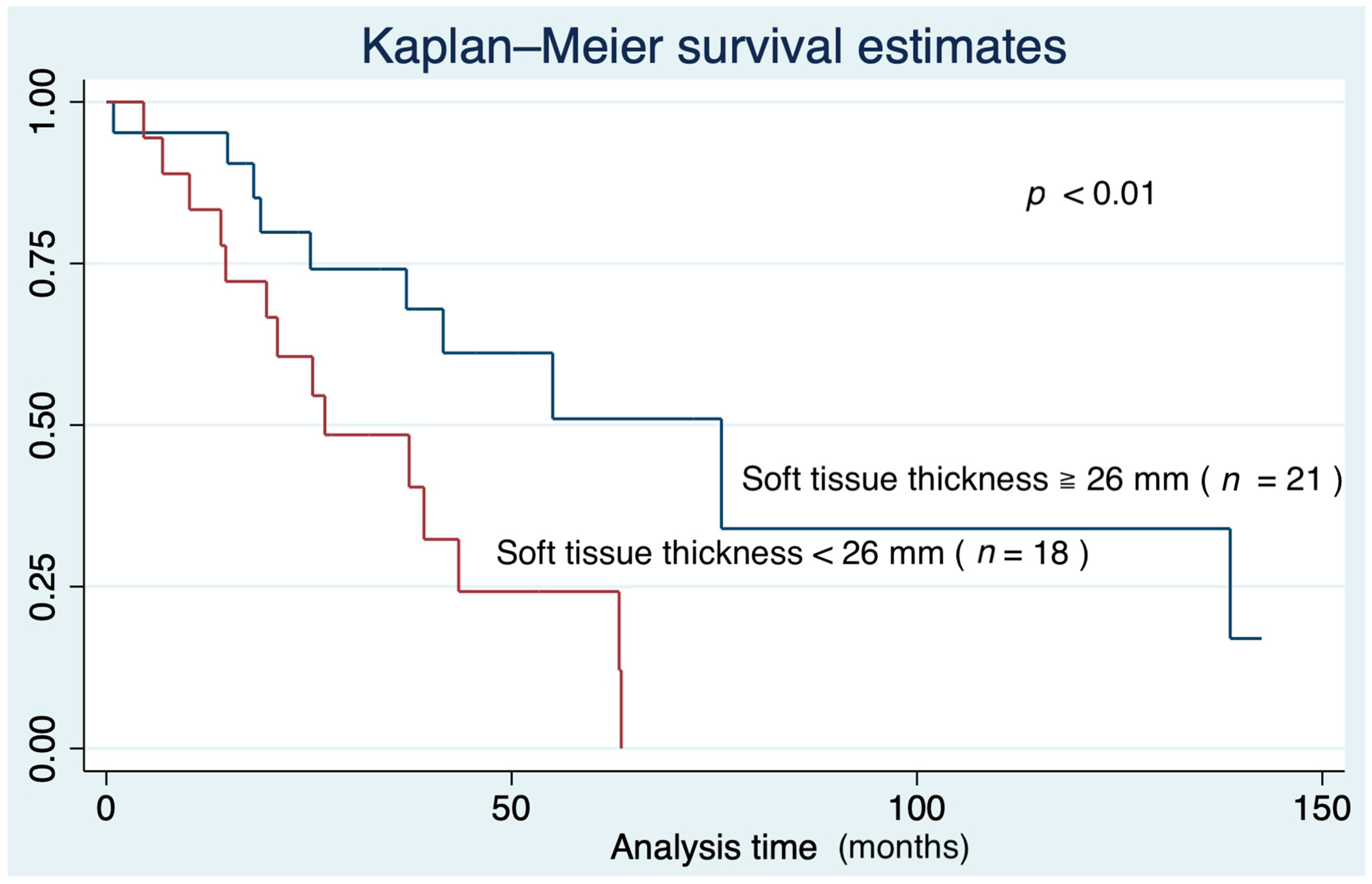
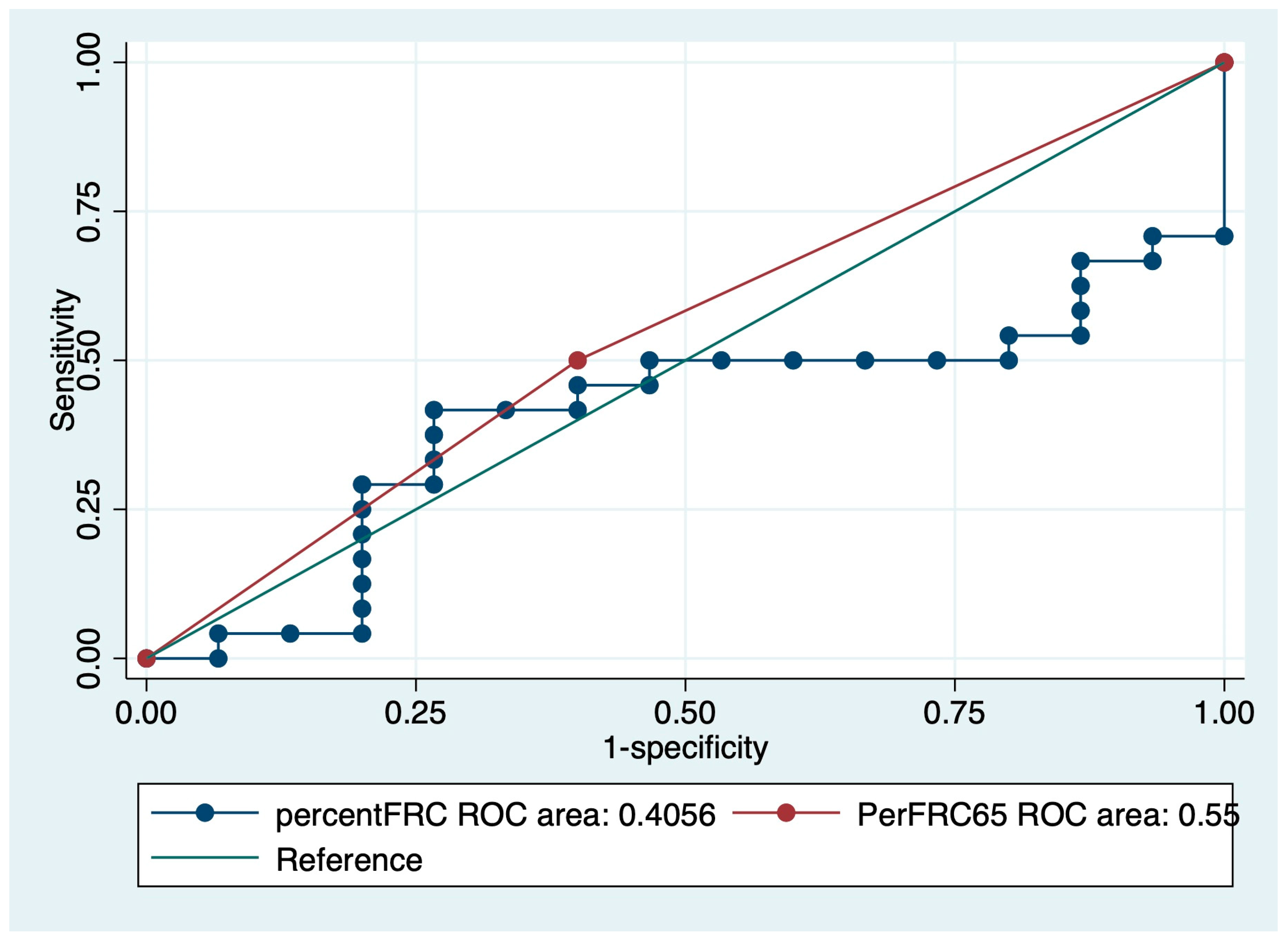
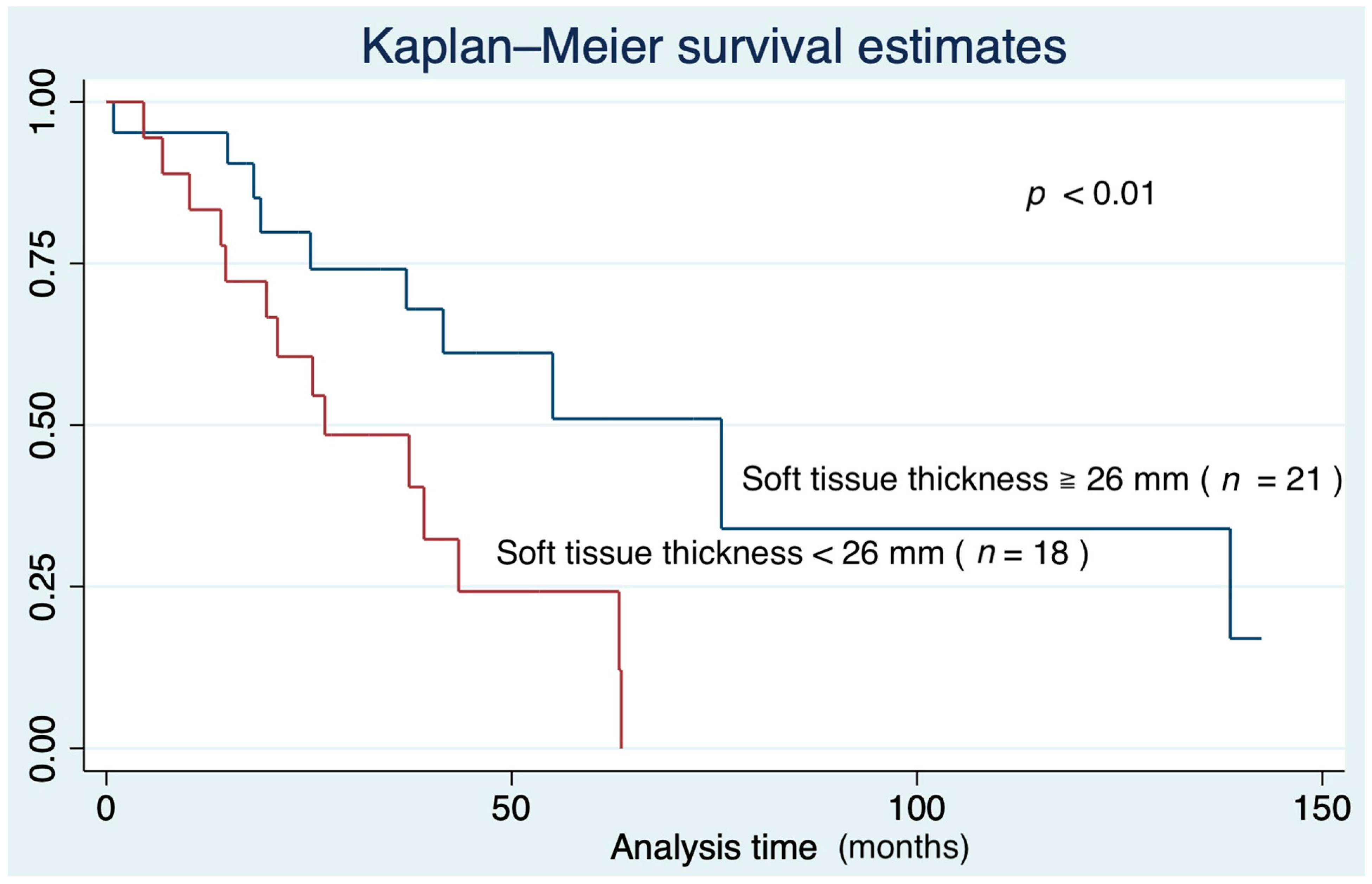
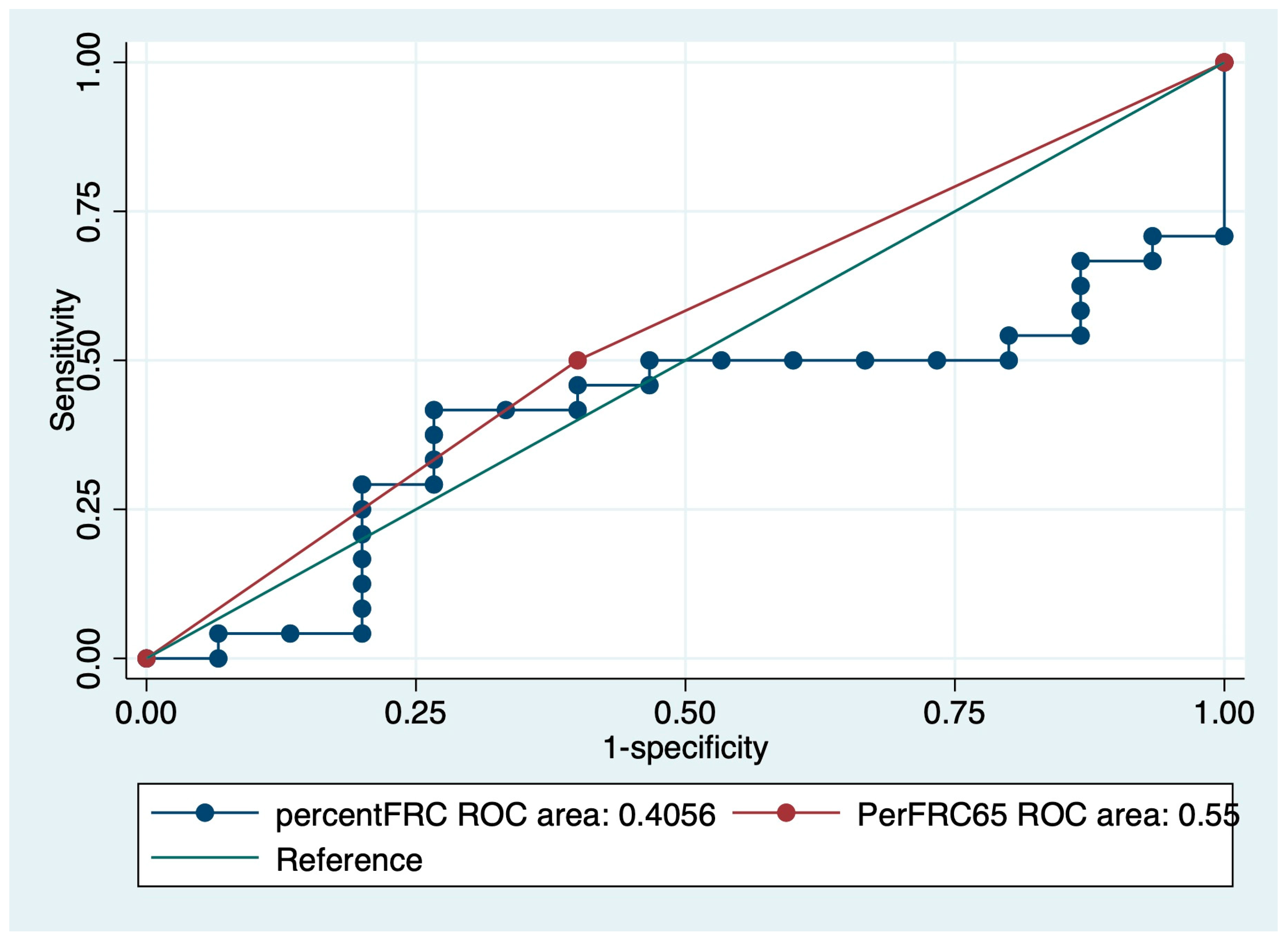
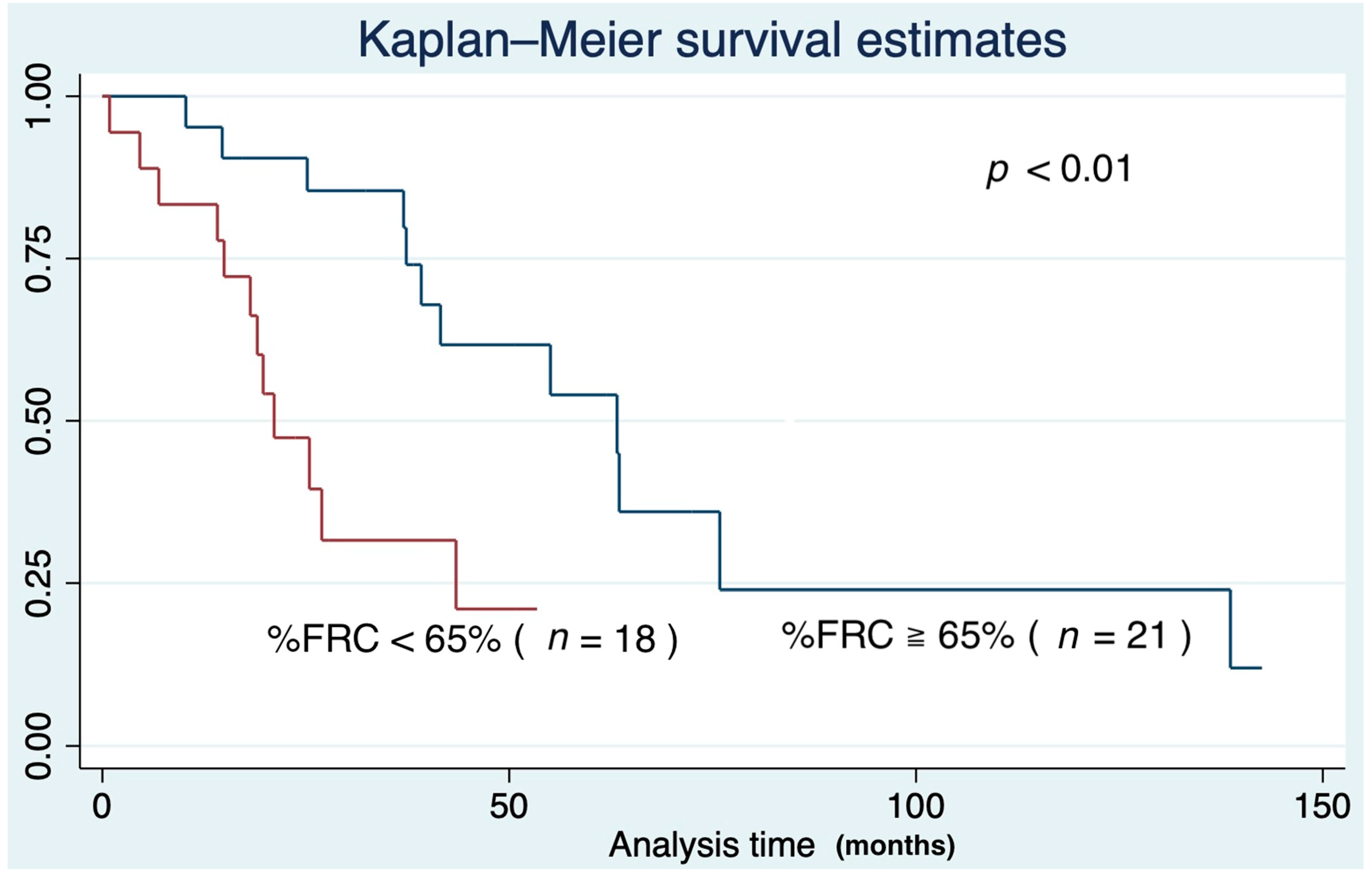
3.4. Survival Curve Based on Predictors of IPF Mortality
4. Discussion
5. Limitations and Strengths
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Thoracic Society. Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. ATS/ERS/JRS/ALAT Committee on idiopathic pulmonary fibrosis: An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [CrossRef]
- Robalo-Cordeiro, C.; Morais, A. Translating idiopathic pulmonary fibrosis guidelines into clinical practice. Pulmonology 2021, 27, 7–13. [Google Scholar] [CrossRef]
- Gross, T.J.; Hunninghake, G.W. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2001, 345, 517. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Azuma, A.; Inase, N.; Taniguchi, H.; Ogura, T.; Inoue, E.; Takeuchi, M.; Yoshizawa, Y.; Sugiyama, Y.; Kudoh, S.; et al. Cyclosporine A combined with low-dose corticosteroid treatment in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2015, 53, 288–295. [Google Scholar] [CrossRef]
- Kondoh, Y.; Taniguchi, H.; Yokoi, T.; Nishiyama, O.; Ohishi, T.; Kato, T.; Suzuki, K.; Suzuki, R. Cyclophosphamide and low-dose prednisolone in idiopathic pulmonary fibrosis and fibrosing nonspecific interstitial pneumonia. Eur. Respir. J. 2005, 25, 528–533. [Google Scholar] [CrossRef]
- Raghu, G.; Richeldi, L. Current approaches to the management of idiopathic pulmonary fibrosis. Respir. Med. 2017, 129, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Holtze, C.H.; Freiheit, E.A.; Limb, S.L.; Stauffer, J.L.; Raimundo, K.; Pan, W.T.; Flaherty, K.R.; Kim, H.J. Patient and site characteristics associated with pirfenidone and nintedanib use in the United States; an analysis of idiopathic pulmonary fibrosis patients enrolled in the Pulmonary Fibrosis Foundation Patient Registry. Respir. Res. 2020, 21, 48. [Google Scholar] [CrossRef]
