
Implications of SARS-CoV-2 Infection in Systemic Juvenile Idiopathic Arthritis




Abstract
1. Introduction
2. Systemic Juvenile Idiopathic Arthritis
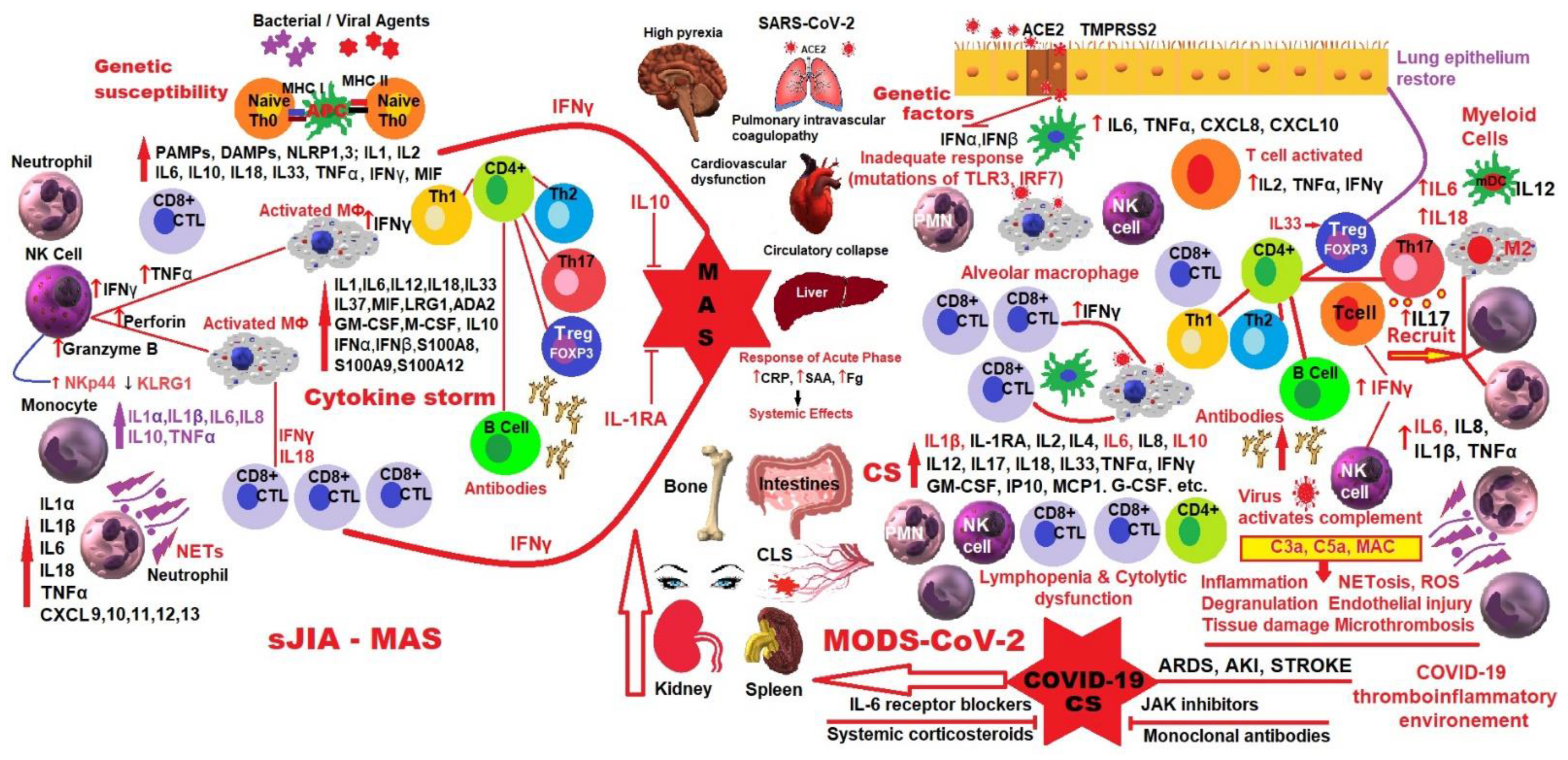
3. Macrophage Activation Syndrome in sJIA
4. MAS in sJIA and Hyperinflammation in COVID-19
5. Interrelationship between SARS-CoV-2 Infection and sJIA
6. Discussions
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AKI | Acute kidney injury |
| ARDS | Acute respiratory distress syndrome |
| ADA2 | Adenosine deaminase 2 |
| AOSD | Adult-onset Still’s disease |
| AGEs | Advanced glycation end products |
| AHSP | Alpha hemoglobin–stabilizing protein |
| ACR | American College of Rheumatology |
| ACE-2 | Angiotensin-converting enzyme 2 |
| ANKRD55 | Ankyrin Repeat Domain 55 |
| APC | Antigen-presenting cell |
| ANA | Antinuclear antibodies |
| AST | Aspartate aminotransferase |
| AID | Autoinflammatory disease |
| BCL6 | B-cell lymphoma 6 protein, encoded by the BCL6 gene |
| bDMARDs | Biologic disease-modifying antirheumatic drugs |
| CP | Calprotectin |
| CLS | Capillary leak syndrome |
| CASP8 | Caspase-8 |
| CCL23 | C-C Motif Chemokine Ligand 23 |
| CDC | Centers for Disease Control and Prevention |
| CXCL1 | Chemokine (C-X-C motif) ligand 1 |
| CCL2 | Chemokine (C-C motif) ligand 2 |
| CID | Clinically inactive disease |
| CD6 | Cluster of Differentiation 6 |
| CD163 | Cluster of Differentiation 163 |
| C3a | Complement component 3a |
| C3b | Complement component 3b |
| cNK | Conventional NK cells |
| CNVs | Copy number variations |
| CRP | C-reactive protein |
| COVID-CS | Cytokine storm COVID-19 |
| CSS | Cytokine storm syndrome |
| CMV | Cytomegalovirus |
| CTL | Cytotoxic T-Lymphocyte |
| CTLA4 | Cytotoxic T-Lymphocyte Associated Protein 4 |
| DAMP | Damage-associated molecular pattern |
| DAMPs | Danger (or damage)-associated molecular patterns |
| DMARDs | Disease-modifying antirheumatic drugs |
| DIC | Disseminated intravascular coagulation |
| ELP | Endogenous lipoid pneumonia |
| EHEC | Enterohemorrhagic Escherichia coli |
| ERA | Enthesitis-related arthritis |
| EBV | Epstein-Barr virus |
| EBV-HLH | Epstein-Barr virus-induced hemophagocytic lymphohistiocytosis |
| ESR | Erythrocyte sedimentation rate |
| ALAS2 | Erythroid-specific 5-aminolevulinate synthase |
| EULAR | European League Against Rheumatism |
| FMF | Familial Mediterranean Fever |
| FasL | Fas ligand |
| Fg | Fibrinogen |
| FDA | Food and Drug Administration |
| GWAS | Genome-wide association study |
| GF | Glycosylated ferritin |
| G-CSF | Granulocyte colony stimulating factor |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| GAS | Group A beta-hemolytic Streptococci |
| HSPB1 | Heat shock protein beta-1 |
| HSC | Hematopoietic stem cell |
| HLH | Hemophagocytic lymphohistiocytosis |
| HMGB1 | High mobility group box-1 |
| hsCRP | High-sensitivity C-reactive protein |
| HIV | Human Immunodeficiency Virus |
| HLA | Human leukocyte antigen |
| IgM-RF | Immunoglobulin M rheumatoid factor |
| H1N1 | Influenza A virus subtype H1N1 |
| ILCs | Innate lymphoid cells |
| ILC1s | Innate lymphoid cells group 1 |
| IFN-γ | Interferon-gamma |
| IRF | Interferon regulatory factors |
| IL6ST | Interleukin 6 Signal Transducer |
| IL-18BP | IL-18 binding protein |
| IV | Intravenous |
| KD | Kawasaki disease |
| KDSS | KD shock syndrome |
| KIRs | Killer cell immunoglobulin-like receptors |
| KLRF1 | Killer cell lectin-like receptor F1 |
| KLRB1 | Killer cell lectin-like receptor subfamily B member 1 |
| KLRG1 | Killer cell lectin-like receptor subfamily G member 1 |
| KITLG | KIT ligand |
| KLF1 | Kruppel-like factor 1 |
| LDH | Lactic acid dehydrogenase |
| LRG | Leucine-rich α2-glycoprotein |
| LPS | Lipopolysaccharide |
| LD | Lung disease |
| TNFSF1 | Lymphotoxin-alpha (LT-α) = tumor necrosis factor-beta (TNF-β) |
| MAS | Macrophage activation syndrome |
| sJIA-MAS | Macrophage activation syndrome in sJIA |
| MIF | Macrophage migration inhibitory factor |
| MHC | Major histocompatibility complex |
| MMP-1 | Matrix metalloproteinase 1 |
| MEFV | Mediterranean fever |
| MAC | Membrane attack complex |
| MTX | Methotrexate |
| MCP1 | Monocyte chemoattractant protein 1 |
| MODS-CoV-2 | Multiple organ dysfunction in SARS-CoV-2 |
| MIS-C | Multisystem inflammatory syndrome in children |
| RMD | Musculoskeletal rheumatic disease |
| MPO-DNA complexes | Myeloperoxidase-DNA complexes |
| mDC | Myeloid dendritic cell |
| MRP | Myeloid-related protein |
| NK | Natural killer |
| NKT cells | Natural killer T cells |
| cNK | Conventional NK |
| trNK | Tissue-resident NK |
| NETs/NETosis | Neutrophil extracellular traps |
| NLR | NOD-like receptor |
| nsJIA | Non-systemic forms of JIA |
| NOD | Nucleotide-binding oligomerization |
| NLRP | Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain |
| NLRP3 | Nucleotide-binding oligomerization domain-like receptor pyrin domain-containing protein 3 |
| NLRs | Nucleotide-binding oligomerization domain-like receptors = NOD-like receptors |
| OSM | Oncostatin M |
| PAMP | Pathogen-associated molecular pattern |
| PRRs | Pattern recognition receptors |
| PIMS-TS | Pediatric Multisystem Inflammatory Syndrome temporarily associated with SARS-CoV-2 |
| PRINTO | Pediatric Rheumatology International Trials Organization |
| PRF1 | Perforin 1 |
| PBMC | Peripheral blood mononuclear cell |
| PMN, PML, or PMNL | Polymorphonuclear leukocytes |
| pHLH | Primary Hemophagocytic Lymphohistiocytosis |
| PCT | Procalcitonin |
| PKC | Protein kinase C |
| PAP | Pulmonary alveolar proteinosis |
| PIC | Pulmonary intravascular coagulopathy |
| RAGE | Receptor for advanced glycation end products |
| rIL-1Ra | Recombinant IL-1 receptor antagonist |
| rr | Reference range |
| RMD | Rheumatic and musculoskeletal disease |
| RUNX1 | Runt-related transcription factor 1 |
| RUNX3 | Runt-related transcription factor 3 |
| sec-HLH | Secondary Hemophagocytic Lymphohistiocytosis |
| SAA | Serum Amyloid A |
| STAT | Signal Transducer and Activator of Transcription |
| STAT4 | Signal Transducer and Activator of Transcription 4 |
| SNP | Single Nucleotide Polymorphism |
| SIRT2 | Sirtuin 2 |
