
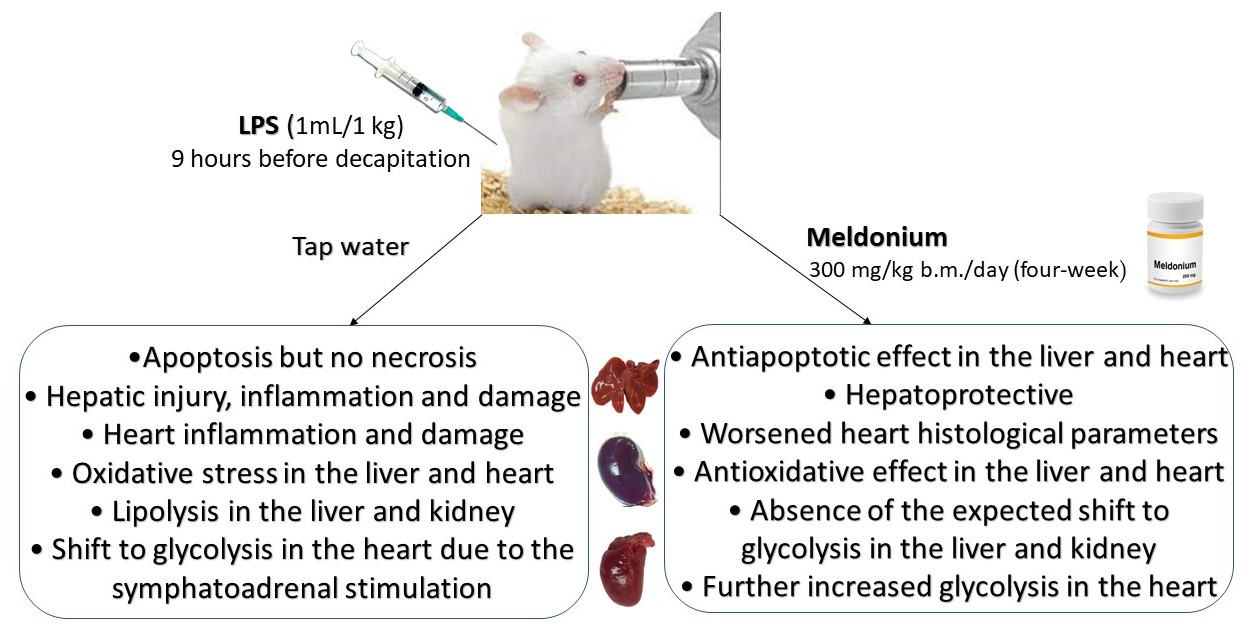
The Effects of a Meldonium Pre-Treatment on the Course of the LPS-Induced Sepsis in Rats

 , , ,
, , ,  ,
,  ,
,  ,
,  ,
, 
 ,
,  ,
, 
Abstract
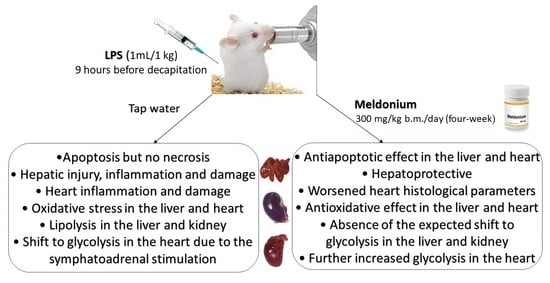
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Animals and Treatments
3.2. Serum and Tissue Collection
3.3. Biochemical Analysis
3.3.1. Serum Analysis
3.3.2. Tissue Analysis of L Carnitine, Lactic Acid, Glucose, Glycerol and Inositol
3.3.3. Lipidomics Tissue Preparation
3.3.4. Tissue Fatty Acids Determination
3.3.5. Tissue Triglycerides Determination
3.4. Determination of Oxidative Stress Biomarkers
3.5. Determination of Adrenaline and Noradrenaline Content
3.5.1. Determination of Adrenaline and Noradrenaline Content in Serum
3.5.2. Determination of Adrenaline and Noradrenaline Content in Adrenal Glands
3.6. Western Immunoblot Analysis
3.7. Histology Analysis
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shrum, B.; Anantha, R.V.; Xu, S.X.; Donnelly, M.; Haeryfar, S.M.; McCormick, J.K.; Mele, T. A robust scoring system to evaluate sepsis severity in an animal model. BMC Res. Notes 2014, 7, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Vincent, J.L.; Jones, G.; David, S.; Olariu, E.; Cadwell, K.K. Frequency and mortality of septic shock in Europe and North America: A systematic review and meta-analysis. Crit. Care 2019, 23, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjakste, N.; Gutcaits, A.; Kalvinsh, I. Mildronate: An antiischemic drug for neurological indications. CNS Drug Rev. 2005, 11, 151–168. [Google Scholar] [CrossRef]
- Durasevic, S.; Ruzicic, A.; Lakic, I.; Tosti, T.; Durovic, S.; Glumac, S.; Pavlovic, S.; Borkovic-Mitic, S.; Grigorov, I.; Stankovic, S.; et al. The Effects of a Meldonium Pre-Treatment on the Course of the Faecal-Induced Sepsis in Rats. Int. J. Mol. Sci. 2021, 22, 9698. [Google Scholar] [CrossRef] [PubMed]
- Durasevic, S.; Stojkovic, M.; Sopta, J.; Pavlovic, S.; Borkovic-Mitic, S.; Ivanovic, A.; Jasnic, N.; Tosti, T.; Durovic, S.; Dordevic, J.; et al. The effects of meldonium on the acute ischemia/reperfusion liver injury in rats. Sci. Rep. 2021, 11, 1305. [Google Scholar] [CrossRef]
- Durasevic, S.; Stojkovic, M.; Bogdanovic, L.; Pavlovic, S.; Borkovic-Mitic, S.; Grigorov, I.; Bogojevic, D.; Jasnic, N.; Tosti, T.; Durovic, S.; et al. The Effects of Meldonium on the Renal Acute Ischemia/Reperfusion Injury in Rats. Int. J. Mol. Sci. 2019, 20, 5747. [Google Scholar] [CrossRef] [Green Version]
- Berlato, D.G.; Bairros, A.V. Meldonium: Pharmacological, toxicological, and analytical aspects. Toxicol. Res. Appl. 2020, 4, 2397847320915143. [Google Scholar] [CrossRef]
- Porter, C.; Constantin-Teodosiu, D.; Constantin, D.; Leighton, B.; Poucher, S.M.; Greenhaff, P.L. Muscle carnitine availability plays a central role in regulating fuel metabolism in the rodent. J. Physiol. 2017, 595, 5765–5780. [Google Scholar] [CrossRef]
- Dambrova, M.; Makrecka-Kuka, M.; Vilskersts, R.; Makarova, E.; Kuka, J.; Liepinsh, E. Pharmacological effects of meldonium: Biochemical mechanisms and biomarkers of cardiometabolic activity. Pharmacol. Res. 2016, 113, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Wasyluk, W.; Zwolak, A. Metabolic Alterations in Sepsis. J. Clin. Med. 2021, 10, 2412. [Google Scholar] [CrossRef]