- Suzuki, Y.; Mori, K.; Aono, Y.; Kono, M.; Hasegawa, H.; Yokomura, K.; Naoi, H.; Hozumi, H.; Karayama, M.; Furuhashi, K.; et al. Switching antifibrotics in patients with idiopathic pulmonary fibrosis: A multi-center retrospective cohort study. BMC Pulm. Med. 2021, 21, 221. [Google Scholar] [CrossRef] [PubMed]
- Glaspole, I.; Bonella, F.; Bargagli, E.; Glassberg, M.K.; Caro, F.; Stansen, W.; Quaresma, M.; Orsatti, L.; Bendstrup, E. Efficacy and safety of nintedanib in patients with idiopathic pulmonary fibrosis who are elderly or have comorbidities. Respir Res. 2021, 22, 125. [Google Scholar] [CrossRef] [PubMed]
- Mononen, M.E.; Kettunen, H.P.; Suoranta, S.K.; Kärkkäinen, M.S.; Selander, T.A.; Purokivi, M.K.; Kaarteenaho, R.L. Several specific high-resolution computed tomography patterns correlate with survival in patients with idiopathic pulmonary fibrosis. J. Thorac Dis. 2021, 13, 2319–2330. [Google Scholar] [CrossRef]
- Jacob, J.; Aksman, L.; Mogulkoc, N.; Procter, A.J.; Gholipour, B.; Cross, G.; Barnett, J.; Brereton, C.J.; Jones, M.G.; van Moorsel, C.H.; et al. Serial CT analysis in idiopathic pulmonary fibrosis: Comparison of visual features that determine patient outcome. Thorax 2020, 75, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Poletti, V.; Vancheri, C.; Albera, C.; Harari, S.; Pesci, A.; Metella, R.R.; Campolo, B.; Crespi, G.; Rizzoli, S.; FIBRONET Study Group. Clinical course of IPF in Italian patients during 12 months of observation: Results from the FIBRONET observational study. Respir. Res. 2021, 22, 66. [Google Scholar] [CrossRef]