| sJIA-LD | sJIA-associated lung disease |
| sRAGE | Soluble receptor for advanced glycation end products |
| SULT1A1 | Sulfotransferase 1A1 |
| SLE | Systemic lupus erythematosus |
| SURFS | Systemic undifferentiated recurring fever syndromes |
| TIM-3 | T-cell immunoglobulin mucin-3 |
| TMA | Thrombotic microangiopathy |
| trNK cells | Tissue-resident (tr)NK cells |
| TWEAK | TNF-like weak inducer of apoptosis |
| TCZ | Tocilizumab |
| TLR | Toll-like receptor |
| TGF-β | Transforming growth factor beta |
| TMPRSS2 | Transmembrane serine protease 2 |
| TAMs | Tumor-associated macrophages |
| TNF-α | Tumor necrosis factor alpha |
| TNFR1 | Tumor necrosis factor receptor 1 |
| TRAF1/C5 | Tumor necrosis factor-receptor associated factor 1 and C5 (complement component 5) |
| TRAPS | Tumor necrosis factor receptor-associated periodic fever syndrome |
| PTPN2 | Tyrosine-protein phosphatase non-receptor type 2 |
| UNC13D | Unc-13 Homolog D |
| WGCNA | Weighted gene co-expression network analysis |
| ↑ | Increased |
| ↓ | Decreased |
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Verikios, G. The dynamic effects of infectious disease outbreaks: The case of pandemic influenza and human coronavirus. Socio-Econ. Plann. Sci. 2020, 71, 100898. [Google Scholar] [CrossRef]
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76, Erratum in: Int. J. Surg. 2020, 77, 217. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef] [PubMed]
- Mélo Silva Júnior, M.L.; Souza, L.; Dutra, R.; Valente, R.; Melo, T.S. Review on therapeutic targets for COVID-19: Insights from cytokine storm. Postgrad. Med. J. 2021, 97, 391–398. [Google Scholar] [CrossRef] [PubMed]
- United Nations. Department of Economic and Social Affairs Social Inclusion. Everyone Included: Social Impact of COVID-19. Available online: https://www.un.org/development/desa/dspd/everyone-included-covid-19.html (accessed on 10 February 2022).
- Niknam, Z.; Jafari, A.; Golchin, A.; Danesh Pouya, F.; Nemati, M.; Rezaei-Tavirani, M.; Rasmi, Y. Potential therapeutic options for COVID-19: An update on current evidence. Eur. J. Med. Res. 2022, 27, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506, Erratum in: Lancet 30 January 2020. [Google Scholar] [CrossRef]
- WHO. Listings of WHO’s Response to COVID-19. Available online: https://www.who.int/news/item/29-06-2020-covidtimeline (accessed on 8 November 2021).
- Greenwood, M. What Mutations of SARS-CoV-2 Are Causing Concern? Available online: https://www.news-medical.net/health/What-Mutations-of-SARS-CoV-2-are-Causing-Concern.aspx (accessed on 9 November 2021).
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int (accessed on 25 February 2022).
- COVID-19 Disease in Children and Adolescents: Scientific Brief, 29 September 2021. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-Sci_Brief-Children_and_adolescents-2021.1 (accessed on 8 February 2022).
- Buonsenso, D.; Munblit, D.; De Rose, C.; Sinatti, D.; Ricchiuto, A.; Carfi, A.; Valentini, P. Preliminary evidence on long COVID in children. Acta Paediatr. 2021, 110, 2208–2211. [Google Scholar] [CrossRef]
- Rodriguez-Smith, J.J.; Verweyen, E.L.; Clay, G.M.; Esteban, Y.M.; de Loizagac, S.R.; Baker, E.J.; Do, T.; Dhakal, S.; Lang, S.M.; Grom, A.A.; et al. Inflammatory biomarkers in COVID-19-associated multisystem inflammatory syndrome in children, Kawasaki disease, and macrophage activation syndrome: A cohort study. Lancet Rheumatol. 2021, 3, e574–e584. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, L.; Du, H.; Zhang, J.; Li, Y.Y.; Qu, J.; Zhang, W.; Wang, Y.; Bao, S.; Li, Y.; et al. SARS-CoV-2 Infection in Children. N. Engl. J. Med. 2020, 382, 1663–1665. [Google Scholar] [CrossRef] [PubMed]
- Verdoni, L.; Mazza, A.; Gervasoni, A.; Martelli, L.; Ruggeri, M.; Ciuffreda, M.; Bonanomi, E.; D’Antiga, L. An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet 2020, 395, 1771–1778. [Google Scholar] [CrossRef]
- Flood, J.; Shingleton, J.; Bennett, E.; Walker, B.; Amin-Chowdhury, Z.; Oligbu, G.; Avis, J.; Lynn, R.M.; Davis, P.; Bharucha, T.; et al. Paediatric multisystem inflammatory syndrome temporally associated with SARS-CoV-2 (PIMS-TS): Prospective, national surveillance, United Kingdom and Ireland, 2020. Lancet Reg. Health Eur. 2021, 3, 100075. [Google Scholar] [CrossRef] [PubMed]
- Pouletty, M.; Borocco, C.; Ouldali, N.; Caseris, M.; Basmaci, R.; Lachaume, N.; Bensaid, P.; Pichard, S.; Kouider, H.; Morelle, G.; et al. Paediatric multisystem inflammatory syndrome temporally associated with SARS-CoV-2 mimicking Kawasaki disease (Kawa-COVID-19): A multicentre cohort. Ann. Rheum. Dis. 2020, 79, 999–1006. [Google Scholar] [CrossRef]
- Godfred-Cato, S.; Bryant, B.; Leung, J.; Oster, M.E.; Conklin, L.; Abrams, J.; Roguski, K.; Wallace, B.; Prezzato, E.; Koumans, E.H.; et al. COVID-19-Associated Multisystem Inflammatory Syndrome in Children—United States, March–July 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1074–1080, Erratum in: MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 1229. [Google Scholar] [CrossRef]
- Diorio, C.; Henrickson, S.E.; Vella, L.A.; McNerney, K.O.; Chase, J.; Burudpakdee, C.; Lee, J.H.; Jasen, C.; Balamuth, F.; Barrett, D.M.; et al. Multisystem inflammatory syndrome in children and COVID-19 are distinct presentations of SARS-CoV-2. J. Clin. Investig. 2020, 130, 5967–5975. [Google Scholar] [CrossRef]
- Consiglio, C.R.; Cotugno, N.; Sardh, F.; Pou, C.; Amodio, D.; Rodriguez, L.; Tan, Z.; Zicari, S.; Ruggiero, A.; Pascucci, G.R. The Immunology of Multisystem Inflammatory Syndrome in Children with COVID-19. Cell 2020, 183, 968–981.e7. [Google Scholar] [CrossRef]
- Crayne, C.; Cron, R.Q. Pediatric macrophage activation syndrome, recognizing the tip of the Iceberg. Eur. J. Rheumatol. 2019, 7, 1–8. [Google Scholar] [CrossRef]
- Go, E.; van Veenendaal, M.; Manlhiot, C.; Schneider, R.; McCrindle, B.W.; Yeung, R.S.M. Kawasaki Disease and Systemic Juvenile Idiopathic Arthritis—Two Ends of the Same Spectrum. Front. Pediatrics 2021, 9, 665815. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Multisystem Inflammatory Syndrome. Available online: https://www.cdc.gov/mis/mis-c/hcp/index.html (accessed on 24 November 2021).