- Ostergaard, L.; Granfeldt, A.; Secher, N.; Tietze, A.; Iversen, N.K.; Jensen, M.S.; Andersen, K.K.; Nagenthiraja, K.; Gutierrez-Lizardi, P.; Mouridsen, K.; et al. Microcirculatory dysfunction and tissue oxygenation in critical illness. Acta Anaesthesiol. Scand. 2015, 59, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Arulkumaran, N.; Deutschman, C.S.; Pinsky, M.R.; Zuckerbraun, B.; Schumacker, P.T.; Gomez, H.; Gomez, A.; Murray, P.; Kellum, J.A.; Workgroup, A.X. Mitochondrial Function in Sepsis. Shock 2016, 45, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Suetrong, B.; Walley, K.R. Lactic Acidosis in Sepsis: It’s Not All Anaerobic: Implications for Diagnosis and Management. Chest 2016, 149, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Gong, C.; Fu, J.; Liu, X.; Bi, H.; Cheng, Y.; Liu, Y.; Tang, Y.; Wang, D. Evaluation of 2 Rat Models for Sepsis Developed by Improved Cecal Ligation/Puncture or Feces Intraperitoneal-Injection. Med. Sci. Monit. 2020, 26, e919054. [Google Scholar] [CrossRef]
- Starr, M.E.; Steele, A.M.; Saito, M.; Hacker, B.J.; Evers, B.M.; Saito, H. A new cecal slurry preparation protocol with improved long-term reproducibility for animal models of sepsis. PLoS ONE 2014, 9, e115705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Zou, Y.; Cao, Z. Comparison of two different models of sepsis induced by cecal ligation and puncture in rats. J. Surg. Res. 2018, 229, 277–282. [Google Scholar] [CrossRef]
- Osuchowski, M.F.; Ayala, A.; Bahrami, S.; Bauer, M.; Boros, M.; Cavaillon, J.M.; Chaudry, I.H.; Coopersmith, C.M.; Deutschman, C.S.; Drechsler, S.; et al. Minimum Quality Threshold in Pre-Clinical Sepsis Studies (MQTiPSS): An International Expert Consensus Initiative for Improvement of Animal Modeling in Sepsis. Shock 2018, 50, 377–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli-de-Figueiredo, L.F.; Garrido, A.G.; Nakagawa, N.; Sannomiya, P. Experimental models of sepsis and their clinical relevance. Shock 2008, 30 (Suppl. 1), 53–59. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.J.; Seymour, C.W.; Rosengart, M.R. Current Murine Models of Sepsis. Surg. Infect. 2016, 17, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Korneev, K.V. Mouse Models of Sepsis and Septic Shock. Mol. Biol. 2019, 53, 799–814. [Google Scholar] [CrossRef]
- Lepper, P.M.; Held, T.K.; Schneider, E.M.; Bolke, E.; Gerlach, H.; Trautmann, M. Clinical implications of antibiotic-induced endotoxin release in septic shock. Intensive Care Med. 2002, 28, 824–833. [Google Scholar] [CrossRef]
- Vilskersts, R.; Kigitovica, D.; Korzh, S.; Videja, M.; Vilks, K.; Cirule, H.; Skride, A.; Makrecka-Kuka, M.; Liepinsh, E.; Dambrova, M. Protective Effects of Meldonium in Experimental Models of Cardiovascular Complications with a Potential Application in COVID-19. Int. J. Mol. Sci. 2021, 23, 45. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Stevens, N.E.; Chapman, M.J.; Fraser, C.K.; Kuchel, T.R.; Hayball, J.D.; Diener, K.R. Therapeutic targeting of HMGB1 during experimental sepsis modulates the inflammatory cytokine profile to one associated with improved clinical outcomes. Sci. Rep. 2017, 7, 5850. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. Regulation of NF-kappaB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuele, S.; Celesia, A.; D’Anneo, A.; Lauricella, M.; Carlisi, D.; De Blasio, A.; Giuliano, M. The Good and Bad of Nrf2: An Update in Cancer and New Perspectives in COVID-19. Int. J. Mol. Sci. 2021, 22, 7963. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.H.C.; Sharma, N.; Kwong, A.C.; Dwivedi, D.J.; Khan, M.; Grin, P.M.; Fox-Robichaud, A.E.; Liaw, P.C. Body temperature and mouse scoring systems as surrogate markers of death in cecal ligation and puncture sepsis. Intensive Care Med. Exp. 2018, 6, 20. [Google Scholar] [CrossRef] [Green Version]
- Hall, P.; Cash, J. What is the real function of the liver ‘function’ tests? Ulster Med. J. 2012, 81, 30–36. [Google Scholar]
- Pandya, D.; Nagrajappa, A.K.; Ravi, K.S. Assessment and Correlation of Urea and Creatinine Levels in Saliva and Serum of Patients with Chronic Kidney Disease, Diabetes and Hypertension- A Research Study. J. Clin. Diagn. Res. 2016, 10, ZC58–ZC62. [Google Scholar] [CrossRef] [PubMed]