- Perez, T.; Burgel, P.R.; Paillasseur, J.L.; Caillaud, D.; Deslée, G.; Chanez, P.; Roche, N.; INITIATIVES BPCO Scientific Committee. Modified Medical Research Council scale vs Baseline Dyspnea Index to evaluate dyspnea in chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis. 2015, 1, 1663–1672. [Google Scholar]
- Wells, A.U.; Desai, S.R.; Rubens, M.B.; Goh, N.S.; Cramer, D.; Nicholson, A.G.; Colby, T.V.; du Bois, R.M.; Hansell, D.M. Idiopathic pulmonary fibrosis: A composite physiologic index derived from disease extent observed by computed tomography. Am. J. Respir. Crit. Care Med. 2003, 167, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Ley, B.; Ryerson, C.J.; Vittinghoff, E.; Ryu, J.H.; Tomassetti, S.; Lee, J.S.; Poletti, V.; Buccioli, M.; Elicker, B.M.; Jones, K.D.; et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann. Intern. Med. 2012, 156, 684–691. [Google Scholar] [CrossRef]
- Kishaba, T.; Nagano, H.; Nei, Y.; Yamashiro, S. Body mass index-percent forced vital capacity-respiratory hospitalization: New staging for idiopathic pulmonary fibrosis patients. J. Thorac. Dis. 2016, 8, 3596–3604. [Google Scholar] [CrossRef] [Green Version]
- Nakatsuka, Y.; Handa, T.; Kokosi, M.; Tanizawa, K.; Puglisi, S.; Jacob, J.; Sokai, A.; Ikezoe, K.; Kanatani, K.T.; Kubo, T.; et al. The Clinical Significance of Body Weight Loss in Idiopathic Pulmonary Fibrosis Patients. Respiration 2018, 96, 338–347. [Google Scholar] [CrossRef]
- Gluck, M.C.; Twigg, H.L.; Ball, M.F.; Rhodes, P.G. Shadows bordering the lung on radiographs of normal and obese persons. Thorax 1972, 27, 232–238. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute Exacerbation of Idiopathic Pulmonary Fibrosis. An International Working Group Report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef]
- Nakagawa, H.; Otoshi, R.; Isomoto, K.; Katano, T.; Baba, T.; Komatsu, S.; Hagiwara, E.; Nakano, Y.; Kuwahira, I.; Ogura, T. Relationship of flow-volume curve pattern on pulmonary function test with clinical and radiological features in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2020, 20, 214. [Google Scholar] [CrossRef]
