
2-Pyridine Carboxaldehyde for Semi-Automated Soft Spot Identification in Cyclic Peptides



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
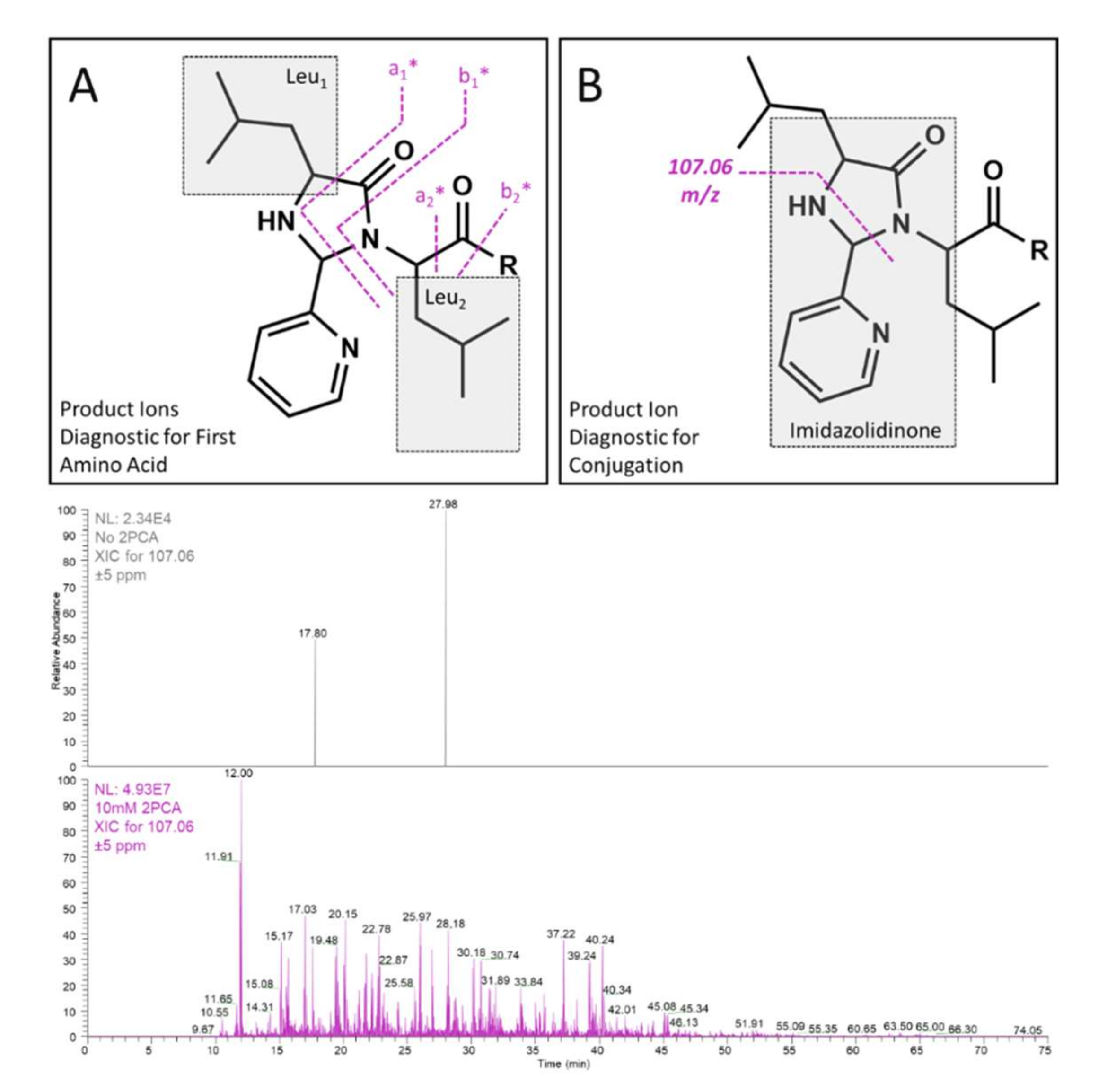
2.1. Selectivity
2.2. Diagnostic Fragmentation
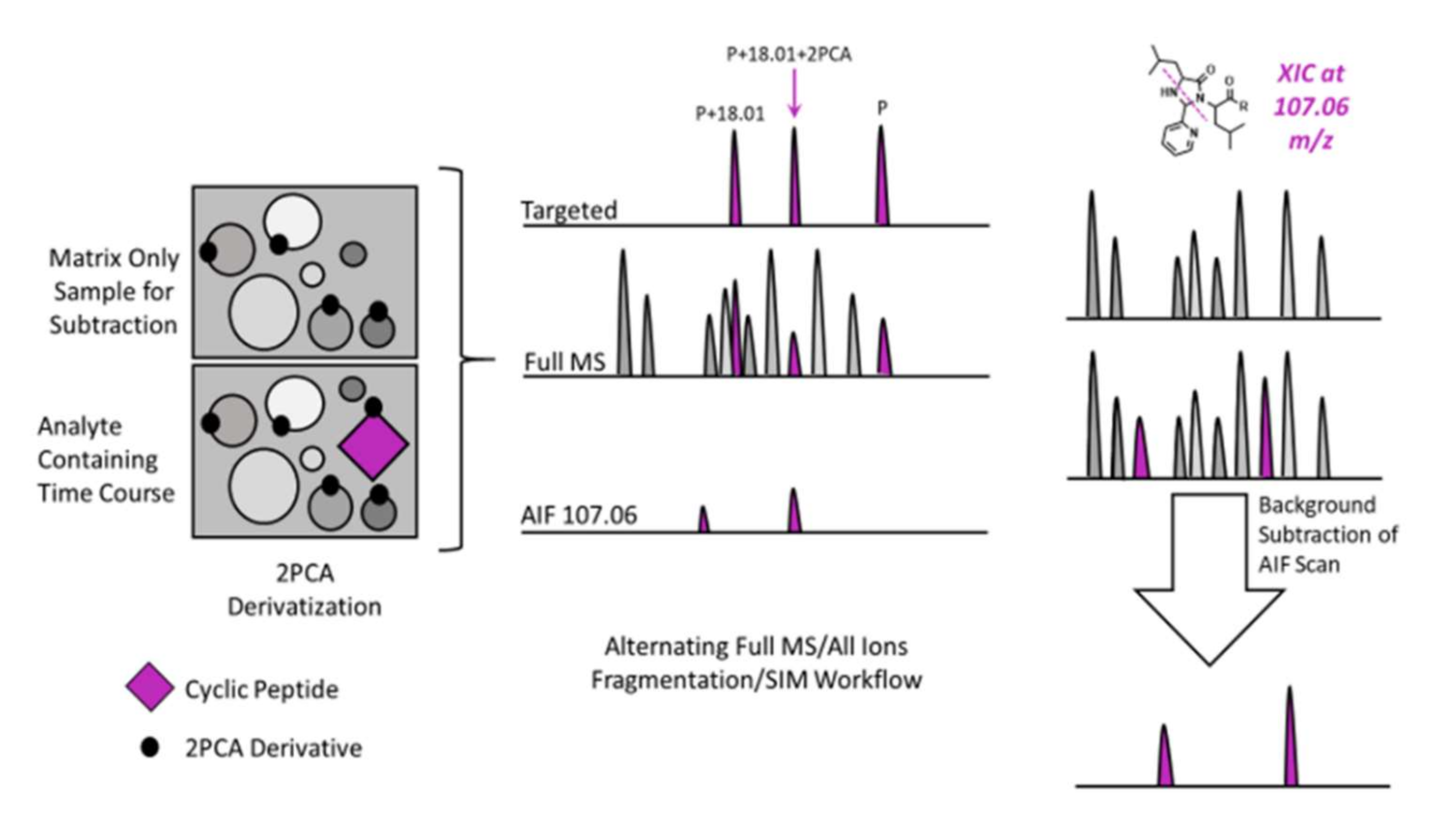
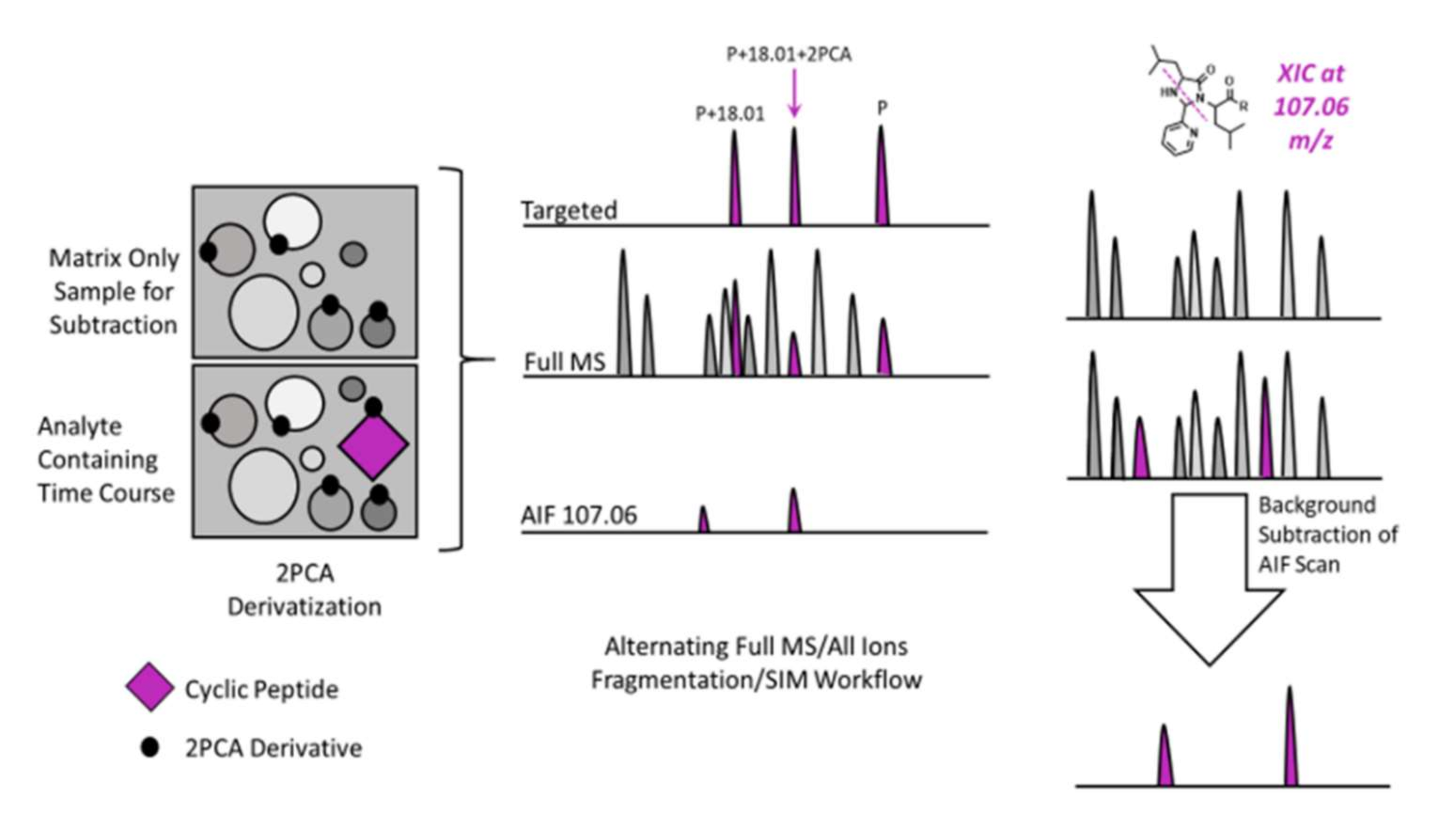
2.3. Background Subtraction and AIF Workflow
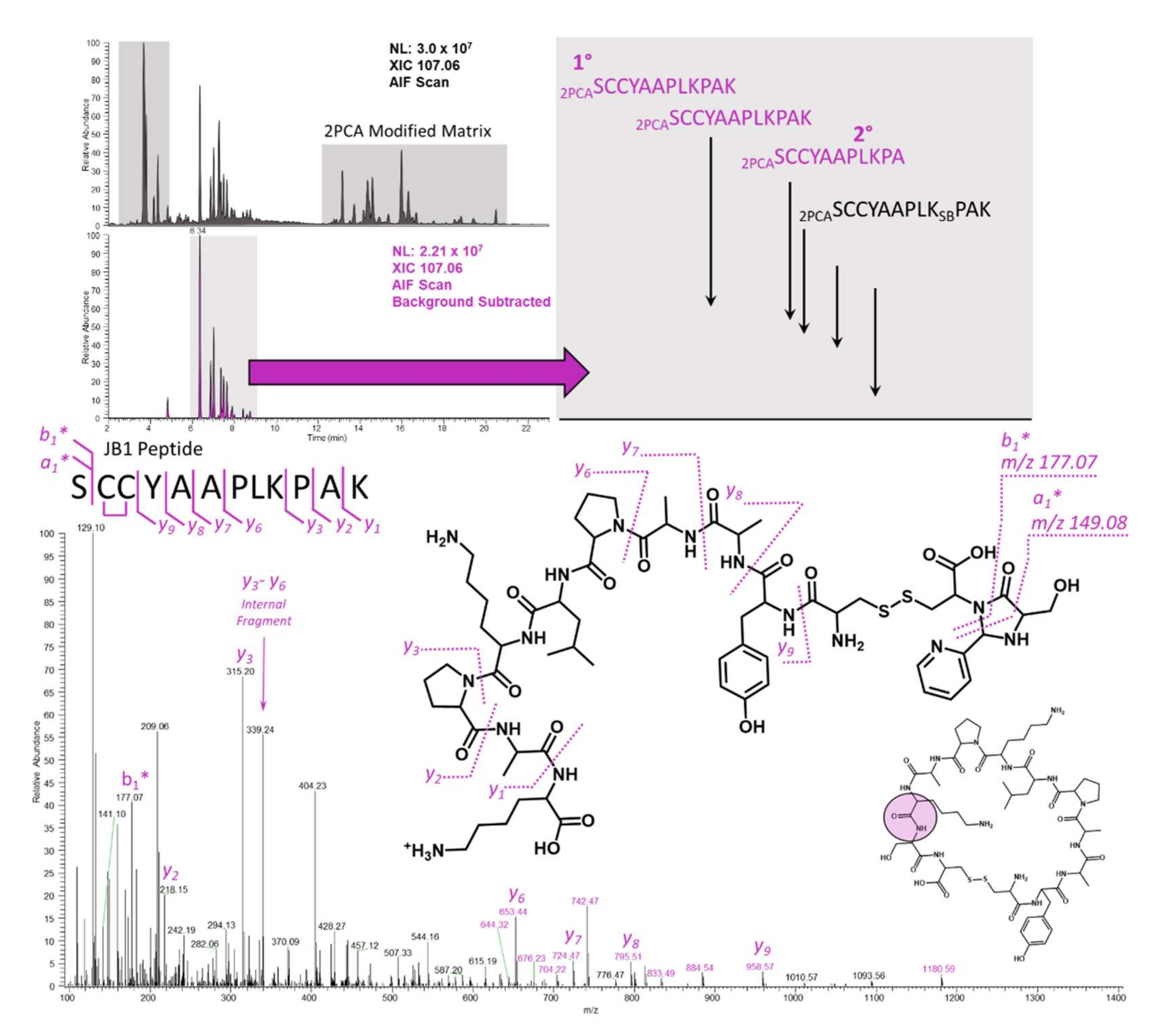
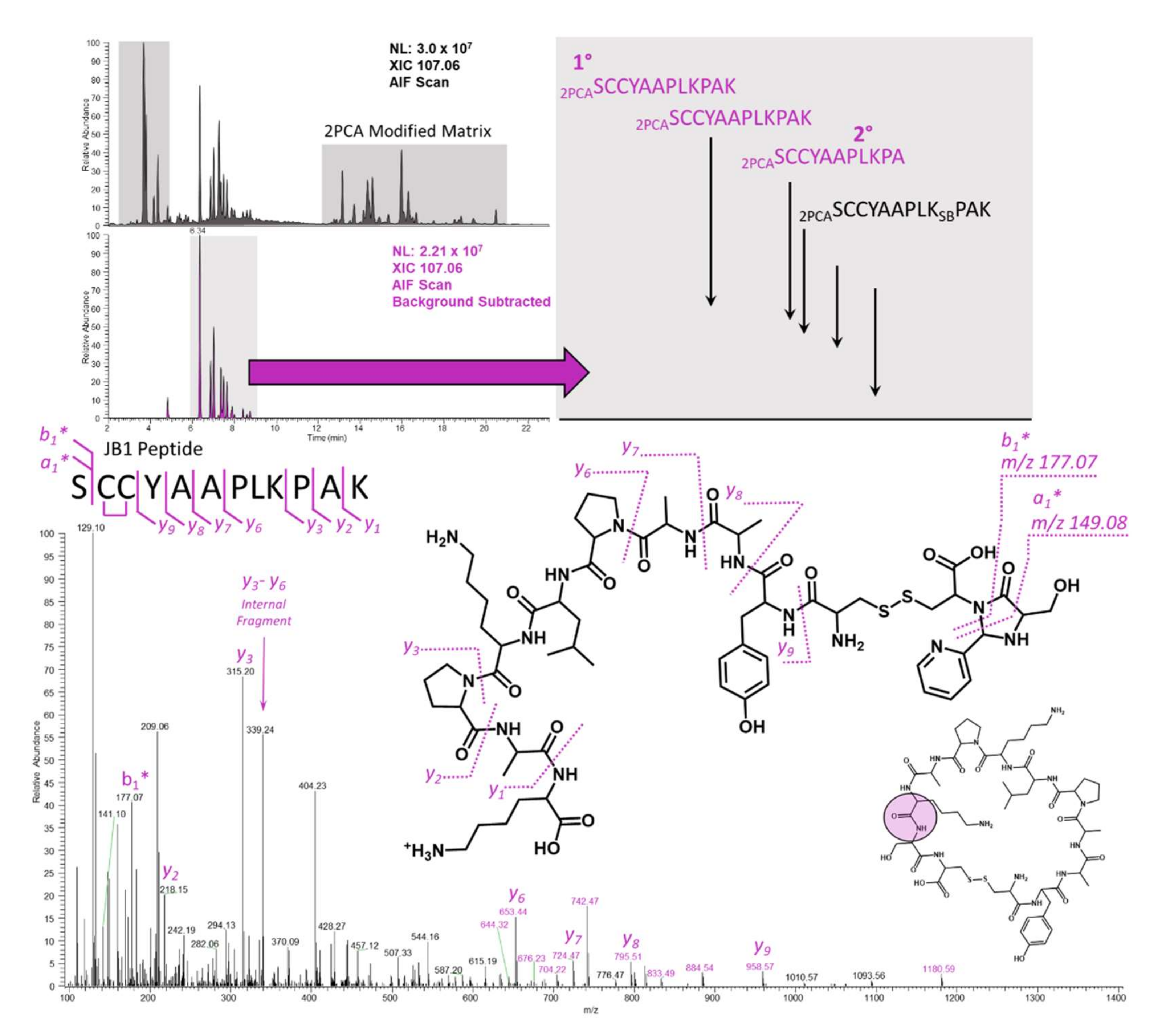
2.4. JB1
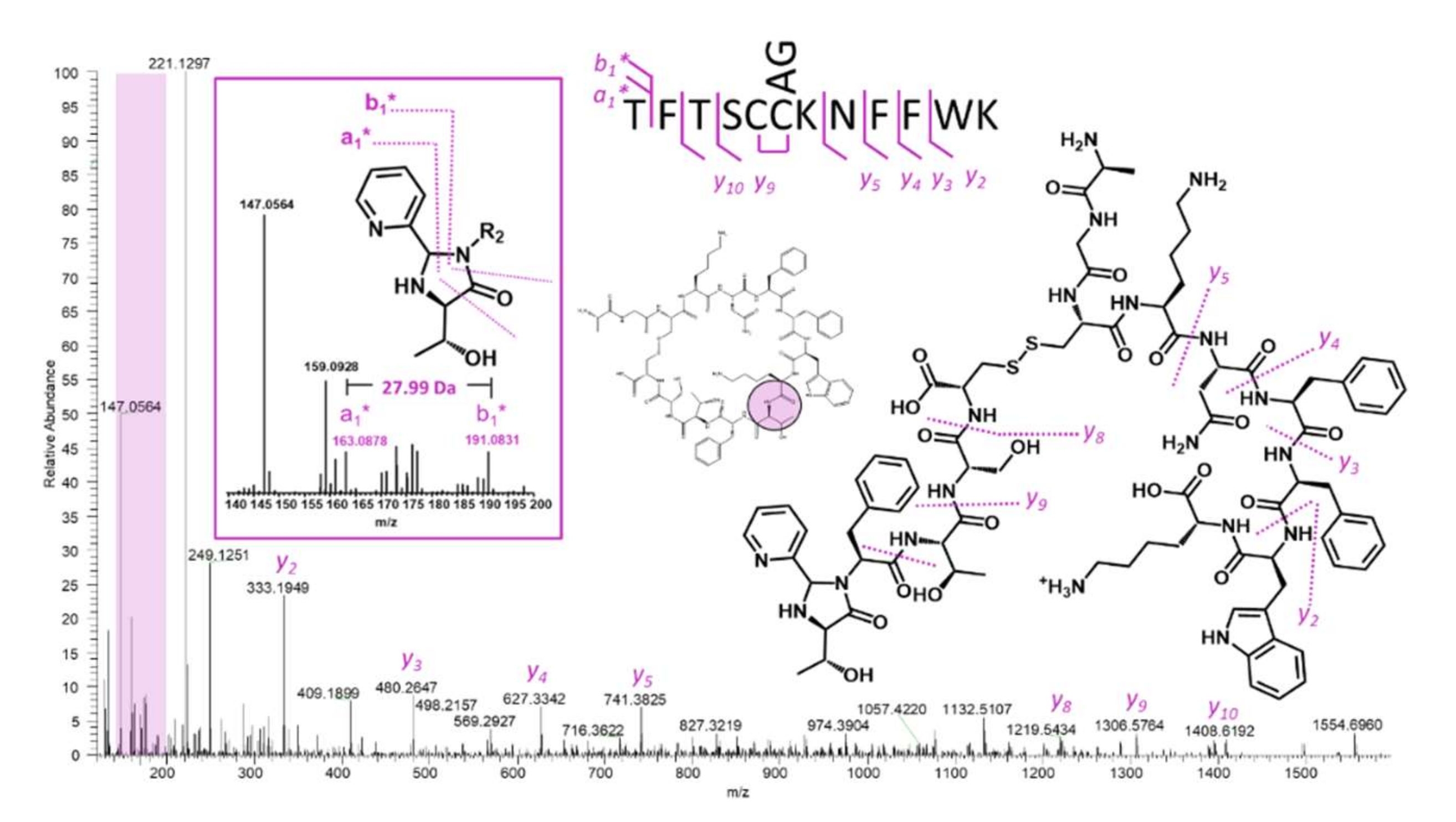
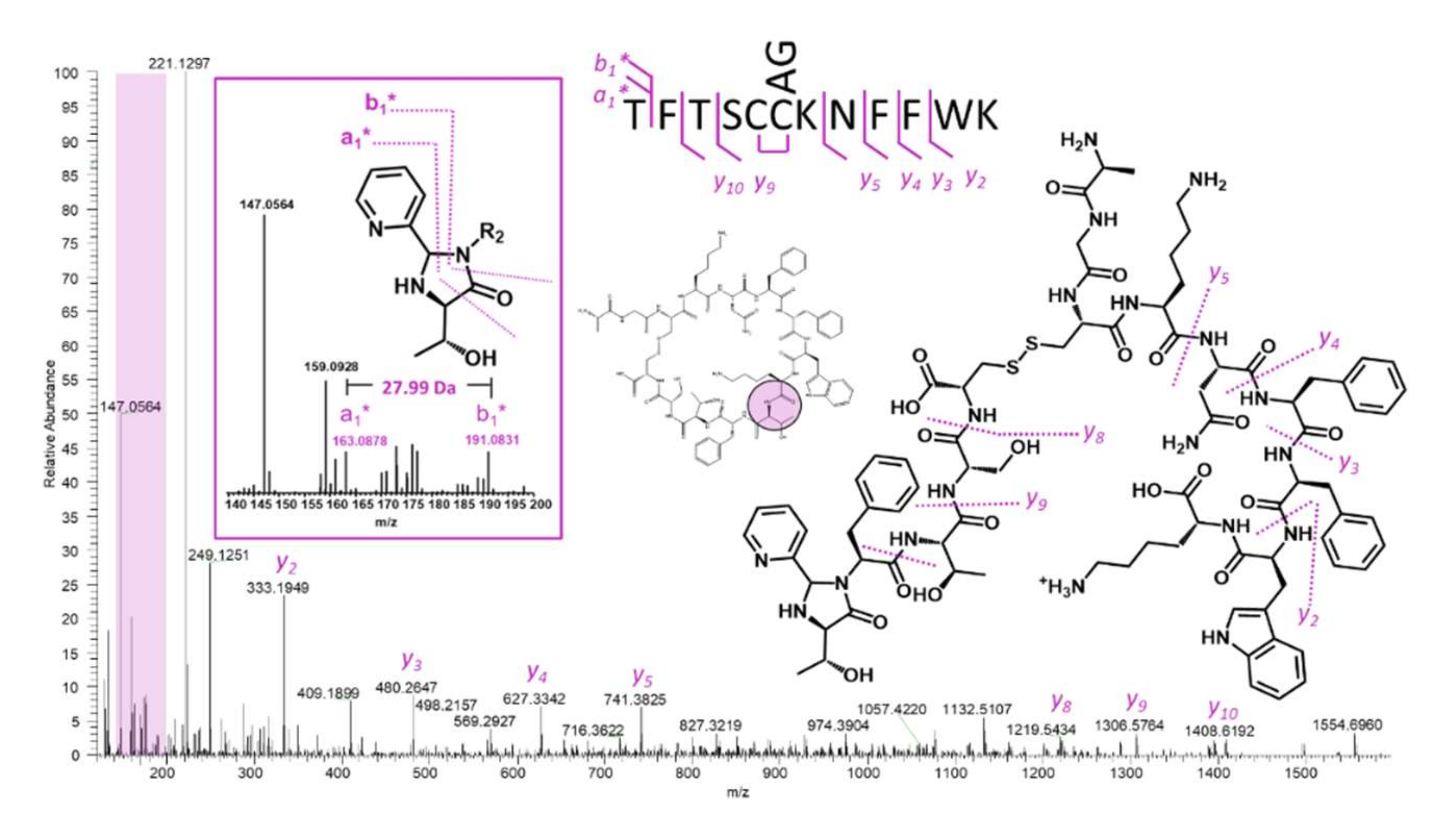
2.5. Somatostatin
3. Materials and Methods
3.1. Materials
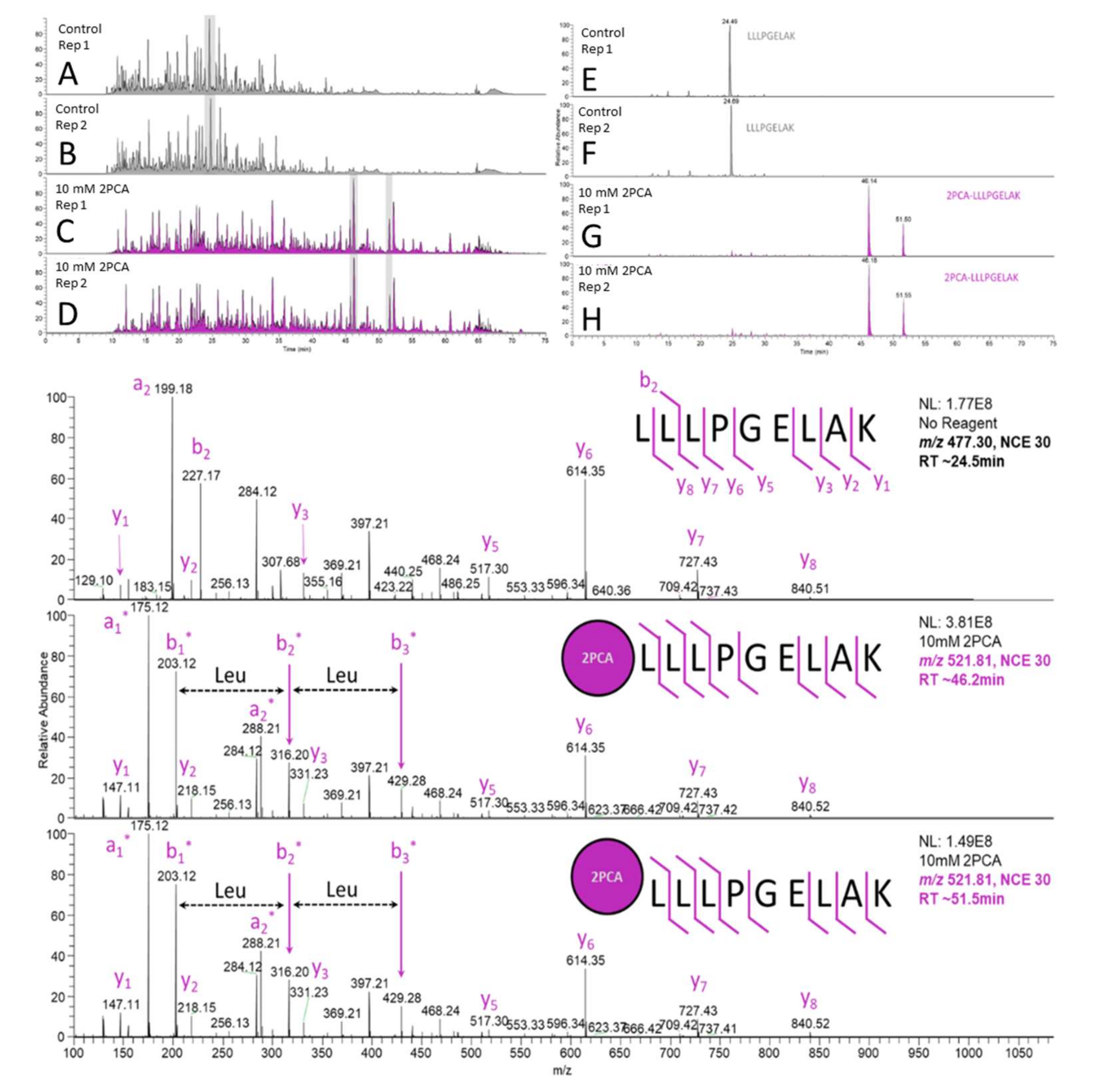
3.2. Complex Mixture Derivatization
3.3. Metabolite Identification Time Course Automated Sample Preparation
3.4. LC-MS/MS
3.5. Background Subtraction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 2021, 184, 3022–3040.e28. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Hipolito, C.J.; Suga, H. Ribosomal production and in vitro selection of natural product-like peptidomimetics: The FIT and RaPID systems. Curr. Opin. Chem. Biol. 