- Schillaci, G.; Reboldi, G.; Verdecchia, P. High-normal serum creatinine concentration is a predictor of cardiovascular risk in essential hypertension. Arch. Intern. Med. 2001, 161, 886–891. [Google Scholar] [CrossRef] [Green Version]
- Fine, L.G.; Salehmoghaddam, S. Proteinuria. In Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed.; Walker, H.K., Hall, W.D., Hurst, J.W., Eds.; Butterworths: Boston, MA, USA, 1990. [Google Scholar]
- Glavind, E.; Aagaard, N.K.; Gronbaek, H.; Moller, H.J.; Orntoft, N.W.; Vilstrup, H.; Thomsen, K.L. Alcoholic Hepatitis Markedly Decreases the Capacity for Urea Synthesis. PLoS ONE 2016, 11, e0158388. [Google Scholar] [CrossRef]
- Nielsen, S.S.; Grofte, T.; Tygstrup, N.; Vilstrup, H. Effect of lipopolysaccharide on in vivo and genetic regulation of rat urea synthesis. Liver Int. 2005, 25, 177–183. [Google Scholar] [CrossRef]
- Kou, X.; Zhu, J.; Xie, X.; Hao, M.; Zhao, Y. The protective effect of glycyrrhizin on hepatic ischemia-reperfusion injury in rats and possible related signal pathway. Iran J. Basic Med. Sci. 2020, 23, 1232–1238. [Google Scholar] [CrossRef]
- Pickkers, P.; Darmon, M.; Hoste, E.; Joannidis, M.; Legrand, M.; Ostermann, M.; Prowle, J.R.; Schneider, A.; Schetz, M. Acute kidney injury in the critically ill: An updated review on pathophysiology and management. Intensive Care Med. 2021, 47, 835–850. [Google Scholar] [CrossRef] [PubMed]
- Tschope, C.; Ammirati, E.; Bozkurt, B.; Caforio, A.L.P.; Cooper, L.T.; Felix, S.B.; Hare, J.M.; Heidecker, B.; Heymans, S.; Hubner, N.; et al. Myocarditis and inflammatory cardiomyopathy: Current evidence and future directions. Nat. Rev. Cardiol. 2021, 18, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhu, S.; Zhou, R.; Li, W.; Sama, A.E. Therapeutic potential of HMGB1-targeting agents in sepsis. Expert Rev. Mol. Med. 2008, 10, e32. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Diener, K.R.; Al-Dasooqi, N.; Lousberg, E.L.; Hayball, J.D. The multifunctional alarmin HMGB1 with roles in the pathophysiology of sepsis and cancer. Immunol. Cell Biol. 2013, 91, 443–450. [Google Scholar] [CrossRef]
- Yu, Y.; Tang, D.; Kang, R. Oxidative stress-mediated HMGB1 biology. Front. Physiol. 2015, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.-L.; Moreno, R.; Takala, J.; Willatts, S.; De Mendonça, A.; Bruining, H.; Reinhart, C.; Suter, P.; Thijs, L.G. The SOFA (Sepsis-Related Organ Failure Assessment) Score to Describe Organ Dysfunction/Failure; Springer: Berlin/Heidelberg, Germany, 1996. [Google Scholar]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef]
- Old, L.J. Tumor necrosis factor. Sci. Am. 1988, 258, 59–75. [Google Scholar] [CrossRef]
- Waters, J.P.; Pober, J.S.; Bradley, J.R. Tumour necrosis factor in infectious disease. J. Pathol. 2013, 230, 132–147. [Google Scholar] [CrossRef]
- Devin, A.; Lin, Y.; Yamaoka, S.; Li, Z.; Karin, M.; Liu, Z.-G. The α and β subunits of IκB kinase (IKK) mediate TRAF2-dependent IKK recruitment to tumor necrosis factor (TNF) receptor 1 in response to TNF. Mol. Cell. Biol. 2001, 21, 3986–3994. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Chang, A.; Hack, B.K.; Eadon, M.T.; Alper, S.L.; Cunningham, P.N. TNF-mediated damage to glomerular endothelium is an important determinant of acute kidney injury in sepsis. Kidney Int. 2014, 85, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.L.; Shi, Y.Z.; Chen, G.G.; Wang, L.L.; Zheng, M.Z.; Jin, H.F.; Chen, Y.Y. TNF-α induces Drp1-mediated mitochondrial fragmentation during inflammatory cardiomyocyte injury. Int. J. Mol. Med. 2018, 41, 2317–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Williams-Skipp, C.; Tao, Y.; Schleicher, M.S.; Cano, L.L.; Duke, R.C.; Scheinman, R.I. NF-kappaB functions as both a proapoptotic and antiapoptotic regulatory factor within a single cell type. Cell Death Differ. 1999, 6, 570–582. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.H.B. Release of cardiac troponin from healthy and damaged myocardium. Front. Lab. Med. 2017, 1, 144–150. [Google Scholar] [CrossRef]