- Taha, N.; D’Amato, D.; Hosein, K.; Ranalli, T.; Sergiacomi, G.; Zompatori, M.; Mura, M. Longitudinal functional changes with clinically significant radiographic progression in idiopathic pulmonary fibrosis: Are we following the right parameters? Respir. Res. 2020, 21, 119. [Google Scholar] [CrossRef] [PubMed]
- Kishaba, T. Practical management of Idiopathic Pulmonary Fibrosis. Sarcoidosis Vasc. Diffus. Lung Dis. 2015, 32, 90–98. [Google Scholar]
- Kishaba, T. Evaluation and management of Idiopathic Pulmonary Fibrosis. Respir. Investig. 2019, 57, 300–311. [Google Scholar] [CrossRef]
- Munchel, J.K.; Shea, B.S. Diagnosis and Management of Idiopathic Pulmonary Fibrosis. Rhode Isl. Med. J. (2013) 2021, 104, 26–29. [Google Scholar]
- Kaya, F.; Özgül, E.; Balcı, A. Quantitative and visual analysis of idiopathic pulmonary fibrosis with different methods: The relationship between clinical correlation and mortality risk model. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 3254–3263. [Google Scholar] [PubMed]
- Kwon, B.S.; Choe, J.; Do, K.H.; Hwang, H.S.; Chae, E.J.; Song, J.W. Computed tomography patterns predict clinical course of idiopathic pulmonary fibrosis. Respir. Res. 2020, 21, 295. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Liu, X.; He, L.; Zhou, P.K. Radiation Exposure Associated with Computed Tomography in Childhood and the Subsequent Risk of Cancer: A Meta-Analysis of Cohort Studies. Dose Response 2020, 18, 1559325820923828. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.C. Nutrition and Muscle in Cirrhosis. J. Clin. Exp. Hepatol. 2017, 7, 340–357. [Google Scholar] [CrossRef]