2012, 16, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Bessho, Y.; Wei, K.; Szostak, J.W.; Suga, H. Ribozyme-catalyzed tRNA aminoacylation. Nat. Struct. Biol. 2000, 7, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Nagano, M.; Huang, Y.; Obexer, R.; Suga, H. One-Pot In Vitro Ribosomal Synthesis of Macrocyclic Depsipeptides. J. Am. Chem. Soc. 2021, 143, 4741–4750. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.M.; Passioura, T.; Suga, H. Nonproteinogenic deep mutational scanning of linear and cyclic peptides. Proc. Natl. Acad. Sci. USA 2018, 115, 10959–10964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, T.; Goto, Y.; Suga, H. In Vitro Selection of Thioether-Closed Macrocyclic Peptide Ligands by Means of the RaPID System. Methods Mol. Biol. 2022, 2371, 247–259. [Google Scholar] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2017, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Fridman, A.; Bagchi, A.; Xu, S.; Kwasnjuk, K.A.; Lu, P.; Cancilla, M.T. Metabolite Identification of Therapeutic Peptides and Proteins by Top-down Differential Mass Spectrometry and Metabolite Database Matching. Anal. Chem. 2020, 92, 8298–8305. [Google Scholar] [CrossRef] [PubMed]
- Mørtz, E.; O’Connor, P.B.; Roepstorff, P.; Kelleher, N.L.; Wood, T.D.; McLafferty, F.W.; Mann, M. Sequence tag identification of intact proteins by matching tanden mass spectral data against sequence data bases. Proc. Natl. Acad. Sci. USA 1996, 93, 8264–8267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Shiyanov, P.; Green, K.B. Top-down mass spectrometry of intact phosphorylated β-casein: Correlation between the precursor charge state and internal fragments. Biol. Mass Spectrom. 2019, 54, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Chin, S.; Chen, T.; Hannoush, R.N.; Crittenden, C.M. Tracking internal and external ions for constrained peptides leads to enhanced sequence coverage and disulfide bond deciphering. J. Pharm. Biomed Anal. 2021, 195, 113893. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.; Dolan, N.S.; Wucherer, K.; Munch, H.K.; Francis, M.B. Site-Selective Protein Immobilization on Polymeric Supports through N-Terminal Imidazolidinone Formation. Biomacromolecules 2019, 20, 3933–3939. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.I.; Munch, H.F.; Moore, T.; Francis, M.B. One-step site-specific modification of native proteins with 2-pyridinecarboxyaldehydes. Nat. Chem. Biol. 2015, 11, 326–331. [Google Scholar] [CrossRef]
- Zhang, H.; Gan, J.; Shu, Y.-Z.; Humphreys, W.G. High-Resolution Mass Spectrometry-Based Background Subtraction for Identifying Protein Modifications in a Complex Biological System: Detection of Acetaminophen-Bound Microsomal Proteins Including Argininosuccinate Synthetase. Chem. Res. Toxicol. 2015, 28, 775–781. [Google Scholar] [CrossRef]
- Zhang, H.; Grubb, M.; Wu, W.; Josephs, J.; Humphreys, W.G. Algorithm for Thorough Background Subtraction of High-Resolution LC/MS Data: Application to Obtain Clean Product Ion Spectra from Nonselective Collision-Induced Dissociation Experiments. Anal. Chem. 2009, 81, 2695–2700. [Google Scholar] [CrossRef]
- Zhang, H.; Ma, L.; He, K.; Zhu, M. An algorithm for thorough background subtraction from high-resolution LC/MS data: Application to the detection of troglitazone metabolites in rat plasma, bile, and urine. J. Mass Spectrom. 2008, 43, 1191–1200. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, Y. An algorithm for thorough background subtraction from high-resolution LC/MS data: Application for detection of glutathione-trapped reactive metabolites. J. Mass Spectrom. 2008, 43, 1181–1190. [Google Scholar] [CrossRef]
- Zhang, H.; Patrone, L.; Kozlosky, J.; Tomlinson, L.; Cosma, G.; Horvath, J. Pooled sample strategy in conjunction with high-resolution liquid chromatography-mass spectrometry-based background subtraction to identify toxicological markers in dogs treated with ibipinabant. Anal. Chem. 2010, 82, 3834–3839. [Google Scholar] [CrossRef]
- Arnesen, T.; Van Damme, P.; Polevoda, B.; Helsens, K.; Evjenth, R.; Colaert, N.; Varhaug, J.E.; Vandekerckhove, J.; Lillehaug, J.R.; Sherman, F.; et al. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Nat. Acad. Sci. USA 2009, 106, 8157–8162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magin, R.; Deng, S.; Zhang, H.; Cooperman, B.; Marmorstein, R. Probing the interaction between NatA and the ribosome for co-translational protein acetylation. PLoS ONE 2017, 12, e0186278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGee, W.M.; McLuckey, S.A. The ornithine effect in peptide cation dissociation. Biol. Mass Spectrom. 2013, 48, 856–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foreman, D.J.; Dziekonski, E.T.; McLuckey, S.A. Maximizing Selective Cleavages at Aspartic Acid and Proline Residues for the Identification of Intact Proteins. J. Am. Soc. Mass Spectrom. 2018, 30, 34–44. [Google Scholar] [CrossRef]
- Foreman, D.J.; Parsley, N.C.; Lawler, J.T.; Aryal, U.K.; Hicks, L.M.; McLuckey, S.A. Gas-Phase Sequencing of Cyclotides: Introduction of Selective Ring Opening at Dehydroalanine via Ion/Ion Reaction. Anal. Chem. 2019, 91, 15608–15616. [Google Scholar] [CrossRef]
- Zenaidee, M.A.; Wei, B.; Lantz, C.; Wu, H.T.; Lambeth, T.R.; Diedrich, J.K.; Loo, R.R.O.; Julian, R.R.; Loo, J.A. Internal Fragments Generated from Different Top-Down Mass Spectrometry Fragmentation Methods Extend Protein Sequence Coverage. J. Am. Soc. Mass Spectrom. 2021, 32, 1752–1758. [Google Scholar] [CrossRef]
- Schmitt, N.D.; Berger, J.M.; Conway, J.B.; Agar, J.N. Increasing Top-Down Mass Spectrometry Sequence Coverage by an Order of Magnitude through Opti-mized Internal Fragment Generation and Assignment. Anal. Chem. 2021, 93, 6355–6362. [Google Scholar] [CrossRef]
- Bouchoux, G. From the mobile proton to wandering hydride ion: Mechanistic aspects of gas-phase ion chemistry. Biol. Mass Spectrom. 2013, 48, 505–518. [Google Scholar] [CrossRef]
- Shaw, J.B.; Li, W.; Holden, D.D.; Zhang, Y.; Griep-Raming, J.; Fellers, R.T.; Early, B.P.; Thomas, P.M.; Kelleher, N.L.; Brodbelt, J.S. Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J. Am. Chem. Soc. 2013, 135, 12646–12651. [Google Scholar] [CrossRef] [Green Version]