- Hammarsten, O.; Mair, J.; Möckel, M.; Lindahl, B.; Jaffe, A.S. Possible mechanisms behind cardiac troponin elevations. Biomarkers 2018, 23, 725–734. [Google Scholar] [CrossRef] [Green Version]
- Gunne, S.; Heinicke, U.; Parnham, M.J.; Laux, V.; Zacharowski, K.; von Knethen, A. Nrf2-A Molecular Target for Sepsis Patients in Critical Care. Biomolecules 2020, 10, 1688. [Google Scholar] [CrossRef]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thimmulappa, R.K.; Scollick, C.; Traore, K.; Yates, M.; Trush, M.A.; Liby, K.T.; Sporn, M.B.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem. Biophys. Res. Commun. 2006, 351, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schobersberger, W.; Dunnwald, T.; Gmeiner, G.; Blank, C. Story behind meldonium-from pharmacology to performance enhancement: A narrative review. Br. J. Sports Med. 2017, 51, 22–25. [Google Scholar] [CrossRef]
- Su, H.; Ma, Z.; Guo, A.; Wu, H.; Yang, X. Salvianolic acid B protects against sepsis-induced liver injury via activation of SIRT1/PGC-1alpha signaling. Exp. Ther. Med. 2020, 20, 2675–2683. [Google Scholar] [CrossRef] [PubMed]
- Di Cristo, F.; Finicelli, M.; Digilio, F.A.; Paladino, S.; Valentino, A.; Scialò, F.; D’Apolito, M.; Saturnino, C.; Galderisi, U.; Giordano, A. Meldonium improves Huntington’s disease mitochondrial dysfunction by restoring peroxisome proliferator-activated receptor γ coactivator 1α expression. J. Cell. Physiol. 2019, 234, 9233–9246. [Google Scholar] [CrossRef]
- Zhang, H.; Feng, Y.W.; Yao, Y.M. Potential therapy strategy: Targeting mitochondrial dysfunction in sepsis. Mil. Med. Res. 2018, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.L.; Ping, D.; Boss, J.M. Tumor necrosis factor alpha and interleukin-1beta regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-beta and NF-kappaB. Mol. Cell. Biol. 1997, 17, 6970–6981. [Google Scholar] [CrossRef] [Green Version]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besancon, F. NF-kappaB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFalpha-treated Ewing sarcoma cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Rojo, A.I.; Salinas, M.; Martin, D.; Perona, R.; Cuadrado, A. Regulation of Cu/Zn-superoxide dismutase expression via the phosphatidylinositol 3 kinase/Akt pathway and nuclear factor-kappaB. J. Neurosci. 2004, 24, 7324–7334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.Z.; Johnson, A.P.; Rando, T.A. NF kappa B and AP-1 mediate transcriptional responses to oxidative stress in skeletal muscle cells. Free Radic. Biol. Med. 2001, 31, 1405–1416. [Google Scholar] [CrossRef]
- Schreiber, J.; Jenner, R.G.; Murray, H.L.; Gerber, G.K.; Gifford, D.K.; Young, R.A. Coordinated binding of NF-kappaB family members in the response of human cells to lipopolysaccharide. Proc. Natl. Acad. Sci. USA 2006, 103, 5899–5904. [Google Scholar] [CrossRef] [Green Version]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Durasevic, S.; Jasnic, N.; Prokic, M.; Grigorov, I.; Martinovic, V.; Dordevic, J.; Pavlovic, S. The protective role of virgin coconut oil on the alloxan-induced oxidative stress in the liver, kidneys and heart of diabetic rats. Food Funct. 2019, 10, 2114–2124. [Google Scholar] [CrossRef]
- Lei, X.G.; Cheng, W.H.; McClung, J.P. Metabolic regulation and function of glutathione peroxidase-1. Annu. Rev. Nutr. 2007, 27, 41–61. [Google Scholar] [CrossRef]
