De novo NIPBL Mutations in Vietnamese Patients with Cornelia de Lange Syndrome


Abstract
1. Introduction
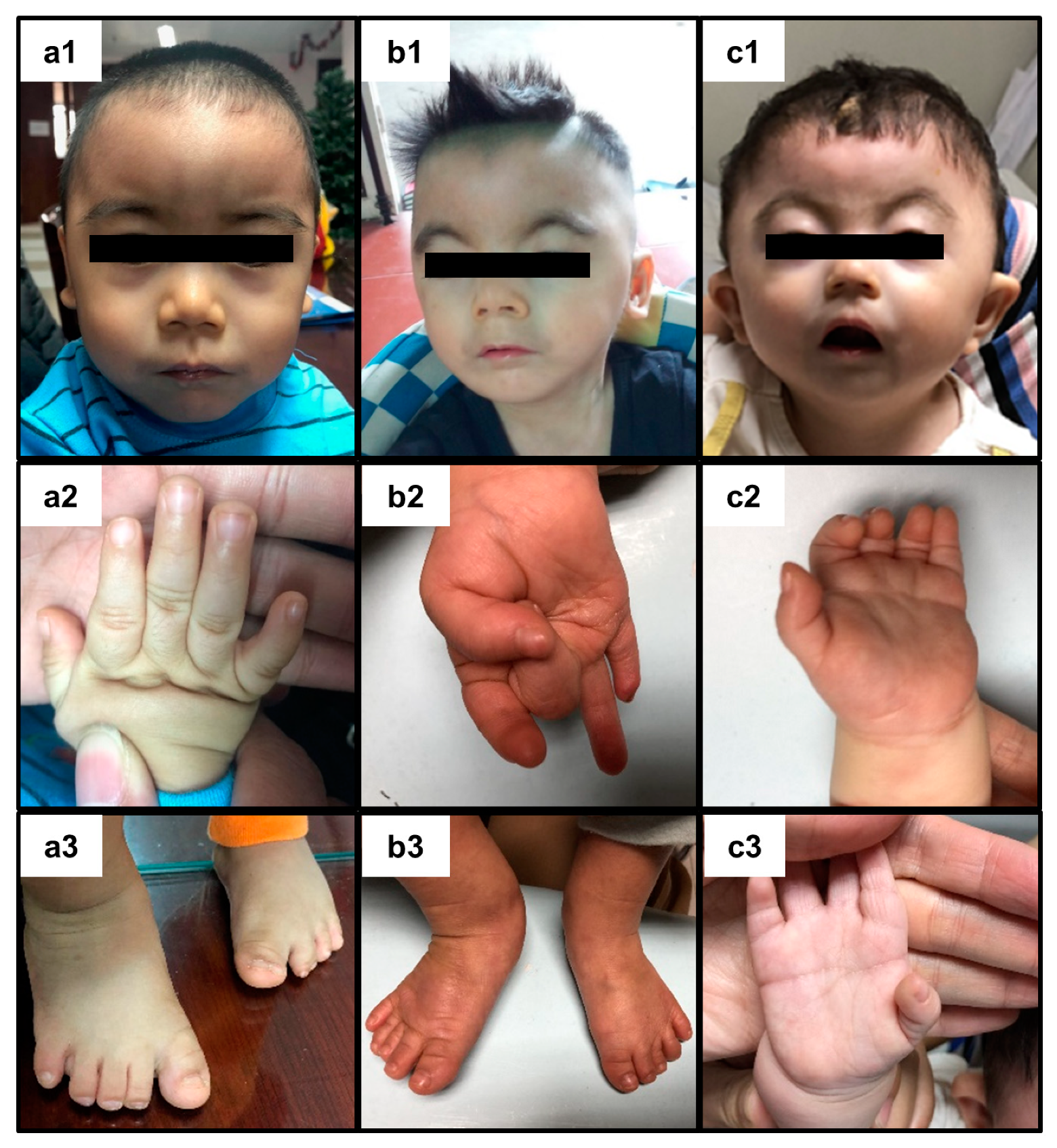
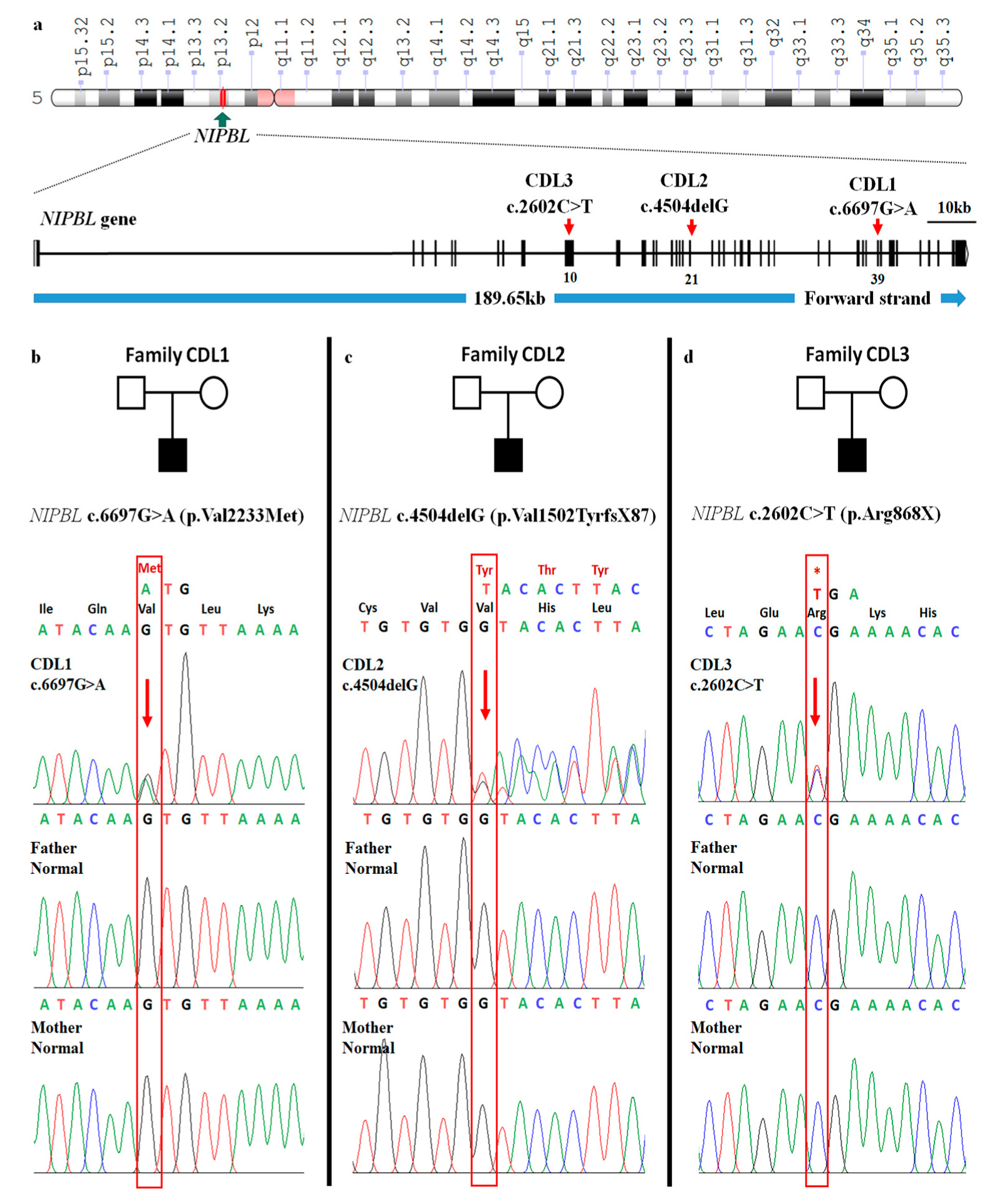
2. Presentation of Case Reports
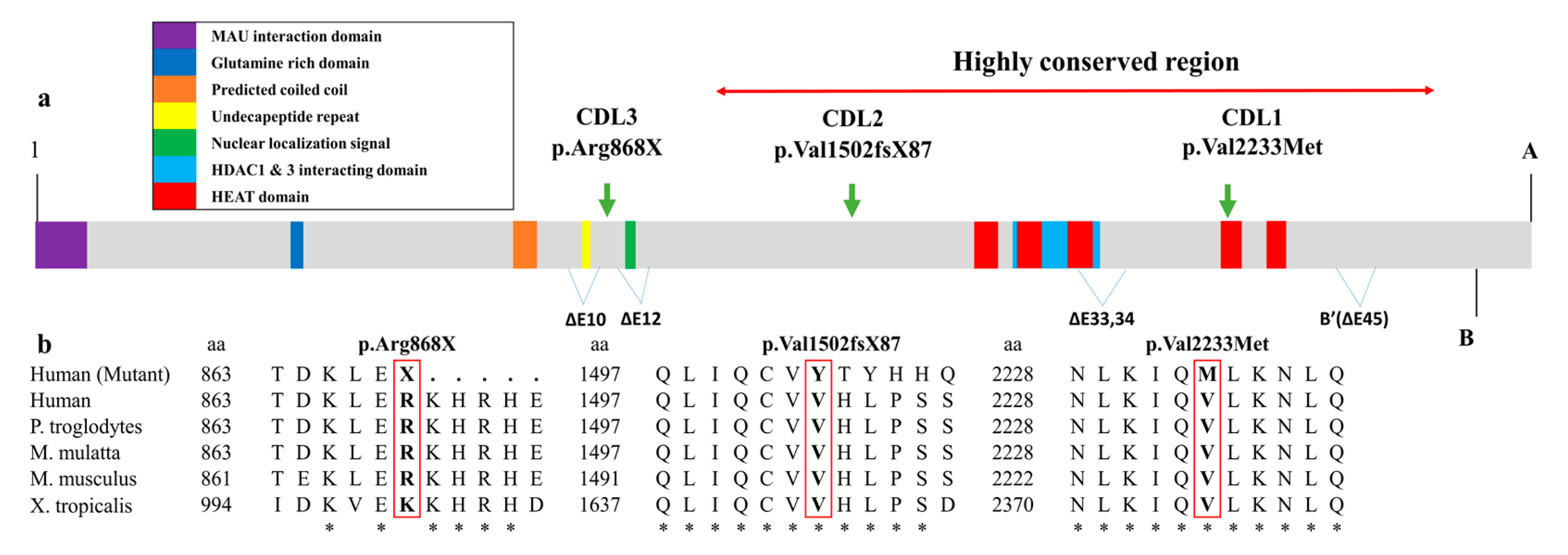
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Krantz, I.D.; McCallum, J.; DeScipio, C.; Kaur, M.; Gillis, L.A.; Yaeger, D.; Jukofsky, L.; Wasserman, N.; Bottani, A.; Morris, C.A.; et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004, 36, 631–635. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.; Kline, A.D.; Barr, M.A.; Koch, S. de Lange syndrome: A clinical review of 310 individuals. Am. J. Med. Genet. 1993, 47, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Kline, A.D.; Moss, J.F.; Selicorni, A.; Bisgaard, A.-M.; Deardorff, M.A.; Gillett, P.M.; Ishman, S.L.; Kerr, L.M.; Levin, A.V.; Mulder, P.A.; et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat. Rev. Genet. 2018, 19, 649–666. [Google Scholar] [CrossRef]
- Dowsett, L.; Porras, A.R.; Kruszka, P.; Davis, B.; Hu, T.; Honey, E.; Badoe, E.; Thong, M.-K.; Leon, E.; Girisha, K.M.; et al. Cornelia de Lange syndrome in diverse populations. Am. J. Med. Genet. A 2019, 179, 150–158. [Google Scholar] [CrossRef]
- Ireland, M.; Donnai, D.; Burn, J. Brachmann-de Lange syndrome. Delineation of the clinical phenotype. Am. J. Med. Genet. 1993, 47, 959–964. [Google Scholar] [CrossRef]
- Allanson, J.E.; Hennekam, R.C.; Ireland, M. De Lange syndrome: subjective and objective comparison of the classical and mild phenotypes. J. Med. Genet. 1997, 34, 645–650. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Kaur, M.; Yaeger, D.; Rampuria, A.; Korolev, S.; Pie, J.; Gil-Rodríguez, C.; Arnedo, M.; Loeys, B.; Kline, A.D.; et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007, 80, 485–494. [Google Scholar] [CrossRef]