- Nawatha, M.; Rogers, J.M.; Bonn, S.M.; Livneh, I.; Lemma, B.; Mali, S.M.; Vamisetti, G.B.; Sun, H.; Bercovich, B.; Huang, Y.; et al. De novo macrocyclic peptides that specifically modulate Lys48-linked ubiquitin chains. Nat. Chem. 2019, 11, 644–652. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Chacko, S.; Cannon, J.R. 2-Pyridine Carboxaldehyde for Semi-Automated Soft Spot Identification in Cyclic Peptides. Int. J. Mol. Sci. 2022, 23, 4269. https://doi.org/10.3390/ijms23084269
Zhang H, Chacko S, Cannon JR. 2-Pyridine Carboxaldehyde for Semi-Automated Soft Spot Identification in Cyclic Peptides. International Journal of Molecular Sciences. 2022; 23(8):4269. https://doi.org/10.3390/ijms23084269
Chicago/Turabian StyleZhang, Haiying, Silvi Chacko, and Joe R. Cannon. 2022. "2-Pyridine Carboxaldehyde for Semi-Automated Soft Spot Identification in Cyclic Peptides" International Journal of Molecular Sciences 23, no. 8: 4269. https://doi.org/10.3390/ijms23084269
APA StyleZhang, H., Chacko, S., & Cannon, J. R. (2022). 2-Pyridine Carboxaldehyde for Semi-Automated Soft Spot Identification in Cyclic Peptides. International Journal of Molecular Sciences, 23(8), 4269. https://doi.org/10.3390/ijms23084269