- Hong, Y.A.; Park, C.W. Catalytic Antioxidants in the Kidney. Antioxidants 2021, 10, 130. [Google Scholar] [CrossRef]
- Chu, Y.; Lan, R.S.; Huang, R.; Feng, H.; Kumar, R.; Dayal, S.; Chan, K.S.; Dai, D.F. Glutathione peroxidase-1 overexpression reduces oxidative stress, and improves pathology and proteome remodeling in the kidneys of old mice. Aging Cell 2020, 19, e13154. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.W.; Kuo, M.C.; Kuo, H.T.; Chang, J.M.; Guh, J.Y.; Lai, Y.H.; Chen, H.C. Alterations of glomerular and extracellular levels of glutathione peroxidase in patients and experimental rats with diabetic nephropathy. J. Lab. Clin. Med. 2005, 145, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Buttari, B.; Panieri, E.; Profumo, E.; Saso, L. An Overview of Nrf2 Signaling Pathway and Its Role in Inflammation. Molecules 2020, 25, 5474. [Google Scholar] [CrossRef] [PubMed]
- Preau, S.; Vodovar, D.; Jung, B.; Lancel, S.; Zafrani, L.; Flatres, A.; Oualha, M.; Voiriot, G.; Jouan, Y.; Joffre, J.; et al. Energetic dysfunction in sepsis: A narrative review. Ann. Intensive Care 2021, 11, 104. [Google Scholar] [CrossRef]
- Cerra, F.B.; Siegel, J.H.; Coleman, B.; Border, J.R.; McMenamy, R.R. Septic autocannibalism. A failure of exogenous nutritional support. Ann. Surg. 1980, 192, 570–580. [Google Scholar] [CrossRef]
- Spanaki, A.M.; Tavladaki, T.; Dimitriou, H.; Kozlov, A.V.; Duvigneau, J.C.; Meleti, E.; Weidinger, A.; Papakonstantinou, E.; Briassoulis, G. Longitudinal Profiles of Metabolism and Bioenergetics Associated with Innate Immune Hormonal Inflammatory Responses and Amino-Acid Kinetics in Severe Sepsis and Systemic Inflammatory Response Syndrome in Children. JPEN J. Parenter. Enteral Nutr. 2018, 42, 1061–1074. [Google Scholar] [CrossRef]
- Simkhovich, B.Z.; Shutenko, Z.V.; Meirena, D.V.; Khagi, K.B.; Mezapuke, R.J.; Molodchina, T.N.; Kalvins, I.J.; Lukevics, E. 3-(2,2,2-Trimethylhydrazinium)propionate (THP)-a novel gamma-butyrobetaine hydroxylase inhibitor with cardioprotective properties. Biochem. Pharmacol. 1988, 37, 195–202. [Google Scholar] [CrossRef]
- Kuwajima, M.; Harashima, H.; Hayashi, M.; Ise, S.; Sei, M.; Lu, K.-M.; Kiwada, H.; Sugiyama, Y.; Shima, K. Pharmacokinetic analysis of the cardioprotective effect of 3-(2, 2, 2-trimethylhydrazinium) propionate in mice: Inhibition of carnitine transport in kidney. J. Pharmacol. Exp. Ther. 1999, 289, 93–102. [Google Scholar]
- Van Wyngene, L.; Vanderhaeghen, T.; Timmermans, S.; Vandewalle, J.; Van Looveren, K.; Souffriau, J.; Wallaeys, C.; Eggermont, M.; Ernst, S.; Van Hamme, E.; et al. Hepatic PPARalpha function and lipid metabolic pathways are dysregulated in polymicrobial sepsis. EMBO Mol. Med. 2020, 12, e11319. [Google Scholar] [CrossRef] [PubMed]
- De Montmollin, E.; Aboab, J.; Mansart, A.; Annane, D. Bench-to-bedside review: Beta-adrenergic modulation in sepsis. Crit. Care 2009, 13, 230. [Google Scholar] [CrossRef] [Green Version]
- Romero Mdel, M.; Sabater, D.; Fernandez-Lopez, J.A.; Remesar, X.; Alemany, M. Glycerol Production from Glucose and Fructose by 3T3-L1 Cells: A Mechanism of Adipocyte Defense from Excess Substrate. PLoS ONE 2015, 10, e0139502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hibuse, T.; Maeda, N.; Nakatsuji, H.; Tochino, Y.; Fujita, K.; Kihara, S.; Funahashi, T.; Shimomura, I. The heart requires glycerol as an energy substrate through aquaporin 7, a glycerol facilitator. Cardiovasc. Res. 2009, 83, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Gladka, M.; el Azzouzi, H.; De Windt, L.J.; da Costa Martins, P.A. Aquaporin 7: The glycerol aquaeductus in the heart. Cardiovasc. Res. 2009, 83, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambert, S.; Helies-Toussaint, C.; Grynberg, A. Regulation of intermediary metabolism in rat cardiac myocyte by extracellular glycerol. Biochim. Biophys. Acta 2005, 1736, 152–162. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Ishihama, S.; Yoshida, S.; Yoshida, T.; Mori, Y.; Ouchi, N.; Eguchi, S.; Sakaguchi, T.; Tsuda, T.; Kato, K.; Shimizu, Y.; et al. LPL/AQP7/GPD2 promotes glycerol metabolism under hypoxia and prevents cardiac dysfunction during ischemia. FASEB J. 2021, 35, e22048. [Google Scholar] [CrossRef] [PubMed]