- Zhang, Q.Y.; Xu, B.W.; Du, J.B. Similarities and differences between multiple inflammatory syndrome in children associated with COVID-19 and Kawasaki disease: Clinical presentations, diagnosis, and treatment. World J. Pediatrics WJP 2021, 17, 335–340. [Google Scholar] [CrossRef]
- Henderson, L.A.; Canna, S.W.; Friedman, K.G.; Gorelik, M.; Lapidus, S.K.; Bassiri, H.; Behrens, E.M.; Ferris, A.; Kernan, K.F.; Schulert, G.S.; et al. American College of Rheumatology Clinical Guidance for Multisystem Inflammatory Syndrome in Children Associated With SARS–CoV-2 and Hyperinflammation in Pediatric COVID-19: Version 1. Arthritis Rheumatol. 2020, 72, 1791–1805. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.A.; Canna, S.W.; Friedman, K.G.; Gorelik, M.; Lapidus, S.K.; Bassiri, H.; Behrens, E.M.; Ferris, A.; Kernan, K.F.; Schulert, G.S.; et al. American College of Rheumatology Clinical Guidance for Multisystem Inflammatory Syndrome in Children Associated With SARS–CoV-2 and Hyperinflammation in Pediatric COVID-19: Version 2. Arthritis Rheumatol. 2021, 73, e13–e29. [Google Scholar] [CrossRef]
- Payne, A.B.; Gilani, Z.; Godfred-Cato, S.; Belay, E.D.; Feldstein, L.R.; Patel, M.M.; Randolph, A.G.; Newhams, M.; Thomas, D.; Magleby, R.; et al. Incidence of Multisystem Inflammatory Syndrome in Children Among US Persons Infected With SARS-CoV-2. JAMA Netw. Open 2021, 4, e2116420. [Google Scholar] [CrossRef] [PubMed]
- Alunno, A.; Carubbi, F.; Rodríguez-Carrio, J. Storm, typhoon, cyclone or hurricane in patients with COVID-19? Beware of the same storm that has a different origin. RMD Open 2020, 6, e001295. [Google Scholar] [CrossRef]
- Rowley, A.H. Understanding SARS-CoV-2-related multisystem inflammatory syndrome in children. Nat. Rev. Immunol. 2020, 20, 453–454. [Google Scholar] [CrossRef] [PubMed]
- Belhadjer, Z.; Méot, M.; Bajolle, F.; Khraiche, D.; Legendre, A.; Abakka, S.; Auriau, J.; Grimaud, M.; Oualha, M.; Beghetti, M.; et al. Acute Heart Failure in Multisystem Inflammatory Syndrome in Children in the Context of Global SARS-CoV-2 Pandemic. Circulation 2020, 142, 429–436. [Google Scholar] [CrossRef]
- Panigrahy, N.; Policarpio, J.; Ramanathan, R. Multisystem inflammatory syndrome in children and SARS-CoV-2: A scoping review. J. Pediatric Rehabil. Med. 2020, 13, 301–316, Erratum in J. Pediatric Rehabil. Med. 2021, 14, 137. [Google Scholar] [CrossRef] [PubMed]
- Petty, R.E.; Southwood, T.R.; Manners, P.; Baum, J.; Glass, D.N.; Goldenberg, J.; He, X.; Maldonado-Cocco, J.; Orozco-Alcala, J.; Prieur, A.M.; et al. International League of Associations for Rheumatology. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: Second revision, Edmonton, 2001. J. Rheumatol. 2004, 31, 390–392. [Google Scholar] [PubMed]
- Martini, A.; Ravelli, A.; Avcin, T.; Beresford, M.W.; Burgos-Vargas, R.; Cuttica, R.; Ilowite, N.T.; Khubchandani, R.; Laxer, R.M.; Lovell, D.J.; et al. Pediatric Rheumatology International Trials Organization (PRINTO). Toward New Classification Criteria for Juvenile Idiopathic Arthritis: First Steps, Pediatric Rheumatology International Trials Organization International Consensus. J. Rheumatol. 2019, 46, 190–197. [Google Scholar] [CrossRef]
- Thierry, S.; Fautrel, B.; Lemelle, I.; Guillemin, F. Prevalence and incidence of juvenile idiopathic arthritis: A systematic review. Joint Bone Spine 2014, 81, 112–117. [Google Scholar] [CrossRef]
- Behrens, E.M.; Beukelman, T.; Gallo, L.; Spangler, J.; Rosenkranz, M.; Arkachaisri, T.; Ayala, R.; Groh, B.; Finkel, T.H.; Cron, R.Q. Evaluation of the presentation of systemic onset juvenile rheumatoid arthritis: Data from the Pennsylvania Systemic Onset Juvenile Arthritis Registry (PASOJAR). J. Rheumatol. 2008, 35, 343–348. [Google Scholar] [PubMed]
- Costello, R.; McDonagh, J.; Hyrich, K.L.; Humphreys, J.H. Incidence and prevalence of juvenile idiopathic arthritis in the United Kingdom, 2000–2018: Results from the Clinical Practice Research Datalink. Rheumatology 2021, keab714. [Google Scholar] [CrossRef] [PubMed]
- Al-Mayouf, S.M.; Al Mutairi, M.; Bouayed, K.; Habjoka, S.; Hadef, D.; Lotfy, H.M.; Scott, C.; Sharif, E.M.; Tahoun, N. Epidemiology and demographics of juvenile idiopathic arthritis in Africa and Middle East. Pediatric Rheumatol. 2021, 19, 166. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Laxer, R.M. Systemic onset juvenile rheumatoid arthritis. Bailliere’s Clin. Rheumatol. 1998, 12, 245–271. [Google Scholar] [CrossRef]
- Ter Haar, N.M.; Jansen, M.; Frenkel, J.F.; Vastert, S.J. How autoinflammation may turn into autoimmune inflammation: Insights from monogenetic and complex IL-1 mediated auto-inflammatory diseases. Clin. Immunol. 2020, 219, 108538. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Ohta, A.; Tsunematsu, T.; Kasukawa, R.; Mizushima, Y.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Ota, T. Preliminary criteria for classification of adult Still’s disease. J. Rheumatol. 1992, 199, 424–430. [Google Scholar]
- Yang, J.W.; Lee, E.; Seo, J.Y.; Jung, J.Y.; Suh, C.H.; Kim, H.A. Application of the international league against rheumatism classification criteria for systemic juvenile idiopathic arthritis as a prognostic factor in patients with adults-onset Still’s disease. Pediatric Rheumatol. Online J. 2018, 16, 9. [Google Scholar] [CrossRef]
- Silva, J.R.; Brito, I. Systemic juvenile idiopathic arthritis versus adult-onset Still’s disease: The pertinence of changing the current classification criteria. Acta Reumatol. Port. 2020, 45, 150–151. [Google Scholar]
- Ailioaie, L.M.; Litscher, G. Molecular and Cellular Mechanisms of Arthritis in Children and Adults: New Perspectives on Applied Photobiomodulation. Int. J. Mol. Sci. 2020, 21, 6565. [Google Scholar] [CrossRef]
- Albaker, A.R. Current Review of Systemic Juvenile Idiopathic Arthritis: What Do Paediatricians Need to Know? Open J. Pediatrics 2020, 10, 769–801. [Google Scholar] [CrossRef]
- Gattorno, M.; Hofer, M.; Federici, S.; Vanoni, F.; Bovis, F.; Aksentijevich, I.; Anton, J.; Arostegui, J.I.; Barron, K.; Ben-Cherit, E. Eurofever Registry and the Paediatric Rheumatology International Trials Organisation (PRINTO). Classification criteria for autoinflammatory recurrent fevers. Ann. Rheum. Dis. 2019, 78, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Dispenzieri, A.; Fajgenbaum, D.C. Overview of Castleman disease. Blood 2020, 135, 1353–1364. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.D.; Job, A.V.; Cho, W. Differential Diagnosis of Juvenile Idiopathic Arthritis. J. Rheum. Dis. 2017, 24, 131–137. [Google Scholar] [CrossRef]
- Ailioaie, C. Idiopathic Juvenile Arthritis Family Guide; CERMI Technical, Scientific and Didactic Publishing House: Iaşi, Romania, 2005; pp. 36–41. ISBN 973-667-109-7. [Google Scholar]
- Ailioaie, C.; Ailioaie, L.M. Juvenile idiopathic arthritis. In Management of Chronic Rheumatic Pain; PIM Publishing House: Iasi, Romania, 2008; pp. 129–146. [Google Scholar]
- Hemke, R.; Herregods, N.; Jaremko, J.L.; Åström, G.; Avenarius, D.; Becce, F.; Bielecki, D.K.; Boesen, M.; Dalili, D.; Giraudo, C.; et al. Imaging assessment of children presenting with suspected or known juvenile idiopathic arthritis: ESSR-ESPR points to consider. Eur. Radiol. 2020, 30, 5237–5249. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Schneider, R. Systemic Juvenile Idiopathic Arthritis. Pediatric Clin. N. Am. 2018, 65, 691–709. [Google Scholar] [CrossRef]
- Boyarchuk, O.; Kovalchuk, T.; Kovalchuk, N.; Chubata, O. Clinical variability of the systemic juvenile idiopathic arthritis course: Literature review based on case series. Reumatologia 2020, 58, 436–443. [Google Scholar] [CrossRef]
- Wang, M.Y.; Jia, J.C.; Yang, C.D.; Hu, Q.Y. Pathogenesis, disease course, and prognosis of adult-onset Still disease: An update and review. Chin. Med. J. 2019, 132, 2856–2864. [Google Scholar] [CrossRef]
- Hinks, A.; Cobb, J.; Marion, M.C.; Prahalad, S.; Sudman, M.; Bowes, J.; Martin, P.; Comeau, M.E.; Sajuthi, S.; Andrews, R.; et al. Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat. Genet. 2013, 45, 664–669. [Google Scholar] [CrossRef]
- Choo, S.Y. The HLA system: Genetics, immunology, clinical testing, and clinical implications. Yonsei Med. J. 2007, 48, 11–23. [Google Scholar] [CrossRef]