- Kline, A.D.; Grados, M.; Sponseller, P.; Levy, H.P.; Blagowidow, N.; Schoedel, C.; Rampolla, J.; Clemens, D.K.; Krantz, I.; Kimball, A.; et al. Natural history of aging in Cornelia de Lange syndrome. Am. J. Med. Genet. C 2007, 145C, 248–260. [Google Scholar] [CrossRef]
- Tonkin, E.T.; Wang, T.-J.; Lisgo, S.; Bamshad, M.J.; Strachan, T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 2004, 36, 636–641. [Google Scholar] [CrossRef]
- Musio, A.; Selicorni, A.; Focarelli, M.L.; Gervasini, C.; Milani, D.; Russo, S.; Vezzoni, P.; Larizza, L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 2006, 38, 528–530. [Google Scholar] [CrossRef]
- Peters, J.-M.; Tedeschi, A.; Schmitz, J. The cohesin complex and its roles in chromosome biology. Genes Dev. 2008, 22, 3089–3114. [Google Scholar] [CrossRef] [PubMed]
- Dorsett, D.; Krantz, I.D. On the Molecular Etiology of Cornelia de Lange Syndrome. Ann. N. Y. Acad. Sci. 2009, 1151, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.J.; Puisac, B.; Baquero-Montoya, C.; Gil-Rodríguez, M.C.; Bueno, I.; Deardorff, M.A.; Hennekam, R.C.; Kaiser, F.J.; Krantz, I.D.; Musio, A.; et al. Clinical utility gene card for: Cornelia de Lange syndrome. Eur. J. Hum. Genet. 2015, 23, 1431. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange Syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef]
- Kline, A.D.; Krantz, I.D.; Sommer, A.; Kliewer, M.; Jackson, L.G.; FitzPatrick, D.R.; Levin, A.V.; Selicorni, A. Cornelia de Lange syndrome: Clinical review, diagnostic and scoring systems, and anticipatory guidance. Am. J. Med. Genet. A 2007, 143A, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Okamoto, N.; Stark, Z.; Nabetani, M.; Tsurusaki, Y.; Nakashima, M.; Miyake, N.; Mizuguchi, T.; Ohtake, A.; Saitsu, H.; et al. ANKRD11 variants cause variable clinical features associated with KBG syndrome and Coffin-Siris-like syndrome. J. Hum. Genet. 