- Schwebel, C.; Pin, I.; Barnoud, D.; Devouassoux, G.; Brichon, P.; Chaffanjon, P.; Chavanon, O.; Sessa, C.; Blin, D.; Guignier, M.; et al. Prevalence and consequences of nutritional depletion in lung transplant candidates. Eur. Respir. J. 2000, 16, 1050–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jouneau, S.; Crestani, B.; Thibault, R.; Lederlin, M.; Vernhet, L.; Valenzuela, C.; Wijsenbeek, M.; Kreuter, M.; Stansen, W.; Quaresma, M.; et al. Analysis of body mass index, weight loss and progression of idiopathic pulmonary fibrosis. Respir. Res. 2020, 21, 312. [Google Scholar] [CrossRef] [PubMed]
- White, E.S.; Lazar, M.H.; Thannickal, V.J. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J. Pathol. 2003, 201, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Zavaletta, V.A.; Bartholmai, B.; Robb, R.A. High Resolution Multidetector CT-Aided Tissue Analysis and Quantification of Lung Fibrosis. Acad. Radiol. 2007, 14, 772–787. [Google Scholar] [CrossRef]
- Shih, A.R.; Nitiwarangkul, C.; Little, B.P.; Roop, B.W.; Nandy, S.; Szabari, M.V.; Mercaldo, N.; Mercaldo, S.; Montesi, S.B.; Muniappan, A.; et al. Practical application and validation of the 2018 ATS/ERS/JRS/ALAT and Fleischner Society guidelines for the diagnosis of idiopathic pulmonary fibrosis. Respir. Res. 2021, 22, 124. [Google Scholar] [CrossRef]
- Nicholson, A.G. Classification of idiopathic interstitial pneumonias: Making sense of the alphabet soup. Histopathology 2002, 41, 381–391. [Google Scholar] [CrossRef]
- Ricci, F.; Cavallo, A.U.; Luca, P.; Vincenzo, S.; Monia, P.; D’Errico, F.; Benelli, L.; Paola, R.; Floris, R.; Chiocchi, M. Radiological pitfalls associated with the diagnosis of usual interstitial pneumonia pattern on high-resolution computed tomography and associated findings: Experience from a single Italian center. Acta Radiol. 2021, 62, 619–627. [Google Scholar] [CrossRef]