- Yatskiu, H.A.; Savina, N.V.; Nikitchenko, N.V.; Kuzhir, T.D.; Tchitchko, A.M.; Sukalo, A.V.; Goncharova, R.I. Genetic susceptibility to juvenile idiopathic arthritis in the Belarusian population: Gene-gene interactions analysis. Ecol. Genet. 2019, 17, 65–76. [Google Scholar] [CrossRef]
- Tadaki, H.; Saitsu, H.; Nishimura-Tadak, A.; Imagawa, T.; Kikuchi, M.; Hara, R.; Kaneko, U.; Kishi, T.; Miyamae, T.; Miyake, N. De novo 19q13.42 duplications involving NLRP gene cluster in a patient with systemic-onset juvenile idiopathic arthritis. J. Hum. Genet. 2011, 56, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Arakelyan, A.; Nersisyan, L.; Poghosyan, D.; Khondkaryan, L.; Hakobyan, A.; Löffler-Wirth, H.; Melanitou, E.; Binder, H. Autoimmunity and Autoinflammation: A Systems View on Signaling Pathway Dysregulation Profiles. PLoS ONE 2017, 12, e0187572. [Google Scholar] [CrossRef]
- Zaripova, L.N.; Midgley, A.; Christmas, S.E.; Beresford, M.W.; Baildam, E.M.; Oldershaw, R.A. Juvenile idiopathic arthritis: From aetiopathogenesis to therapeutic approaches. Pediatric Rheumatol. Online J. 2021, 19, 135. [Google Scholar] [CrossRef] [PubMed]
- Cimaz, R. Systemic-onset juvenile idiopathic arthritis. Autoimmun. Rev. 2016, 15, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Szekanecz, Z.; McInnes, I.B.; Schett, G.; Szamosi, S.; Benkő, S.; Szűcs, G. Autoinflammation and autoimmunity across rheumatic and musculoskeletal diseases. Nat. Rev. Rheumatol. 2021, 17, 585–595. [Google Scholar] [CrossRef]
- Vandenhaute, J.; Avau, A.; Filtjens, J.; Malengier-Devlies, B.; Imbrechts, M.; Van den Berghe, N.; Ahmadzadeh, K.; Mitera, T.; Boon, L.; Leclercq, G.; et al. Regulatory Role for NK Cells in a Mouse Model of Systemic Juvenile Idiopathic Arthritis. J. Immunol. 2019, 203, 3339–3348. [Google Scholar] [CrossRef]
- MIF Gene—Macrophage Migration Inhibitory Factor. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=MIF (accessed on 14 December 2021).
- Kim, K.W.; Kim, H.R. Macrophage migration inhibitory factor: A potential therapeutic target for rheumatoid arthritis. Korean J. Intern. Med. 2016, 31, 634–642. [Google Scholar] [CrossRef]
- Topham, N.J.; Hewitt, E.W. Natural killer cell cytotoxicity: How do they pull the trigger? Immunology 2009, 128, 7–15. [Google Scholar] [CrossRef]
- Fu, B.; Tian, Z.; Wei, H. Subsets of human natural killer cells and their regulatory effects. Immunology 2014, 141, 483–489. [Google Scholar] [CrossRef]
- Put, K.; Vandenhaute, J.; Avau, A.; van Nieuwenhuijze, A.; Brisse, E.; Dierckx, T.; Rutgeerts, O.; Garcia-Perez, J.E.; Toelen, J.; Waer, M.; et al. Inflammatory Gene Expression Profile and Defective Interferon-γ and Granzyme K in Natural Killer Cells from Systemic Juvenile Idiopathic Arthritis Patients. Arthritis Rheumatol. 2017, 69, 213–224. [Google Scholar] [CrossRef]
- Prencipe, G.; Bracaglia, C.; De Benedetti, F. Interleukin-18 in Pediatric Rheumatic Diseases. Curr. Opin. Rheumatol. 2019, 31, 421–427. [Google Scholar] [CrossRef]
- Takakura, M.; Shimizu, M.; Yakoyama, T.; Mizuta, M.; Yachie, A. Transient Natural Killer Cell Dysfunction Associated with Interleukin-18 Overproduction in Systemic Juvenile Idiopathic Arthritis. Pediatrics Int. 2018, 60, 984–985. [Google Scholar] [CrossRef] [PubMed]
- Imbrechts, M.; Avau, A.; Vandenhaute, J.; Malengier-Devlies, B.; Put, K.; Mitera, T.; Berghmans, N.; Burton, O.; Junius, S.; Liston, A.; et al. Insufficient IL-10 Production as a Mechanism Underlying the Pathogenesis of Systemic Juvenile Idiopathic Arthritis. J. Immunol. 2018, 201, 2654–2663. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Cheng, G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit. Rev. Immunol. 2012, 32, 23–63. [Google Scholar] [CrossRef] [PubMed]
- Möller, J.C.; Paul, D.; Ganser, G.; Range, U.; Gahr, M.; Kelsch, R.; Rösen-Wolff, A.; Hedrich, C.M. IL10 promoter polymorphisms are associated with systemic onset juvenile idiopathic arthritis (SoJIA). Clin. Exp. Rheumatol. 2010, 28, 912–918. [Google Scholar] [PubMed]
- Peng, Y.; Liu, X.; Duan, Z.; Duan, J.; Zhou, Y. The Association of Serum IL-10 Levels with the Disease Activity in Systemic-Onset Juvenile Idiopathic Arthritis Patients. Mediat. Inflamm. 2021, 2021, 6650928. [Google Scholar] [CrossRef]
- Degboé, Y.; Rauwel, B.; Baron, M.; Boyer, J.F.; Ruyssen-Witrand, A.; Constantin, A.; Davignon, J.L. Polarization of Rheumatoid Macrophages by TNF Targeting Through an IL-10/STAT3 Mechanism. Front. Immunol. 2019, 10, 3. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Yang, H.-Y.; Huang, J.-L.; Lai, J.-H. Signals and Mechanisms Regulating Monocyte and Macrophage Activation in the Pathogenesis of Juvenile Idiopathic Arthritis. Int. J. Mol. Sci. 2021, 22, 7960. [Google Scholar] [CrossRef]
- Dusser, P.; Koné-Paut, I. Still’s Disease in the Constellation of Hyperinflammatory Syndromes: A Link with Kawasaki Disease? J. Clin. Med. 2021, 10, 3244. [Google Scholar] [CrossRef]
- Theodoropoulou, K.; Georgin-Lavialle, S. Arthrite juvénile systémique et maladie de Still de l’adulte [Systemic juvenile onset idiopathic arthritis and adult onset still disease]. Rev. Med. Suisse 2018, 14, 372–377. [Google Scholar]
- Gurion, R.; Lehman, T.J.; Moorthy, L.N. Systemic arthritis in children: A review of clinical presentation and treatment. Int. J. Inflam. 2012, 2012, 271569. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Woo, P. Systemic juvenile idiopathic arthritis: Diagnosis, management, and outcome. Nat. Clin. Pract. Rheumatol. 2006, 2, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.; Alekseev, L.S.; Brewer, E.J., Jr.; Dolgopolova, A.V.; Mudholkar, G.S.; Patel, K. Juvenile rheumatoid arthritis. A comparison of patients from the USSR and USA. Arthritis Rheum. 1980, 23, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Schaller, J.G. Juvenile rheumatoid arthritis: Series 1. Arthritis Rheum. 1977, 20, 165–170. [Google Scholar] [PubMed]
- Ansell, B.M. Prognosis in juvenile arthritis. Adv. Exp. Med. Biol. 1999, 455, 27–33. [Google Scholar] [CrossRef]
- Prieur, A.M.; Chèdeville, G. Prognostic factors in juvenile idiopathic arthritis. Curr. Rheumatol. Rep. 2001, 3, 371–378. [Google Scholar] [CrossRef]
- Yanagimachi, M.; Naruto, T.; Miyamae, T.; Hara, T.; Kikuchi, M.; Hara, R.; Imagawa, T.; Mori, M.; Sato, H.; Goto, H. Association of IRF5 polymorphisms with susceptibility to macrophage activation syndrome in patients with juvenile idiopathic arthritis. J Rheumatol. 2011, 38, 769–774. [Google Scholar] [CrossRef]
- Schneider, R.; Lang, B.A.; Reilly, B.J.; Laxer, R.M.; Silverman, E.D.; Ibanez, D.; Bombardier, C.; Roifman, C.M. Prognostic indicators of joint destruction in systemic-onset juvenile rheumatoid arthritis. J. Pediatrics 1992, 120, 200–205. [Google Scholar] [CrossRef]
- Spiegel, L.R.; Schneider, R.; Lang, B.A.; Birdi, N.; Silverman, E.D.; Laxer, R.M.; Stephens, D.; Feldman, B.M. Early predictors of poor functional outcome in systemic-onset juvenile rheumatoid arthritis: A multicenter cohort study. Arthritis Rheum. 2000, 43, 2402–2409. [Google Scholar] [CrossRef]
- Svantesson, H.; Akesson, A.; Eberhardt, K.; Elborgh, R. Prognosis in juvenile rheumatoid arthritis with systemic onset. A follow-up study. Scand. J. Rheumatol. 1983, 12, 139–144. [Google Scholar] [CrossRef]
- Sawhney, S.; Woo, P.; Murray, K.J. Macrophage activation syndrome: A potentially fatal complication of rheumatic disorders. Arch. Dis. Child. 2001, 85, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.I.; Talukder, M.K.; Islam, M.M.; Laila, K.; Rahman, S.A. Macrophage Activation Syndrome in Paediatric Rheumatic Diseases. Mymensingh Med. J. 2017, 26, 356–363. [Google Scholar] [PubMed]
- Stoeber, E. Prognosis in juvenile chronic arthritis. Follow-up of 433 chronic rheumatic children. Eur. J. Pediatrics 1981, 135, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Immonen, K.; Savolainen, A.; Kautiainen, H.; Hakala, M. Longterm outcome of amyloidosis associated with juvenile idiopathic arthritis. J. Rheumatol. 2008, 35, 907–912. [Google Scholar] [PubMed]