2017, 62, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Olley, G.; Ansari, M.; Bengani, H.; Grimes, G.R.; Rhodes, J.; von Kriegsheim, A.; Blatnik, A.; Stewart, F.J.; Wakeling, E.; Carroll, N.; et al. BRD4 interacts with NIPBL and BRD4 is mutated in a Cornelia de Lange–like syndrome. Nat. Genet. 2018, 50, 329–332. [Google Scholar] [CrossRef]
- Oliveira, J.; Dias, C.; Redeker, E.; Costa, E.; Silva, J.; Lima, M.R.; den Dunnen, J.T.; Santos, R. Development of NIPBL Locus-Specific Database Using LOVD: From Novel Mutations to Further Genotype–Phenotype Correlations in Cornelia de Lange Syndrome. Hum. Mutat. 2010, 31, 1216–1222. [Google Scholar] [CrossRef]
- Bettini, L.R.; Locatelli, L.; Mariani, M.; Cianci, P.; Giussani, C.; Canonico, F.; Cereda, A.; Russo, S.; Gervasini, C.; Biondi, A.; et al. Cervical spine malformation in cornelia de lange syndrome: a report of three patients. Am. J. Med. Genet. A 2014, 164A, 1520–1524. [Google Scholar] [CrossRef]
- Teresa-Rodrigo, M.E.; Eckhold, J.; Puisac, B.; Dalski, A.; Gil-Rodríguez, M.C.; Braunholz, D.; Baquero, C.; Hernández-Marcos, M.; De Karam, J.C.; Ciero, M.; et al. Functional Characterization of NIPBL Physiological Splice Variants and Eight Splicing Mutations in Patients with Cornelia de Lange Syndrome. Int. J. Mol. Sci. 2014, 15, 10350–10364. [Google Scholar] [CrossRef]
- Deardorff, M.A.; Noon, S.E.; Krantz, I.D. Cornelia de Lange Syndrome; University of Washington: Seattle, DC, USA, 2016. [Google Scholar]
- Strachan, T. Cornelia de Lange Syndrome and the link between chromosomal function, DNA repair and developmental gene regulation. Curr. Opin. Genet. Dev. 2005, 15, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Chao, W.C.H.; Murayama, Y.; Muñoz, S.; Jones, A.W.; Wade, B.O.; Purkiss, A.G.; Hu, X.-W.; Borg, A.; Snijders, A.P.; Uhlmann, F.; et al. Structure of the cohesin loader Scc2. Nat. Commun. 2017, 8, 13952. [Google Scholar] [CrossRef] [PubMed]