- Touil, I.; Keskes Boudawara, N.; Bouchareb, S.; Ben Saad, A.; Migaou, A.; Cheikh Mhamed, S.; Fahem, N.; Mribah, H.; Knani, J.; Boussoffara, L.; et al. Facteurs pronostiques au cours de la fibrose pulmonaire idiopathique: Étude d’une cohorte tunisienne [Prognostic factors in idiopathic pulmonary fibrosis in a tunisian cohort]. Rev. Mal. Respir. 2021, 38, 681–688. [Google Scholar] [CrossRef]
- Nathan, S.D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Du Bois, R.M.; Fagan, E.A.; Fishman, R.S.; Glaspole, I.; Glassberg, M.K.; Glasscock, K.; et al. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: Analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax 2016, 71, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermudo, G.; Suarez-Cuartin, G.; Rivera-Ortega, P.; Rodriguez-Portal, J.A.; Sauleda, J.; Nuñez, B.; Castillo, D.; Aburto, M.; Portillo, K.; Balcells, E.; et al. Different Faces of Idiopathic Pulmonary Fibrosis With Preserved Forced Vital Capacity. Arch. Bronconeumol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kishaba, T.; Hozumi, H.; Fujisawa, T.; Nei, Y.; Enomoto, N.; Sugiura, H.; Kitani, M.; Suda, T. Predictors of acute exacerbation in biopsy-proven idiopathic pulmonary fibrosis. Respir. Investig. 2020, 58, 177–184. [Google Scholar] [CrossRef]
- Du Bois, R.M.; Weycker, D.; Albera, C.; Bradford, W.Z.; Costabel, U.; Kartashov, A.; King, T.E., Jr.; Lancaster, L.; Noble, P.W.; Sahn, S.A.; et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: Test properties and minimal clinically important difference. Am. J. Respir. Crit. Care Med. 2011, 184, 1382–1389. [Google Scholar] [CrossRef]