- Fair, D.C.; Rodriguez, M.; Knight, A.M.; Rubinstein, T. Depression and Anxiety in Patients with Juvenile Idiopathic Arthritis: Current Insights and Impact On Quality Of Life, A Systematic Review. Open Access Rheumatol. 2019, 11, 237–252. [Google Scholar] [CrossRef]
- David, J.; Cooper, C.; Hickey, L.; Lloyd, J.; Doré, C.; McCullough, C.; Woo, P. The functional and psychological outcomes of juvenile chronic arthritis in young adulthood. Br. J. Rheumatol. 1994, 33, 876–881. [Google Scholar] [CrossRef]
- Sözeri, B.; Demir, F.; Kalın, S.; Hasbal Akkuş, C.; Salı, E.; Çakır, D. SARS-CoV-2 infection in children with rheumatic disease: Experience of a tertiary referral center. Arch. Rheumatol. 2021, 36, 381–388. [Google Scholar] [CrossRef]
- Barut, K.; Adrovic, A.; Sahin, S.; Tarcin, G.; Tahaoglu, G.; Koker, O.; Yildiz, M.; Kasapcopur, O. Prognosis, complications and treatment response in systemic juvenile idiopathic arthritis patients: A single-center experience. Int. J. Rheum. Dis. 2019, 22, 1661–1669. [Google Scholar] [CrossRef]
- Schulert, G.S.; Yasin, S.; Carey, B.; Chalk, C.; Do, T.; Schapiro, A.H.; Husami, A.; Watts, A.; Brunner, H.I.; Huggins, J.; et al. Systemic Juvenile Idiopathic Arthritis-Associated Lung Disease: Characterization and Risk Factors. Arthritis Rheumatol. 2019, 71, 1943–1954. [Google Scholar] [CrossRef]
- Saper, V.E.; Chen, G.; Deutsch, G.H.; Guillerman, R.P.; Birgmeier, J.; Jagadeesh, K.; Canna, S.; Schulert, G.; Deterding, R.; Xu, J.; et al. Emergent high fatality lung disease in systemic juvenile arthritis. Ann. Rheum. Dis. 2019, 78, 1722–1731. [Google Scholar] [CrossRef]
- Moradinejad, M.H.; Ziaee, V. The incidence of macrophage activation syndrome in children with rheumatic disorders. Minerva Pediatrica 2011, 63, 459–466. [Google Scholar] [PubMed]
- Minoia, F.; Davì, S.; Horne, A.; Demirkaya, E.; Bovis, F.; Li, C.; Lehmberg, K.; Weitzman, S.; Insalaco, A.; Wouters, C.; et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: A multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014, 66, 3160–3169. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Weiner, L. Systemic Juvenile Idiopathic Arthritis Complicated by Macrophage Activation Syndrome. Pediatric Ann. 2015, 44, e142-7. [Google Scholar] [CrossRef] [PubMed]
- Boom, V.; Anton, J.; Lahdenne, P.; Quartier, P.; Ravelli, A.; Wulffraat, N.M.; Vastert, S.J. Evidence-based diagnosis and treatment of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Pediatric Rheumatol. Online J. 2015, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Crayne, C.B.; Albeituni, S.; Nichols, K.E.; Cron, R.Q. The Immunology of Macrophage Activation Syndrome. Front. Immunol. 2019, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.; Minoia, F.; Davì, S.; Horne, A.; Bovis, F.; Pistorio, A.; Aricò, M.; Avcin, T.; Behrens, E.M.; De Benedetti, F.; et al. Paediatric Rheumatology International Trials Organisation; Childhood Arthritis and Rheumatology Research Alliance; Pediatric Rheumatology Collaborative Study Group; Histiocyte Society. 2016 Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Arthritis Rheumatol. 2016, 68, 566–576. [Google Scholar] [CrossRef]
- Lin, Y.T.; Wang, C.T.; Gershwin, M.E.; Chiang, B.L. The pathogenesis of oligoarticular/polyarticular vs. systemic juvenile idiopathic arthritis. Autoimmun. Rev. 2011, 10, 482–489. [Google Scholar] [CrossRef]
- Nada, D.W.; Moghazy, A.; Allam, A.E.-S.; Alunno, A.; Ibrahim, A.M. Short-Term Outcomes and Predictors of Effectiveness of Tocilizumab in Systemic Juvenile Idiopathic Arthritis: A Prospective Cohort Study. Front. Med. 2021, 8, 665028. [Google Scholar] [CrossRef]
- Bracaglia, C.; Prencipe, G.; De Benedetti, F. Macrophage Activation Syndrome: Different Mechanisms Leading to a One Clinical Syndrome. Pediatric Rheumatol. 2017, 15, 5. [Google Scholar] [CrossRef]
- Mizuta, M.; Shimizu, M.; Inoue, N.; Nakagishi, Y.; Yachie, A. Clinical Significance of Serum CXCL9 Levels as a Biomarker for Systemic Juvenile Idiopathic Arthritis Associated Macrophage Activation Syndrome. Cytokine 2019, 119, 182–187. [Google Scholar] [CrossRef]
- Zandstra, J.; Jongerius, I.; Kuijpers, T.W. Future Biomarkers for Infection and Inflammation in Febrile Children. Front. Immunol. 2021, 12, 631308. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Xu, Y.; Qian, X.; Zou, L.; Zheng, R.; Teng, L.; Zheng, Q.; Leung Jung, L.K.; Lu, M. Sudden Hypotension and Increased Serum Interferon-γ and Interleukin-10 Predict Early Macrophage Activation Syndrome in Patients with Systemic Juvenile Idiopathic Arthritis. J. Pediatric. 2021, 235, 203–211.e3. [Google Scholar] [CrossRef] [PubMed]
- Yasin, S.; Schulert, G.S. Systemic juvenile idiopathic arthritis and macrophage activation syndrome: Update on pathogenesis and treatment. Curr. Opin. Rheumatol. 2018, 30, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Vandenhaute, J.; Wouters, C.H.; Matthys, P. Natural Killer Cells in Systemic Autoinflammatory Diseases: A Focus on Systemic Juvenile Idiopathic Arthritis and Macrophage Activation Syndrome. Front. Immunol. 2020, 10, 3089. [Google Scholar] [CrossRef]
- Perera Molligoda Arachchige, A.S. Human NK cells: From development to effector functions. Innate Immun. 2021, 27, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Guo, R.; Wang, Y.F.; Yang, W.; Li, R.; Lu, L. Application of Weighted Gene Coexpression Network Analysis to Identify Key Modules and Hub Genes in Systemic Juvenile Idiopathic Arthritis. Biomed Res. Int. 2021, 2021, 957569. [Google Scholar] [CrossRef]
- Vastert, S.J.; van Wijk, R.; D’Urbano, L.E.; de Vooght, K.M.; de Jager, W.; Ravelli, A.; Magni-Manzoni, S.; Insalaco, A.; Cortis, E.; van Solinge, W.W.; et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology 2010, 49, 441–449. [Google Scholar] [CrossRef]
- Hazen, M.M.; Woodward, A.L.; Hofmann, I.; Degar, B.A.; Grom, A.; Filipovich, A.; Binstadt, B.A. Mutations of the hemophagocytic lymphohistiocytosis-associated gene UNC13D in a patient with systemic juvenile idiopathic arthritis. Arthritis Rheum. 2008, 58, 567–570. [Google Scholar] [CrossRef]
- Morris, G.; Bortolasci, C.C.; Puri, B.K.; Marx, W.; O’Neil, A.; Athan, E.; Walder, K.; Berk, M.; Olive, L.; Carvalho, A.F.; et al. The cytokine storms of COVID-19, H1N1 influenza, CRS and MAS compared. Can one sized treatment fit all? Cytokine 2021, 144, 155593. [Google Scholar] [CrossRef]
- Rauti, R.; Shahoha, M.; Leichtmann-Bardoogo, Y.; Nasser, R.; Paz, E.; Tamir, R.; Miller, V.; Babich, T.; Shaked, K.; Ehrlich, A.; et al. Effect of SARS-CoV-2 proteins on vascular permeability. Elife 2021, 10, e69314. [Google Scholar] [CrossRef]
- Pons, S.; Fodil, S.; Azoulay, E.; Zafrani, L. The vascular endothelium: The cornerstone of organ dysfunction in severe SARS-CoV-2 infection. Crit. Care 2020, 24, 353. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.; Grom, A.A.; Behrens, E.M.; Cron, R.Q. Macrophage activation syndrome as part of systemic juvenile idiopathic arthritis: Diagnosis, genetics, pathophysiology and treatment. Genes Immun. 2012, 13, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Borgia, R.E.; Gerstein, M.; Levy, D.M.; Silverman, E.D.; Hiraki, L.T. Features, treatment, and outcomes of macrophage activation syndrome in childhood-onset systemic lupus erythematosus. Arthritis Rheumatol. 2018, 70, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Avcin, T.; Tse, S.M.; Schneider, R.; Ngan, B.; Silverman, E.D. Macrophage activation syndrome as the presenting manifestation of rheumatic diseases in childhood. J. Pediatrics 2006, 148, 683–686. [Google Scholar] [CrossRef]
- Minoia, F.; Bovis, F.; Davì, S.; Insalaco, A.; Lehmberg, K.; Shenoi, S.; Weitzman, S.; Espada, G.; Gao, Y.J.; Anton, J.; et al. Development and Initial Validation of the Macrophage Activation Syndrome/Primary Hemophagocytic Lymphohistiocytosis Score, a Diagnostic Tool that Differentiates Primary Hemophagocytic Lymphohistiocytosis from Macrophage Activation Syndrome. J. Pediatrics 2017, 189, 72–78.e3. [Google Scholar] [CrossRef]
- Minoia, F.; Bovis, F.; Davì, S.; Horne, A.; Fischbach, M.; Frosch, M.; Huber, A.; Jelusic, M.; Sawhney, S.; McCurdy, D.; et al. Development and initial validation of the MS score for diagnosis of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Ann. Rheum. Dis. 2019, 78, 1357–1362. [Google Scholar] [CrossRef]