- Schoumans, J.; Wincent, J.; Barbaro, M.; Djureinovic, T.; Maguire, P.; Forsberg, L.; Staaf, J.; Thuresson, A.C.; Borg, A.; Nordgren, A.; et al. Comprehensive mutational analysis of a cohort of Swedish Cornelia de Lange syndrome patients. Eur. J. Hum. Genet. 2007, 15, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Huisman, S.A.; Redeker, E.J.W.; Maas, S.M.; Mannens, M.M.; Hennekam, R.C.M. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J. Med. Genet. 2013, 50, 339–344. [Google Scholar] [CrossRef]
- Gillis, L.A.; McCallum, J.; Kaur, M.; DeScipio, C.; Yaeger, D.; Mariani, A.; Kline, A.D.; Li, H.; Devoto, M.; Jackson, L.G.; et al. NIPBL Mutational Analysis in 120 Individuals with Cornelia de Lange Syndrome and Evaluation of Genotype-Phenotype Correlations. Am. J. Med. Genet. 2004, 75, 610–623. [Google Scholar] [CrossRef]
- Mannini, L.; Cucco, F.; Quarantotti, V.; Krantz, I.D.; Musio, A. Mutation Spectrum and Genotype–Phenotype Correlation in Cornelia de Lange Syndrome. Hum. Mutat. 2013, 34, 1589–1596. [Google Scholar] [CrossRef]
- Jahnke, P.; Xu, W.; Wülling, M.; Albrecht, M.; Gabriel, H.; Gillessen-Kaesbach, G.; Kaiser, F.J. The Cohesin loading factor NIPBL recruits histone deacetylases to mediate local chromatin modifications. Nucleic Acids Res. 2008, 36, 6450–6458. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| CDL1 | CDL2 | CDL3 | |
|---|---|---|---|
| Clinical Score Based on Clinical Presentation | |||
| Cardinal features (2 points each if present) | |||
| Synophrys | 2 | 2 | 2 |
| Short nose, concave nasal ridge and/or upturned nasal tip | 0 | 2 | 2 |
| Long and/or smooth philtrum | 2 | 2 | 2 |
| Thin upper lip vermilion and/or downturned corners of the mouth | 2 | 2 | 2 |
| Hand oligodactyly and/or adactyly | 0 | 0 | 0 |
| Congenital diaphragmatic hernia | 0 | 0 | 0 |