- Reichmann, W.M.; Yu, Y.F.; Macaulay, D.; Wu, E.Q.; Nathan, S.D. Change in forced vital capacity and associated subsequent outcomes in patients with newly diagnosed idiopathic pulmonary fibrosis. BMC Pulm. Med. 2015, 15, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lofrese, J.J.; Tupper, C.; Lappin, S.L. Physiology, Residual Volume; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Rorvik, S.; Bo, G. Lung volumes and arterial blood gases in obesity. Scand. J. Respir. Dis. Suppl. 1976, 95, 60–64. [Google Scholar] [PubMed]
- Sesé, L.; Nunes, H.; Cottin, V.; Israel-Biet, D.; Crestani, B.; Guillot-Dudoret, S.; Cadranel, J.; Wallaert, B.; Tazi, A.; Maître, B.; et al. Gender Differences in Idiopathic Pulmonary Fibrosis: Are Men and Women Equal? Front. Med. 2021, 8, 713698. [Google Scholar] [CrossRef] [PubMed]
- Nishikiori, H.; Chiba, H.; Lee, S.H.; Kondoh, S.; Kamo, K.I.; Nakamura, K.; Ikeda, K.; Kuronuma, K.; Chung, M.P.; Kondoh, Y.; et al. A modified GAP model for East-Asian populations with idiopathic pulmonary fibrosis. Respir. Investig. 2020, 58, 395–402. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | 72.9 ± 7.0 (53–85) |
| Gender (Men/Women) | (27/12) |
| Smoker (active/never) | (24/15) |
| Pack-year | 20 (0–100) |
| BMI (kg/mm2) | 25.1 ± 3.9 (17.3–35) |
| mMRC | 1.3 ± 0.8(0–3) |
| Cough (Yes/No) | (30/9) |
| Disease duration (months) | 36.4 ± 30.7(2–120) |
| WBC (μL) | 7816 ± 1859 (4700–11700) |
| LDH (U/L) | 248 ± 47 (173–398) |
| KL-6 (U/mL) | 1615 ± 1503 (332–6682) |
| Soft tissue thickness (mm) | 26.4 ± 8.8 (1.7–52.2) |
| HRCT patten (definite/probable /indeterminate) | (15/16/8) |
| Observation period (months) | 38.6 ± 30.6 (0.9–142.5) |
| Survival time (months) | 32.4 (0.9–142.5) |
| FVC (L) | 1.92 ± 0.56 (0.85–3.26) |
| Precent predicted FVC (%) | 66.8 ± 14.9 (31.5–94.8) |
| TLC (L) | 3.30 ± 0.74 (2.06–5.01) |
| Precent predicted TLC (%) | 71.8 ± 13.7 (45.1–115.7) |
| FRC (L) | 1.56 ± 0.85 (0.71–2.92) |
| Precent predicted FRC (%) | 65.0 ± 39.6 (25.4–143.1) |
| Precent predicted DLco (%) | 64.6 ± 27.9 (28.1–136.1) |