- Weiss, E.S.; Girard-Guyonvarc’h, C.; Holzinger, D.; de Jesus, A.A.; Tariq, Z.; Picarsic, J.; Schiffrin, E.J.; Foell, D.; Grom, A.A.; Ammann, S.; et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 2018, 131, 1442–1455. [Google Scholar] [CrossRef]
- Zhou, T.; Damsky, W.; Weizman, O.-E.; McGeary, M.K.; Hartmann, K.P.; Rosen, C.E.; Fischer, S.; Jackson, R.; Flavell, R.A.; Wang, J.; et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 2020, 583, 609–614. [Google Scholar] [CrossRef]
- Yasin, S.; Fall, N.; Brown, R.A.; Henderlight, M.; Canna, S.W.; Girard-Guyonvarc’hc, C.; Gabay, C.; Grom, A.A.; Schulert, G.S. IL-18 as a biomarker linking systemic juvenile idiopathic arthritis and macrophage activation syndrome. Rheumatology 2020, 59, 361–366. [Google Scholar] [CrossRef]
- Harel, M.; Fauteux-Daniel, S.; Girard-Guyonvarc’h, C.; Gabay, C. Balance between Interleukin-18 and Interleukin-18 binding protein in autoinflammatory diseases. Cytokine 2021, 150, 155781. [Google Scholar] [CrossRef]
- Girard-Guyonvarc’h, C.; Harel, M.; Gabay, C. The Role of Interleukin 18/Interleukin 18-Binding Protein in Adult-Onset Still’s Disease and Systemic Juvenile Idiopathic Arthritis. J. Clin. Med. 2022, 11, 430. [Google Scholar] [CrossRef] [PubMed]
- Yongzhi, X. COVID-19-associated cytokine storm syndrome and diagnostic principles: An old and new Issue. Emerg. Microb. Infect. 2021, 10, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ren, L.; Zhang, L.; Zhong, J.; Xiao, Y.; Jia, Z.; Guo, L.; Yang, J.; Wang, C.; Jiang, S.; et al. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 2020, 27, 883–890.e2. [Google Scholar] [CrossRef] [PubMed]
- Ombrello, M.J.; Schulert, G.S. COVID-19 and cytokine storm syndrome: Are there lessons from macrophage activation syndrome? Transl. Res. 2021, 232, 1–12. [Google Scholar] [CrossRef]
- Goswami, J.; MacArthur, T.A.; Sridharan, M.; Pruthi, R.K.; McBane, R.D., 2nd; Witzig, T.E.; Park, M.S. A Review of Pathophysiology, Clinical Features, and Management Options of COVID-19 Associated Coagulopathy. Shock 2021, 55, 700–716. [Google Scholar] [CrossRef]
- Iba, T.; Connors, J.M.; Levy, J.H. The coagulopathy, endotheliopathy, and vasculitis of COVID-19. Inflamm Res. 2020, 69, 1181–1189. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Kerrigan, S.A.; McInnes, I.B. JAK Inhibitors in Rheumatology: Implications for Paediatric Syndromes? Curr. Rheumatol. Rep. 2018, 20, 83. [Google Scholar] [CrossRef]
- Sadik, N.A.; Shaker, O.G.; Ghanem, H.Z.; Hassan, H.A.; Abdel-Hamid, A.H. Single-nucleotide polymorphism of Toll-like receptor 4 and interleukin-10 in response to interferon-based therapy in Egyptian chronic hepatitis C patients. Arch. Virol. 2015, 160, 2181–2195. [Google Scholar] [CrossRef]
- Kopitar-Jerala, N. The Role of Interferons in Inflammation and Inflammasome Activation. Front. Immunol. 2017, 8, 873. [Google Scholar] [CrossRef]
- Pandolfi, L.; Fossali, T.; Frangipane, V.; Bozzini, S.; Morosini, M.; D’Amato, M.; Lettieri, S.; Urtis, M.; Di Toro, A.; Saracino, L.; et al. Broncho-alveolar inflammation in COVID-19 patients: A correlation with clinical outcome. BMC Pulm. Med. 2020, 20, 301. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The role of cytokines including Interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef] [PubMed]
- Parkc, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Pereda, R.; González, D.; Rivero, H.B.; Rivero, J.C.; Pérez, A.; Lopez, L.D.R.; Mezquia, N.; Venegas, R.; Betancourt, J.R.; Domínguez, R.E.; et al. Therapeutic Effectiveness of Interferon Alpha 2b Treatment for COVID-19 Patient Recovery. J. Interferon Cytokine Res. 2020, 40, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Sodeifian, F.; Nikfarjam, M.; Kian, N.; Mohamed, K.; Rezaei, N. The role of type I interferon in the treatment of COVID-19. J. Med. Virol. 2022, 94, 63–81. [Google Scholar] [CrossRef]
- Adachi, S.; Sonoda, M.; Ishimura, M.; Eguchi, K.; Tanaka, T.; Motomura, Y.; Ohga, S. Optimal biologics for juvenile idiopathic arthritis in an infection with SARS-CoV-2 α-variant. Pediatric Allergy Immunol. 2022, 33, e13686. [Google Scholar] [CrossRef]
- Koker, O.; Demirkan, F.G.; Kayaalp, G.; Cakmak, F.; Tanatar, A.; Karadag, S.; Sonmez, H.E.; Omeroglu, R.; Ayaz, N.A. Does immunosuppressive treatment entail an additional risk for children with rheumatic diseases? A survey-based study in the era of COVID-19. Rheumatol. Int. 2020, 40, 1613–1623. [Google Scholar] [CrossRef]
- Demir, F.; Ulu, K.; Çağlayan, Ş.; Coşkuner, T.; Sözeri, B. Clinical course of COVID-19 in children with rheumatic disease under biologic therapy. Clin. Exp. Rheumatol. 2021, 39, 36–37. [Google Scholar] [CrossRef]
- Corominas, H.; Castellví, I.; Pomar, V.; Antonijoan, R.; Mur, I.; Matas, L.; Gich, I.; de Benito, N.; Laiz, A.; Castillo, D.; et al. Effectiveness and safety of intravenous tocilizumab to treat COVID-19-associated hyperinflammatory syndrome: Covizumab-6 observational cohort. Clin. Immunol. 2021, 223, 108631. [Google Scholar] [CrossRef]
- Aydın, F.; Çelikel, E.; Ekici Tekin, Z.; Coşkun, S.; Sezer, M.; Karagöl, C.; Kaplan, M.M.; Tekgöz, N.; Kurt, T.; Özcan, S.; et al. Comparison of baseline laboratory findings of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis and multisystem inflammatory syndrome in children. Int. J. Rheum. Dis. 2021, 24, 542–547. [Google Scholar] [CrossRef]
- Dushnicky, M.J.; Campbell, C.; Beattie, K.A.; Berard, R.; Cellucci, T.; Chan, M.; Gerschman, T.; Johnson, N.; Lim, L.; Luca, N.; et al. Impact of the COVID-19 Pandemic on Juvenile Idiopathic Arthritis Presentation and Research Recruitment: Results from the CAPRI Registry. Rheumatology 2021, keab812. [Google Scholar] [CrossRef] [PubMed]
- Makowska, J.; Styrzyński, F. Between COVID-19 severity and its prevention—What should rheumatologists be aware of? Reumatologia 2021, 59, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Spokes, P.; He, W.; Kaldor, J. High risk groups for severe COVID-19 in a whole of population cohort in Australia. BMC Infect. Dis. 2021, 21, 685. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.W.; Zachariah, P.; Gorelik, M.; Boneparth, A.; Kernie, S.G.; Orange, J.S.; Milner, J.D. Multisystem Inflammatory Syndrome Related to COVID-19 in Previously Healthy Children and Adolescents in New York City. JAMA 2020, 324, 294–296. [Google Scholar] [CrossRef]
- Feldstein, L.R.; Rose, E.B.; Horwitz, S.M.; Collins, J.P.; Newhams, M.M.; Son, M.; Newburger, J.W.; Kleinman, L.C.; Heidemann, S.M.; Martin, A.A.; et al. Multisystem Inflammatory Syndrome in U.S. Children and Adolescents. N. Engl. J. Med. 2020, 383, 334–346. [Google Scholar] [CrossRef]
- Walters, H.M.; Mian, Z.; Thomas, L.; Cerise, J.; Eberhard, B.A.; Pagano, E.; Gottlieb, B.S.; Steigerwald, K.; Hui-Yuen, J.S. Seroprevalence and Clinical Outcomes of SARS-CoV-2 in Paediatric Patients with Rheumatic Disease. Rheumatology 2021, keab730. [Google Scholar] [CrossRef]
- Sozeri, B.; Ulu, K.; Kaya-Akça, U.; Haslak, F.; Pac-Kisaarslan, A.; Otar-Yener, G.; Baba, O.; Altug-Gucenmez, O.; Sahin, N.; Bağlan, E.; et al. The clinical course of SARS-CoV-2 infection among children with rheumatic disease under biologic therapy: A retrospective and multicenter study. Rheumatol Int. 2022, 42, 469–475. [Google Scholar] [CrossRef]
- Boyarchuk, O.; Predyk, L.; Yuryk, I. COVID-19 in patients with juvenile idiopathic arthritis: Frequency and severity. Reumatologia 2021, 59, 197–199. [Google Scholar] [CrossRef]
- Sengler, C.; Eulert, S.; Minden, K.; Niewerth, M.; Horneff, G.; Kuemmerle-Deschner, J.; Siemer, C.; Berendes, R.; Girschick, H.; Hühn, R.; et al. Clinical manifestations and outcome of SARS-CoV-2 infections in children and adolescents with rheumatic musculoskeletal diseases: Data from the National Paediatric Rheumatology Database in Germany. RMD Open 2021, 7, e001687. [Google Scholar] [CrossRef]
- WHO. Coronavirus Disease (COVID-19): Vaccines. Available online: https://www.who.int/news-room/questions-and-answers/item/coronavirus-disease-(covid-19)vaccines?gclid=CjwKCAiAzrWOBhBjEiwAq85QZ1XAka0xjLaH94Y7aJxKSjjV_QcQTzq7WZQVQ6nyuiBvvBeFSSeDjBoCnQIQAvD_BwE&topicsurvey=v8kj13 (accessed on 30 December 2021).