- Gambert, S.; Helies-Toussaint, C.; Grynberg, A. Extracellular glycerol regulates the cardiac energy balance in a working rat heart model. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1600–H1606. [Google Scholar] [CrossRef] [Green Version]
- Endo, M.; Oyadomari, S.; Suga, M.; Mori, M.; Gotoh, T. The ER stress pathway involving CHOP is activated in the lungs of LPS-treated mice. J. Biochem. 2005, 138, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Han, L.; Gao, Y.; Li, L.; Shang, X.; Hu, W.; Xue, C. The endoplasmic reticulum stress-mediated apoptosis signal pathway is involved in sepsis-induced abnormal lymphocyte apoptosis. Eur. Surg. Res. 2008, 41, 219–225. [Google Scholar] [CrossRef]
- Esposito, V.; Grosjean, F.; Tan, J.; Huang, L.; Zhu, L.; Chen, J.; Xiong, H.; Striker, G.E.; Zheng, F. CHOP deficiency results in elevated lipopolysaccharide-induced inflammation and kidney injury. Am. J. Physiol. Renal Physiol. 2013, 304, F440–F450. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Mao, L.; Yang, L.; Zou, J.; Liu, K.; Liu, M.; Zhang, H.; Xiao, X.; Wang, K. Resveratrol protects against early polymicrobial sepsis-induced acute kidney injury through inhibiting endoplasmic reticulum stress-activated NF-kappaB pathway. Oncotarget 2017, 8, 36449–36461. [Google Scholar] [CrossRef]
- Sun, X.; Song, L.; Feng, S.; Li, L.; Yu, H.; Wang, Q.; Wang, X.; Hou, Z.; Li, X.; Li, Y.; et al. Fatty Acid Metabolism is Associated With Disease Severity After H7N9 Infection. EBioMedicine 2018, 33, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, H.T.; Chyuan, I.T.; Chen, W.Y. Shaping of Innate Immune Response by Fatty Acid Metabolite Palmitate. Cells 2019, 8, 1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLennan, P.L. Cardiac physiology and clinical efficacy of dietary fish oil clarified through cellular mechanisms of omega-3 polyunsaturated fatty acids. Eur. J. Appl. Physiol. 2014, 114, 1333–1356. [Google Scholar] [CrossRef] [Green Version]
- Orr, S.K.; Palumbo, S.; Bosetti, F.; Mount, H.T.; Kang, J.X.; Greenwood, C.E.; Ma, D.W.; Serhan, C.N.; Bazinet, R.P. Unesterified docosahexaenoic acid is protective in neuroinflammation. J. Neurochem. 2013, 127, 378–393. [Google Scholar] [CrossRef] [Green Version]
- Brenna, J.T.; Salem, N., Jr.; Sinclair, A.J.; Cunnane, S.C.; International Society for the Study of Fatty, A.; Lipids, I. alpha-Linolenic acid supplementation and conversion to n-3 long-chain polyunsaturated fatty acids in humans. Prostaglandins Leukot. Essent. Fatty Acids 2009, 80, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Domenichiello, A.F.; Kitson, A.P.; Bazinet, R.P. Is docosahexaenoic acid synthesis from alpha-linolenic acid sufficient to supply the adult brain? Prog. Lipid Res. 2015, 59, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Park, W.J.; Kothapalli, K.S.; Lawrence, P.; Tyburczy, C.; Brenna, J.T. An alternate pathway to long-chain polyunsaturates: The FADS2 gene product Delta8-desaturates 20:2n-6 and 20:3n-3. J. Lipid Res. 2009, 50, 1195–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepka, A.; Ochocinska, A.; Chojnowska, S.; Borzym-Kluczyk, M.; Skorupa, E.; Knas, M.; Waszkiewicz, N. Potential Role of L-Carnitine in Autism Spectrum Disorder. J. Clin. Med. 2021, 10, 1202. [Google Scholar] [CrossRef]