| Composite physiological index | 59.4 ± 26.7 (0.3–104) |
| GAP score | 4.9 ± 1.8 (1–8) |
| GAP stage (I/II/III) | (9/15/15) |
| Hazard Ratio | 95%CI | p-Value | |
|---|---|---|---|
| Univariate | |||
| Age at PFT, years | 1.07 | 1.01–1.12 | 0.02 |
| BMI (kg/mm2) | 0.89 | 0.79–1.00 | 0.06 |
| LDH (U/L) | 1.00 | 0.99–1.01 | 0.78 |
| KL-6 (U/mL) | 0.99 | 0.99–1.00 | 0.37 |
| Soft tissue thickness (mm) | 0.90 | 0.85–0.96 | <0.01 |
| FVC | 0.74 | 0.37–1.46 | 0.38 |
| %FVC | 0.99 | 0.97–1.01 | 0.44 |
| TLC | 0.66 | 0.24–0.33 | 0.24 |
| %TLC | 0.99 | 0.96–1.02 | 0.44 |
| FRC | 0.38 | 0.22–0.67 | <0.01 |
| %FRC | 0.98 | 0.97–0.99 | <0.01 |
| %DLco | 0.99 | 0.96–1.01 | 0.34 |
| Pirfenidone | 0.88 | 0.76–1.03 | 0.12 |
| Nintedanib | 0.75 | 0.32–1.77 | 0.51 |
| Multivariate | |||
| BMI (kg/mm2) | 0.91 | 0.80–1.03 | 0.13 |
| Soft tissue thickness (mm) | 0.92 | 0.86–0.98 | <0.01 |
| FRC | 0.41 | 0.23–0.72 | <0.01 |
| %FRC | 0.98 | 0.97–0.99 | <0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kishaba, T.; Maeda, A.; Yamazato, S.; Nabeya, D.; Yamashiro, S.; Nagano, H. Radiological and Physiological Predictors of IPF Mortality. Medicina 2021, 57, 1121. https://doi.org/10.3390/medicina57101121
Kishaba T, Maeda A, Yamazato S, Nabeya D, Yamashiro S, Nagano H. Radiological and Physiological Predictors of IPF Mortality. Medicina. 2021; 57(10):1121. https://doi.org/10.3390/medicina57101121
Chicago/Turabian StyleKishaba, Tomoo, Akiko Maeda, Shoshin Yamazato, Daijiro Nabeya, Shin Yamashiro, and Hiroaki Nagano. 2021. "Radiological and Physiological Predictors of IPF Mortality" Medicina 57, no. 10: 1121. https://doi.org/10.3390/medicina57101121
APA StyleKishaba, T., Maeda, A., Yamazato, S., Nabeya, D., Yamashiro, S., & Nagano, H. (2021). Radiological and Physiological Predictors of IPF Mortality. Medicina, 57(10), 1121. https://doi.org/10.3390/medicina57101121