- Parums, D.V. Editorial: Tocilizumab, a Humanized Therapeutic IL-6 Receptor (IL-6R) Monoclonal Antibody, and Future Combination Therapies for Severe COVID-19. Med. Sci. Monit. 2021, 27, e933973. [Google Scholar] [CrossRef]
- Actemra/RoActemra Approved by the European Commission to Treat Patients with Severe COVID-19. Available online: https://www.roche.com/investors/updates/inv-update-2021-12-07.htm (accessed on 6 March 2022).
- Kang, S.; Kishimoto, T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp. Mol. Med. 2021, 53, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; De Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Tassan Din, C.; Boffini, N.; et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e325–e331. [Google Scholar] [CrossRef]
- Mehta, P.; Cron, R.Q.; Hartwell, J.; Manson, J.J.; Tattersall, R.S. Silencing the cytokine storm: The use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. 2020, 2, e358–e367. [Google Scholar] [CrossRef]
- Phadke, O.; Rouster-Stevens, K.; Giannopoulos, H.; Chandrakasan, S.; Prahalad, S. Intravenous administration of anakinra in children with macrophage activation syndrome. Pediatric Rheumatol. Online J. 2021, 19, 98. [Google Scholar] [CrossRef] [PubMed]
- Min, K.H.; Rhee, C.K.; Jung, J.Y.; Suh, M.W. Characteristics of adverse effects when using high dose short term steroid regimen. Korean J. Audiol. 2012, 16, 65–70. [Google Scholar] [CrossRef]
- Batu, E.D. Glucocorticoid treatment in juvenile idiopathic arthritis. Rheumatol. Int. 2019, 39, 13–27. [Google Scholar] [CrossRef]
- Cordtz, R.; Lindhardsen, J.; Soussi, B.G.; Vela, J.; Uhrenholt, L.; Westermann, R.; Kristensen, S.; Nielsen, H.; Torp-Pedersen, C.; Dreyer, L. Incidence and severeness of COVID-19 hospitalization in patients with inflammatory rheumatic disease: A nationwide cohort study from Denmark. Rheumatology 2021, 60, SI59–SI67. [Google Scholar] [CrossRef]
- Licciardi, F.; Giani, T.; Baldini, L.; Favalli, E.G.; Caporali, R.; Cimaz, R. COVID-19 and what pediatric rheumatologists should know: A review from a highly affected country. Pediatric Rheumatol. Online J. 2020, 18, 35. [Google Scholar] [CrossRef]
- Fernández-Sarmiento, J.; De Souza, D.; Jabornisky, R.; Gonzalez, G.A.; Arias López, M.; Palacio, G. Paediatric inflammatory multisystem syndrome temporally associated with COVID-19 (PIMS-TS): A narrative review and the viewpoint of the Latin American Society of Pediatric Intensive Care (SLACIP) Sepsis Committee. BMJ Paediatr. Open 2021, 5, e000894. [Google Scholar] [CrossRef]
- Hügle, B.; Krumrey-Langkammerer, M.; Haas, J.P. Infection with SARS-CoV-2 causes flares in patients with juvenile idiopathic arthritis in remission or inactive disease on medication. Pediatric Rheumatol. 2021, 19, 163. [Google Scholar] [CrossRef]
- Haşlak, F.; Yıldız, M.; Adrovic, A.; Barut, K.; Kasapçopur, Ö. Childhood rheumatic diseases and COVID-19 pandemic: An intriguing linkage and a new horizon. Balkan Med. J. 2020, 37, 184–188. [Google Scholar] [CrossRef] [PubMed]
- Sarzi-Puttini, P.; Giorgi, V.; Sirotti, S.; Marotto, D.; Ardizzone, S.; Rizzardini, G.; Antinori, S.; Galli, M. COVID-19, cytokines and immunosuppression: What can we learn from severe acute respiratory syndrome? Clin. Exp. Rheumatol. 2020, 38, 337–342. [Google Scholar] [PubMed]
- Chen, J.; Jiang, Q.; Xia, X.; Liu, K.; Yu, Z.; Tao, W.; Gong, W.; Han, J.J. Individual variation of the SARS-CoV-2 receptor ACE2 gene expression and regulation. Aging Cell 2020, 19, e13168. [Google Scholar] [CrossRef] [PubMed]
- Haşlak, F.; Ozbey, D.; Yildiz, M.; Adrovic, A.; Sahin, S.; Koker, O.; Aliyeva, A.; Guliyeva, V.; Yalcin, G.; Inanli, G.; et al. Asymptomatic SARS-CoV-2 seropositivity: Patients with childhood-onset rheumatic diseases versus healthy children. Clin. Rheumatol. 2022, 1–11. [Google Scholar] [CrossRef]
- Misra, D.P.; Agarwal, V.; Gasparyan, A.Y.; Zimba, O. Rheumatologists’ perspective on coronavirus disease 19 (COVID-19) and potential therapeutic targets. Clin. Rheumatol. 2020, 39, 2055–2062. [Google Scholar] [CrossRef]
- Eloseily, E.M.; Weiser, P.; Crayne, C.B.; Haines, H.; Mannion, M.L.; Stoll, M.L.; Beukelman, T.; Atkinson, T.P.; Cron, R.Q. Benefit of Anakinra in Treating Pediatric Secondary Hemophagocytic Lymphohistiocytosis. Arthritis Rheumatol. 2020, 72, 326–334. [Google Scholar] [CrossRef]
- De Benedetti, F.; Brogan, P.; Bracaglia, C.; Pardeo, M.; Marucci, G.; Sacco, E.; Eleftheriou, D.; Papadopoulou, C.; Grom, A.; Quartier, P.; et al. Emapalumab (Anti-Interferon-Gamma Monoclonal Antibody) in Patients with Macrophage Activation Syndrome (MAS) Complicating Systemic Juvenile Idiopathic Arthritis (sJIA) [abstract]. Arthritis Rheumatol. 2020, 72 (Suppl. S4). Available online: https://acrabstracts.org/abstract/emapalumab-anti-interferon-gamma-monoclonal-antibody-in-patients-with-macrophage-activation-syndrome-mas-complicating-systemic-juvenile-idiopathic-arthritis-sjia (accessed on 24 January 2022).
- Hermine, O.; Mariette, X.; Tharaux, P.L.; Resche-Rigon, M.; Porcher, R.; Ravaud, P.; CORIMUNO-19 Collaborative Group. Effect of Tocilizumab vs. Usual Care in Adults Hospitalized With COVID-19 and Moderate or Severe Pneumonia: A Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 32–40, Erratum in: JAMA Intern. Med. 2021, 181, 144; Erratum in JAMA Intern. Med. 2021, 181, 1021. [Google Scholar] [CrossRef]
- McGonagle, D.; Ramanan, A.V.; Bridgewood, C. Immune cartography of macrophage activation syndrome in the COVID-19 era. Nat. Rev. Rheumatol. 2021, 17, 145–157. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Z.; Ren, Z.; Li, Y. Reactions Related to CAR-T Cell Therapy. Front. Immunol. 2021, 12, 663201. [Google Scholar] [CrossRef]
- Murira, A.; Lamarre, A. Type-I Interferon Responses: From Friend to Foe in the Battle against Chronic Viral Infection. Front. Immunol. 2016, 7, 609. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Farsalinos, K.; Barbouni, A.; Niaura, R. Systematic review of the prevalence of current smoking among hospitalized COVID-19 patients in China: Could nicotine be a therapeutic option? Intern. Emerg. Med. 2020, 15, 845–852. [Google Scholar] [CrossRef]
- “Cytokine storm syndrome” Merriam-Webster.com Dictionary, Merriam-Webster. Available online: https://www.merriam-webster.com/dictionary/cytokine%20storm%20syndrome (accessed on 25 January 2022).