- Infante, J.P.; Huszagh, V.A. Secondary carnitine deficiency and impaired docosahexaenoic (22:6n-3) acid synthesis: A common denominator in the pathophysiology of diseases of oxidative phosphorylation and beta-oxidation. FEBS Lett. 2000, 468, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, M.; Wu, H.N.; Tanaka, M.; Tsuda, N.; Tantengco, O.A.G.; Matsushima, T.; Nakao, T.; Ishibe, T.; Sakata, I.; Yanagihara, I. A case series of the dynamics of lipid mediators in patients with sepsis. Acute Med. Surg. 2019, 6, 413–418. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Chen, H.; Vaziri, N.D.; Mao, J.R.; Zhang, L.; Bai, X.; Zhao, Y.Y. Metabolomic Signatures of Chronic Kidney Disease of Diverse Etiologies in the Rats and Humans. J. Proteome Res. 2016, 15, 3802–3812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Moraes, I.M.; Magno, F.; Campbell, C.; Estevam, P.; Araújo, C.; Bozza, P.; Gonçalves-de-Albuquerque, C.; Silva, A.; Neto, H.C.F. Increased survival after a cecal ligation and puncture-induced sepsis in mice consuming oleic acid. Critical Care 2010, 14, P26. [Google Scholar] [CrossRef] [Green Version]
- Goncalves-de-Albuquerque, C.F.; Medeiros-de-Moraes, I.M.; Oliveira, F.M.; Burth, P.; Bozza, P.T.; Castro Faria, M.V.; Silva, A.R.; Castro-Faria-Neto, H.C. Omega-9 Oleic Acid Induces Fatty Acid Oxidation and Decreases Organ Dysfunction and Mortality in Experimental Sepsis. PLoS ONE 2016, 11, e0153607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazaee, R.; Vinokurtseva, A.; McCaig, L.A.; Yamashita, C.; Hardy, D.B.; Arany, E.; Veldhuizen, R.A.W. The impact of maternal protein restriction during perinatal life on the response to a septic insult in adult rats. J. Dev. Orig. Health Dis. 2020, 12, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Tuck, M.K.; Chan, D.W.; Chia, D.; Godwin, A.K.; Grizzle, W.E.; Krueger, K.E.; Rom, W.; Sanda, M.; Sorbara, L.; Stass, S.; et al. Standard operating procedures for serum and plasma collection: Early detection research network consensus statement standard operating procedure integration working group. J. Proteome Res. 2009, 8, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerard-Monnier, D.; Erdelmeier, I.; Regnard, K.; Moze-Henry, N.; Yadan, J.C.; Chaudiere, J. Reactions of 1-methyl-2-phenylindole with malondialdehyde and 4-hydroxyalkenals. Analytical applications to a colorimetric assay of lipid peroxidation. Chem. Res. Toxicol. 1998, 11, 1176–1183. [Google Scholar] [CrossRef]
- Misra, H.P.; Fridovich, I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [CrossRef]
- Beutler, E. Red Cell Metabolism: A Manual of Biochemical Methods, 3rd ed.; Grune & Stratton: Orlando, FL, USA, 1984; p. xviii. 188p. [Google Scholar]
- Salbitani, G.; Bottone, C.; Carfagna, S. Determination of Reduced and Total Glutathione Content in Extremophilic Microalga Galdieria phlegrea. Bio Protoc. 2017, 7, e2372. [Google Scholar] [CrossRef] [Green Version]
- Paglia, D.E.; Valentine, W.N. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J. Lab. Clin. Med. 1967, 70, 158–169. [Google Scholar]
- Suzuki, S.; Toledo-Pereyra, L.H.; Rodriguez, F.J.; Cejalvo, D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation 1993, 55, 1265–1272. [Google Scholar] [CrossRef]
- Gregorini, M.; Corradetti, V.; Pattonieri, E.F.; Rocca, C.; Milanesi, S.; Peloso, A.; Canevari, S.; De Cecco, L.; Dugo, M.; Avanzini, M.A.; et al. Perfusion of isolated rat kidney with Mesenchymal Stromal Cells/Extracellular Vesicles prevents ischaemic injury. J. Cell. Mol. Med. 2017, 21, 3381–3393. [Google Scholar] [CrossRef] [PubMed]
- Aretz, H.T. Myocarditis: The Dallas criteria. Hum. Pathol. 1987, 18, 619–624. [Google Scholar] [CrossRef]
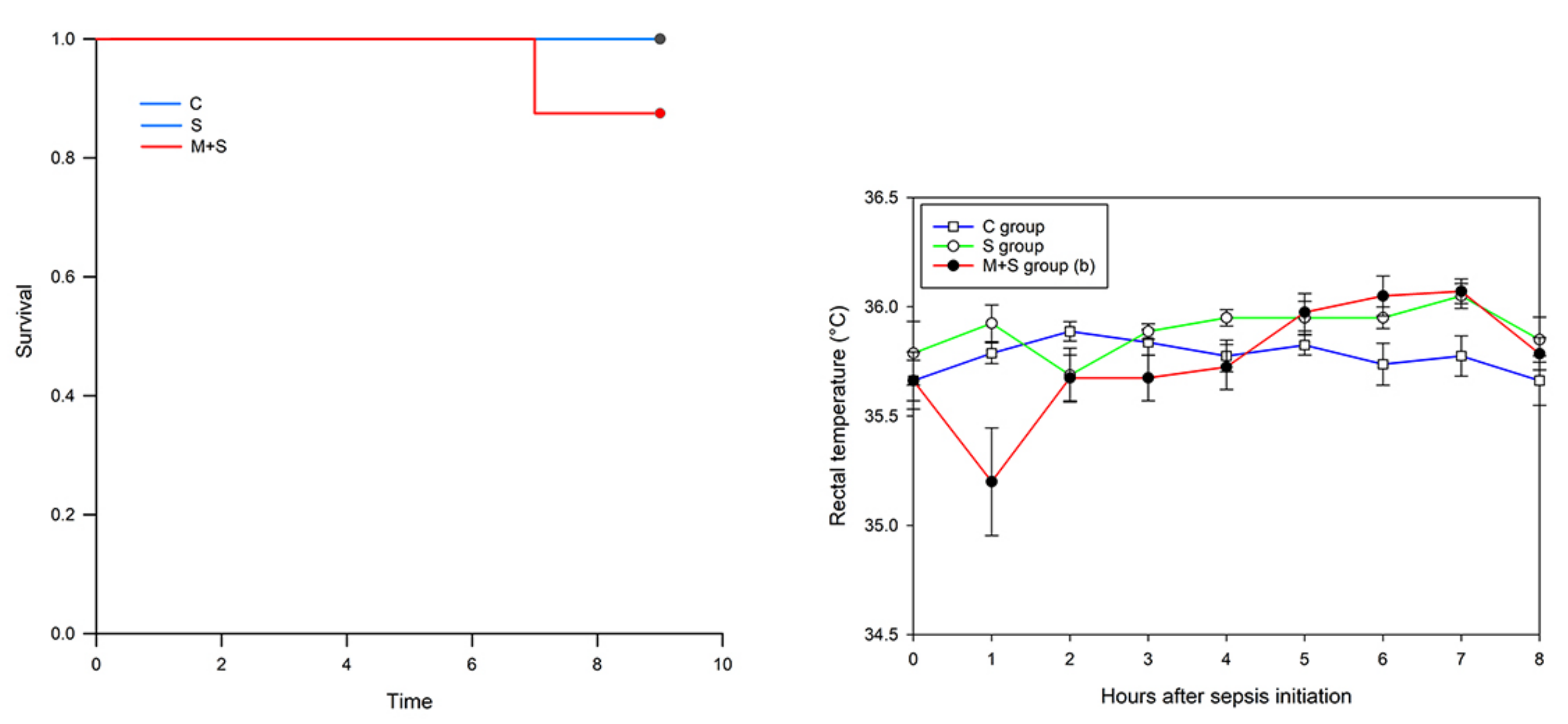
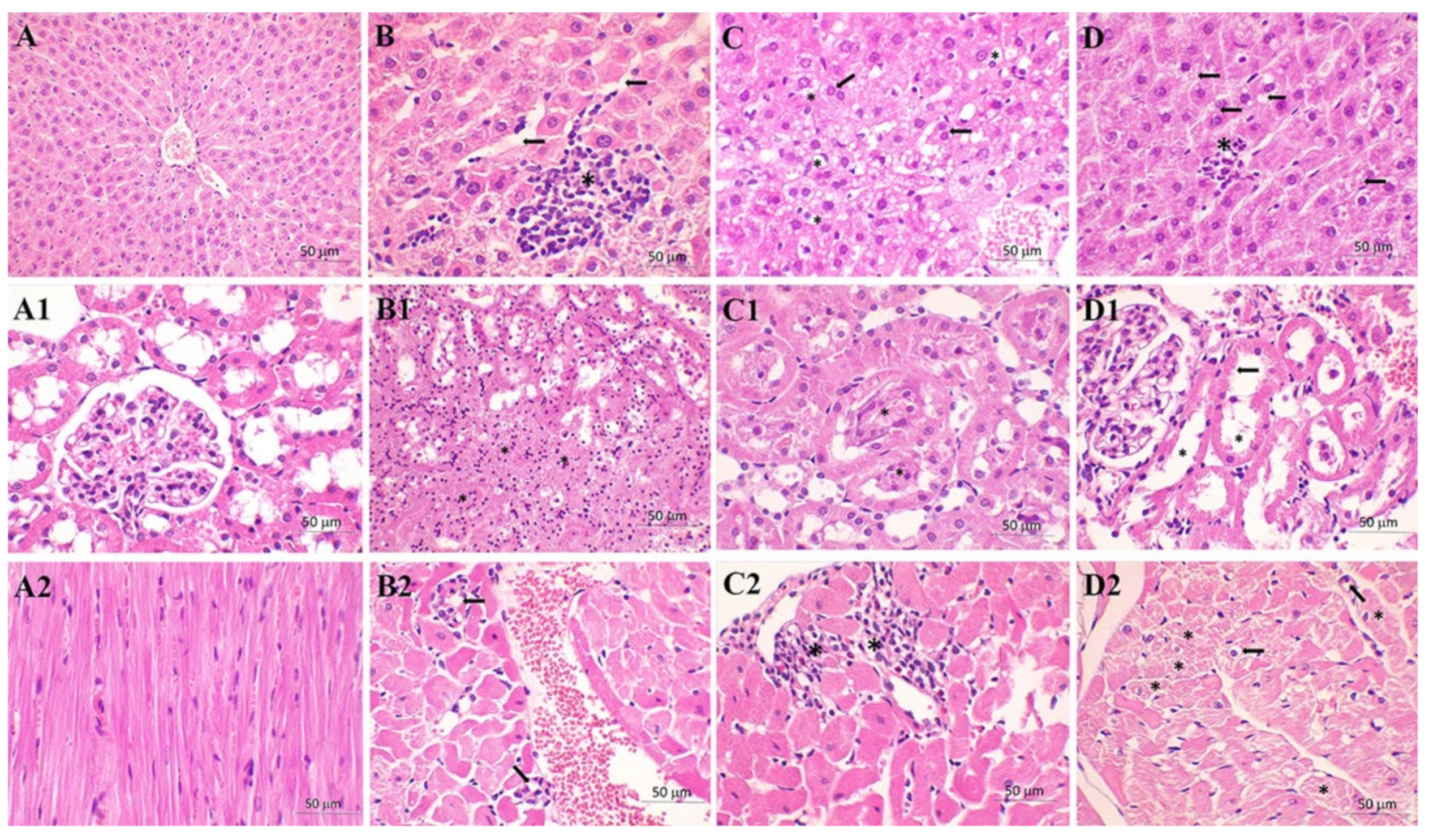
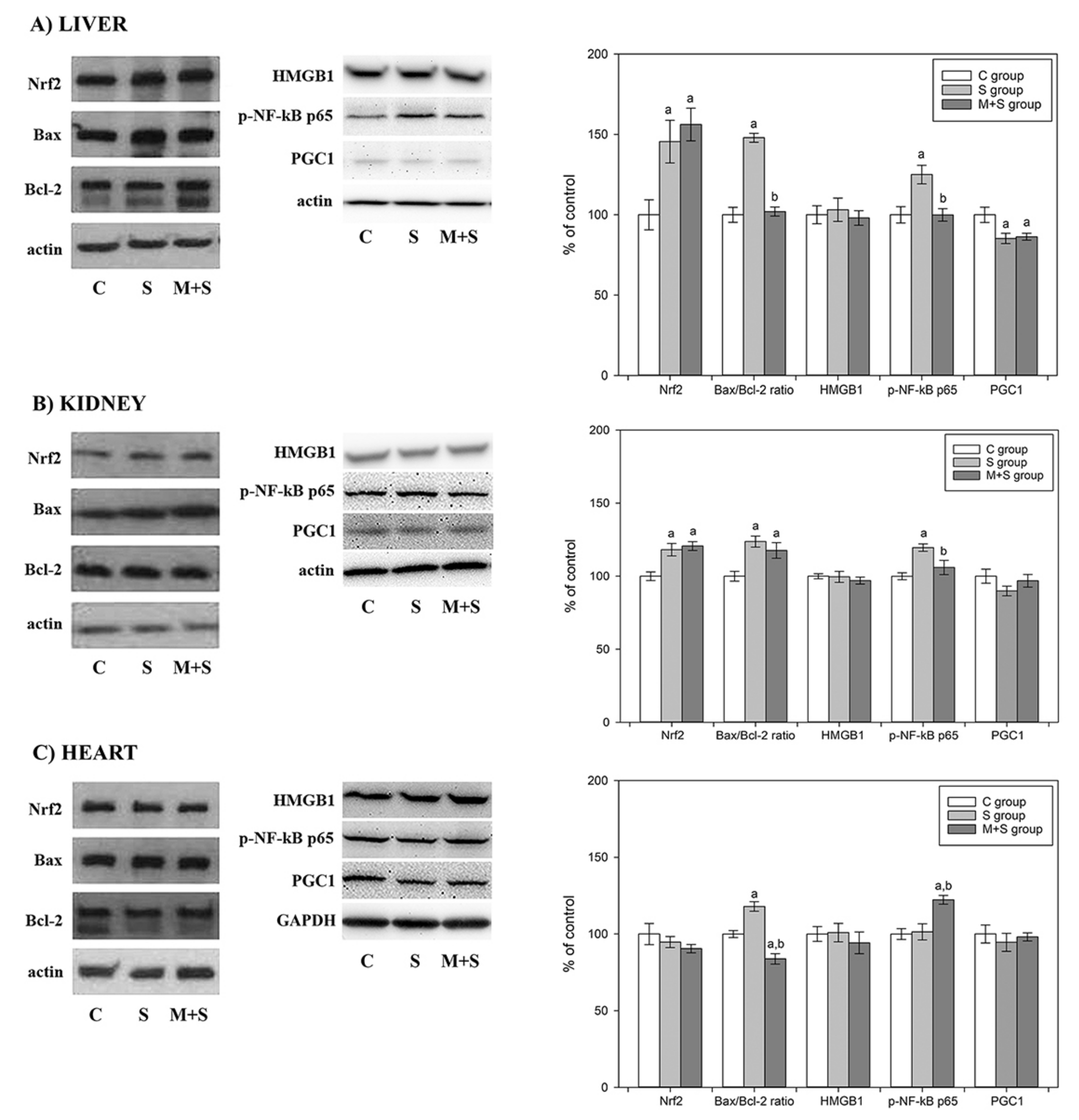