| Suggestive features (1 point each if present) | |||
| Global developmental delay and/or intellectual disability | 1 | 1 | 1 |
| Prenatal growth retardation (<2 SD) | 0 | 0 | 0 |
| Postnatal growth retardation (<2 SD) | 1 | 1 | 1 |
| Microcephaly (prenatally and/or postnatally) | 1 | 0 | 1 |
| Small hands and/or feet | 1 | 1 | 1 |
| Short fifth finger | 1 | 1 | 1 |
| Hirsutism | 1 | 0 | 0 |
| Total clinical score | 12 | 12 | 13 |
| Molecular genetic analyses | |||
| Gene | NIPBL | NIPBL | NIPBL |
| Exon | 39/47 | 21/47 | 10/47 |
| c.DNA mutation | c.6697G>A | c.4504delG | c.2602C>T |
| Protein change | p.Val2233Met | p.Val1502TyrfsX87 | p.Arg868X |
| SIFT | 0.002 | _ | 0 |
| PolyPhen-2 | 0.999 | _ | 0.988 |
| Zygosity | HET | HET | HET |
| Effect | missense | frameshift | stop-gained |
| ClinVar database | VCV000644755.1 | _ | VCV000096337.2 |
| dbSNP database | _ | Novel | rs398124466 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thanh, D.C.; Ngoc, C.T.B.; Nguyen, N.-L.; Vu, C.D.; Tung, N.V.; Nguyen, H.H. De novo NIPBL Mutations in Vietnamese Patients with Cornelia de Lange Syndrome. Medicina 2020, 56, 76. https://doi.org/10.3390/medicina56020076
Thanh DC, Ngoc CTB, Nguyen N-L, Vu CD, Tung NV, Nguyen HH. De novo NIPBL Mutations in Vietnamese Patients with Cornelia de Lange Syndrome. Medicina. 2020; 56(2):76. https://doi.org/10.3390/medicina56020076
Chicago/Turabian StyleThanh, Duong Chi, Can Thi Bich Ngoc, Ngoc-Lan Nguyen, Chi Dung Vu, Nguyen Van Tung, and Huy Hoang Nguyen. 2020. "De novo NIPBL Mutations in Vietnamese Patients with Cornelia de Lange Syndrome" Medicina 56, no. 2: 76. https://doi.org/10.3390/medicina56020076
APA StyleThanh, D. C., Ngoc, C. T. B., Nguyen, N.-L., Vu, C. D., Tung, N. V., & Nguyen, H. H. (2020). De novo NIPBL Mutations in Vietnamese Patients with Cornelia de Lange Syndrome. Medicina, 56(2), 76. https://doi.org/10.3390/medicina56020076