- Dimopoulos, G.; de Mast, Q.; Markou, N.; Theodorakopoulou, M.; Komnos, A.; Mouktaroudi, M.; Netea, M.G.; Spyridopoulos, T.; Verheggen, R.J.; Hoogerwerf, J.; et al. Favorable Anakinra Responses in Severe Covid-19 Patients with Secondary Hemophagocytic Lymphohistiocytosis. Cell Host Microbe 2020, 28, 117–123.e1. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, S.; Fautrel, B. Clinical Phenotypes of Adult-Onset Still’s Disease: New Insights from Pathophysiology and Literature Findings. J. Clin. Med. 2021, 10, 2633. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Matthay, M.A.; Calfee, C.S. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern. Med. 2020, 180, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020, 2, e437–e445. [Google Scholar] [CrossRef]
- Han, H.; Ma, Q.; Li, C.; Liu, R.; Zhao, L.; Wang, W.; Zhang, P.; Liu, X.; Gao, G.; Liu, F.; et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg. Microbes Infect. 2020, 9, 1123–1130. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, A.; Wan, Y.; Liu, X.; Qiu, C.; Xi, X.; Ren, Y.; Wang, J.; Dong, Y.; Bao, M.; et al. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 769–774. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Gupta, N.; Mayer, D. Interaction of JAK with steroid receptor function. JAKSTAT 2013, 2, e24911. [Google Scholar] [CrossRef] [PubMed]
- Recovery Collaborative Group; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Yener, G.O.; Kısaarslan, A.P.; Ulu, K.; Atalay, E.; Haşlak, F.; Özdel, S.; Yücel, B.B.; Yıldırım, D.G.; Çakmak, F.; Öztürk, K.; et al. Differences and similarities of multisystem inflammatory syndrome in children, Kawasaki disease and macrophage activating syndrome due to systemic juvenile idiopathic arthritis: A comparative study. Rheumatol. Int. 2021, 1–11. [Google Scholar] [CrossRef]
- Çakan, M.; Karadağ, Ş.G.; Tanatar, A.; Ayaz, N.A. The frequency of macrophage activation syndrome and disease course in systemic juvenile idiopathic arthritis. Mod. Rheumatol. 2020, 30, 900–904. [Google Scholar] [CrossRef]
- Sahajpal, N.S.; Lai, C.Y.J.; Hastie, A.; Mondal, A.K.; Dehkordi, S.R.; van der Made, C.I.; Fedrigo, O.; Al-Ajli, F.; Jalnapurkar, S.; Byrska-Bishop, M.; et al. Optical genome mapping identifies rare structural variations as predisposition factors associated with severe COVID-19. iScience 2022, 25, 103760. [Google Scholar] [CrossRef] [PubMed]
- Chiran, D.A.; Weber, M.; Ailioaie, L.M.; Moraru, E.; Ailioaie, C.; Litscher, D.; Litscher, G. Intravenous laser blood irradiation and tocilizumab in a patient with juvenile arthritis. Case Rep. Med. 2014, 2014, 923496. [Google Scholar] [CrossRef] [PubMed]
- Chiran, D.A.; Ailioaie, L.M.; Ailioaie, C. New challenges in treating pediatric rheumatic diseases with lasers in the age of biologic therapy. In Proceedings of the 9th World Association for Laser Therapy Congress (WALT) (Conference: 9th Biennial Congress of the World Asso-ciation for Laser Therapy, Gold Coast, Australia, 28–30 September 2012); Laakso, E.L., Young, C., Eds.; MEDIMOND, Monduzzi Editore, International Proceedings Division MEDIMOND s.r.l.: Editografica: Bologna, Italy, 2013; pp. 25–27. ISBN 978-88-7587-677-7. [Google Scholar]
- Ailioaie, L.M.; Litscher, G.; Weber, M.; Ailioaie, C.; Litscher, D.; Chiran, D.A. Innovations and challenges by applying sublingual laser blood irradiation in juvenile idiopathic arthritis. Int. J. Photoenergy 2014, 2014, 130417. [Google Scholar] [CrossRef]


{kind=link}
| Cardinal Symptoms | Other Systemic Manifestations | Biological Parameters | Life-Threatening Complications for Patients |
|---|---|---|---|
| Fever for at least 2 weeks | Mucous lesions (odynophagia) | ↑ ESR and ↑ CRP levels | MAS (10% apparent, 40% subclinical) |
| Skin evanescent rash (diffuse macular, scarlatiniform, urticarial) | Generalized lymph node enlargement | ↑ Serum ferritin ↓ Glycosylate ferritin | Fulminant hepatitis |
| Leukocytes ≥ 10,000 mm3 Neutrophils ≥ 80% | Gastrointestinal symptoms (abdominal pain, hepato-splenomegaly) | ↑ D-dimer | Disseminated intravascular coagulation |
| Arthritis in one or more joints or Arthralgia | Heart involvement (pericarditis, myoca2-glycoproteinrditis, coronary aneurysm) | ↓ Fg | Thrombotic microangiopathy |
| Lung damage (pneumonia, pleurisy) | ↑ LDH, ↑ AST, ↑ ALT | Cardiac tamponade/myocarditis | |
| Myalgia/Myositis | Coagulation disorders | Acute respiratory failure syndrome | |
| Hepatitis | |||
| Uveitis (is quite rare) |
| Major criteria | Evanescent rash Arthritis | |||
| Minor criteria | Generalized adenomegaly and/or splenomegaly and/or hepatomegaly | Arthralgia | Serositis (pleural, pericardial or peritoneal) | Leukocytosis (≥15,000/mm3) with neutrophilia |
| Differential diagnosis for sJIA subtypes | (a) Psoriasis or a history of psoriasis in the patient or in a first-degree relative. (b) Arthritis in an HLA-B27-positive boy beginning after the sixth birthday. (c) Ankylosing spondylitis, enthesitis-related arthritis (ERA), sacroiliitis with inflammatory bowel disease or acute anterior uveitis, or a history of one of these disorders in a first-degree relative. (d) Association with 2 positive tests for immunoglobulin M rheumatoid factor (IgM-RF) at least 3 months apart. | |||
| Other entities excluded for positive diagnosis | Infections | Malignancy and autoimmune disease | Vasculitis | Autoinflammatory diseases with periodic familial fevers |
| Generalized infections: septicemia; Deep infections: osteomyelitis or abscesses; Causes of prolonged fever: endocarditis, yersiniosis, tuberculosis, brucellosis; Post-infectious arthritis: Group A beta-hemolytic Streptococci (GAS) Viruses: Epstein-Barr virus (EBV), Cytomegalovirus (CMV), Human Immunodeficiency Virus (HIV) parvovirus B19, hepatitis, measles; human herpesvirus 8 associated multicentric Castleman disease (HHV-8-associated MCD). | Leukemia, lymphoma, neuroblastoma etc. Polyarticular JIA with systemic manifestations or Systemic Lupus Erythematosus (SLE), juvenile dermatomyositis. | Polyarteritis nodosa, KD. | Mediterranean fever (FMF) caused by mutations of MEFV gene; tumor necrosis factor receptor-associated periodic fever syndrome (TRAPS), mevalonate kinase deficiency (MKD) by mutations of the mevalonate kinase gene; cryopyrin-associated periodic syndromes (CAPS), by mutations of NLRP3; periodic fever, aphthosis, pharyngitis and adenitis (PFAPA). | |
| High, Prolonged Fever of Known Origin in a Known or Suspected sJIA Patient may be Considered an Essential Element in the Diagnosis of MAS if Associated with the Following Criteria: | |
|---|---|
| Ferritin > 684 ng/mL and Any Two of the Following Criteria: | |
| Platelet count ≤ 181 × 109/L | |
| Aspartate aminotransferase (AST) > 48 unites/L | |
| Triglycerides > 156 ng/dL | |
| Fibrinogen ≤ 360 ng/dL | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ailioaie, L.M.; Ailioaie, C.; Litscher, G. Implications of SARS-CoV-2 Infection in Systemic Juvenile Idiopathic Arthritis. Int. J. Mol. Sci. 2022, 23, 4268. https://doi.org/10.3390/ijms23084268
Ailioaie LM, Ailioaie C, Litscher G. Implications of SARS-CoV-2 Infection in Systemic Juvenile Idiopathic Arthritis. International Journal of Molecular Sciences. 2022; 23(8):4268. https://doi.org/10.3390/ijms23084268
Chicago/Turabian StyleAilioaie, Laura Marinela, Constantin Ailioaie, and Gerhard Litscher. 2022. "Implications of SARS-CoV-2 Infection in Systemic Juvenile Idiopathic Arthritis" International Journal of Molecular Sciences 23, no. 8: 4268. https://doi.org/10.3390/ijms23084268
APA StyleAilioaie, L. M., Ailioaie, C., & Litscher, G. (2022). Implications of SARS-CoV-2 Infection in Systemic Juvenile Idiopathic Arthritis. International Journal of Molecular Sciences, 23(8), 4268. https://doi.org/10.3390/ijms23084268