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
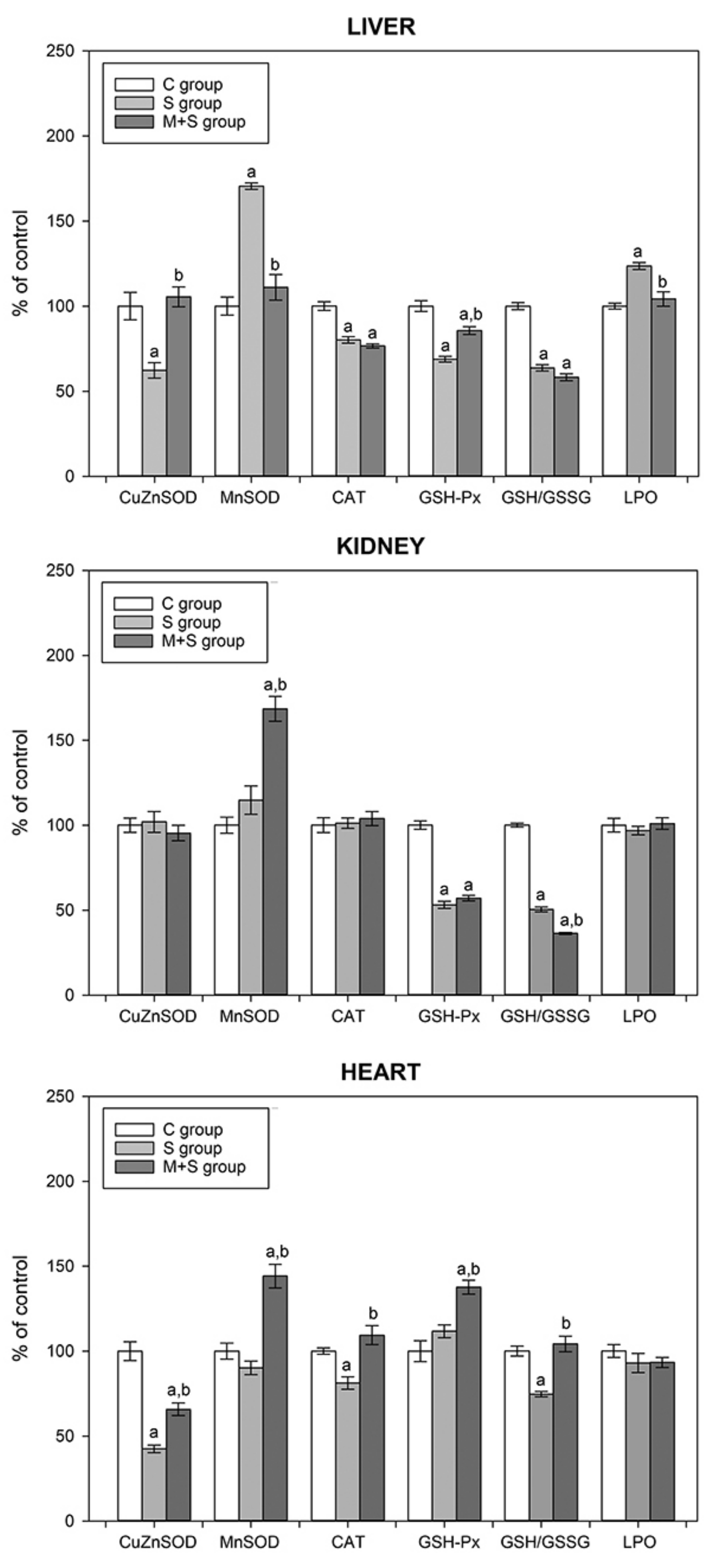
| C Group | S Group | M + S Group | |
|---|---|---|---|
| AST | 271.15 ± 13.93 | 288.10 ± 18.12 a | 212.83 ± 4.11 a,b |
| ALT | 65.18 ± 3.88 | 91.71 ± 2.32 a | 62.45 ± 1.62 b |
| ALP | 122.25 ± 4.49 | 164.50 ± 5.49 a | 166.67 ± 3.86 a |
| Troponin T | 31.25 ± 1.93 | 124.28 ± 2.99 a | 64.66 ± 1.62 a,b |
| Urea | 5.19 ± 0.13 | 11.76 ± 0.42 a | 7.83 ± 0.35 a,b |
| Creatinine | 45.03 ± 0.67 | 50.69 ± 1.62 | 47.88 ± 0.73 |
| TNFα | 11.32 ± 1.73 | 34.13 ± 2.57 a | 17.60 ± 2.11 a,b |
| Liver | C Group | S Group | M + S Group |
|---|---|---|---|
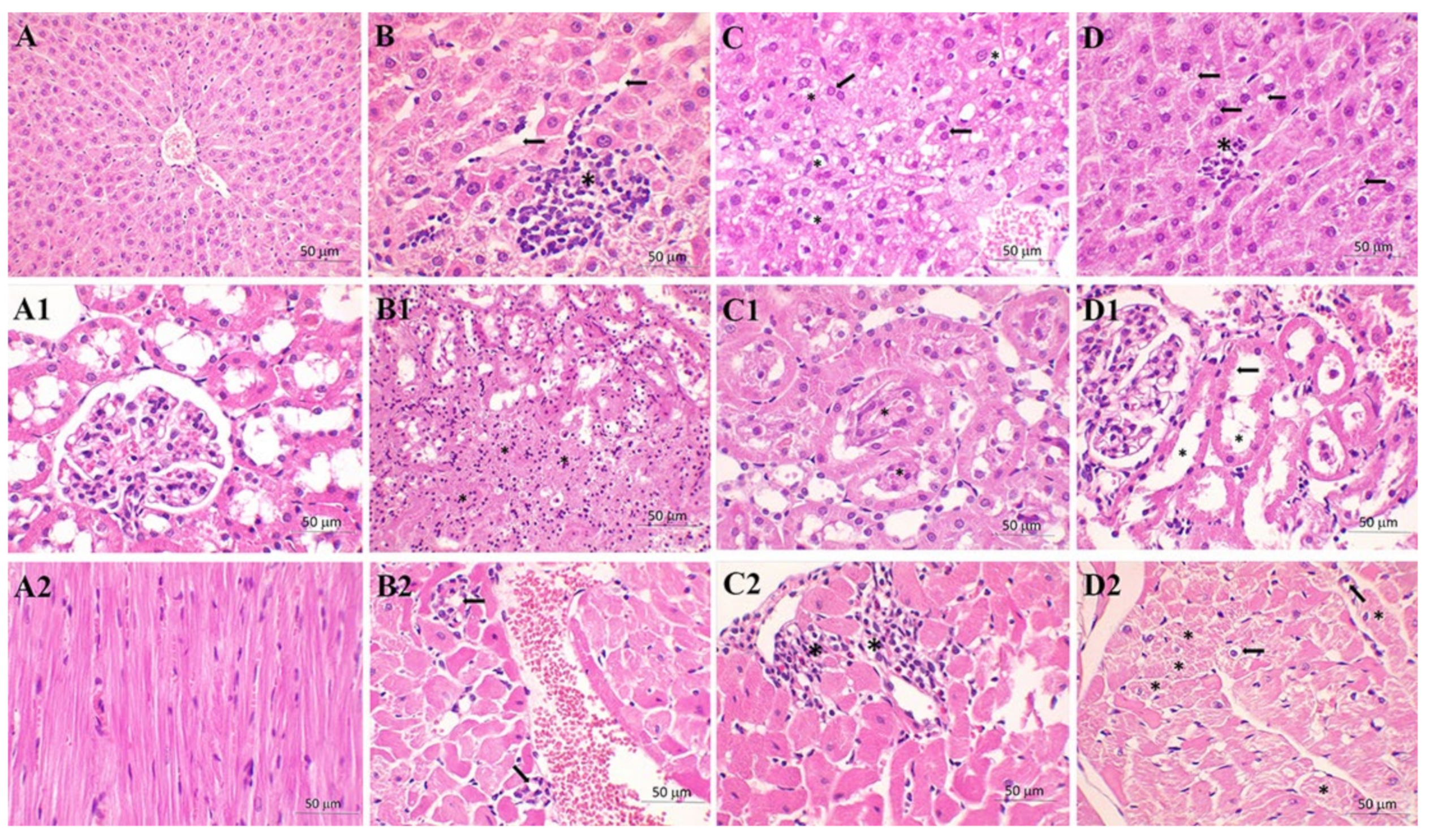
| Congestion | 1 | 2.71 | 1.25 |
| Vacuolization | 0.50 | 2.29 | 1.13 |
| Necrosis | 0.50 | 2.14 | 1.13 |
| Kidney | C group | S group | M+S group |
| TF | 4.25 | 7.57 | 6.38 |
| BBL | 4.13 | 12.29 | 11.50 |
| TN | 1.25 | 8.57 | 7.88 |
| TO | 0.00 | 9.86 | 6.38 |
| Heart | C group | S group | M+S group |
| II | 0.00 | 0.25 | 0.88 |
| Distribution | 0.00 | 0.25 | 0.63 |
| Liver | C Group | S Group | M + S Group |
|---|---|---|---|
| L carnitine | 0.575 ± 0.006 | 0.625 ± 0.005 a | 0.682 ± 0.004 a,b |
| TGAs | 1.054 ± 0.015 | 1.224 ± 0.005 a | 0.644 ± 0.010 a,b |
| Glycerol | 2.269 ± 0.014 | 2.665 ± 0.015 a | 2.908 ± 0.008 a,b |
| Total FFAs | 2.216 ± 0.137 | 2.487 ± 0.090 a | 2.880 ± 0.90 a,b |
| Glucose | 1.217 ± 0.002 | 1.091 ± 0.003 a | 1.078 ± 0.028 a |
| Lactate | 2.626 ± 0.021 | 2.777 ± 0.021a | 2.419 ± 0.009 a,b |
| Inositol | 0.610 ± 0.002 | 1.476 ± 0.013 a | 1.169 ± 0.029 a,b |
| C14:0 | 138.091 ± 0.849 | 147.201 ± 0.530 a | 169.343 ± 0.528 a,b |
| C15:0 | 18.815 ± 0.116 | 27.656 ± 0.099 a | 39.286 ± 0.091 a,b |
| C16:0 | 264.837 ± 1.628 | 231.321 ± 0.833 a | 263.097 ± 0.820 b |
| C16:1 | 60.166 ± 0.370 | 59.043 ± 0.210 | 60.523 ± 0.189 |
| C17:0 | 21.291 ± 0.130 | 29.235 ± 0.105 a | 28.149 ± 0.085 a |
| C17:1 | 15.813 ± 0.097 | 16.153 ± 0.059 | 15.991 ± 0.049 |
| C18:0 | 388.473 ± 2.389 | 425.616 ± 1.424 a | 496.444 ± 1.547 a,b |
| C18:1t | 191.625 ± 1.179 | 224.628 ± 0.772 a | 255.525 ± 0.797 a,b |
| C18:1c | 164.510 ± 1.013 | 191.591 ± 0.762 a | 236.689 ± 0.738 a,b |
| C18:2c+t | 325.322 ± 2.001 | 366.350 ± 1.283 a | 369.451 ± 1.151 a |
| C18:3n3c+t | 58.821 ± 0.361 | 58.657 ± 0.211 | 66.282 ± 0.206 a,b |
| C20 | 220.391 ± 1.355 | 254.474 ± 0.916 a | 296.440 ± 0.924 a,b |
| C20:1 | 34.304 ± 0.211 | 52.799 ± 0.189 a | 51.979 ± 0.162 a |
| C20:2 | 46.596 ± 0.286 | 70.005 ± 0.253 a | 71.599 ± 0.229 a |
| C20:3n3 | 21.751 ± 0.133 | 33.780 ± 0.121 a | 35.362 ± 0.110 a |
| C20:3n4 | 11.974 ± 0.073 | 23.725 ± 0.085 a | 23.824 ± 0.074 a |
| C20:3n6 | 23.334 ± 0.144 | 30.689 ± 0.110 a | 31.024 ± 0.097 a |
| C22:6 | 210.238 ± 1.293 | 275.059 ± 0.990 a | 376.264 ± 1.172 a,b |
| Kidney | C group | S group | M+S group |
| L carnitine | 0.623 ± 0.004 | 0.692 ± 0.001 a | 0.730 ± 0.002 a,b |
| TGAs | 0.492 ± 0.006 | 0.738 ± 0.005 a | 0.164 ± 0.002 a,b |
| Glycerol | 0.355 ± 0.002 | 0.783 ± 0.002 a | 0.977 ± 0.009 a,b |
| FFAs | 1152.978 ± 3.261 | 1330.299 ± 3.053 a | 1603.681 ± 7.077 a,b |
| Glucose | 0.732 ± 0.002 | 0.671 ± 0.003 a | 0.687 ± 0.002 a |
| Lactate | 2.462 ± 0.011 | 3.236 ± 0.009 a | 2.932 ± 0.011 a,b |
| Inositol | 0.362 ± 0.002 | 0.859 ± 0.005 a | 0.695 ± 0.002 a,b |
| C14:0 | 66.987 ± 0.189 | 81.855 ± 0.188 a | 96.851 ± 0.427 a,b |
| C15:0 | 8.126 ± 0.023 | 10.325 ± 0.023 a | 14.555 ± 0.065 a,b |
| C16:0 | 138.451 ± 0.391 | 166.398 ± 0.383 a | 185.056 ± 0.817 a,b |
| C16:1 | 34.176 ± 0.097 | 42.641 ± 0.098 a | 55.385 ± 0.244 a,b |
| C17:0 | 13.323 ± 0.037 | 16.809 ± 0.039 a | 19.824 ± 0.088 a,b |
| C17:1 | 8.669 ± 0.024 | 11.440 ± 0.026 a | 15.863 ± 0.070 a,b |
| C18:0 | 198.424 ± 0.561 | 216.048 ± 0.497 a | 319.694 ± 1.146 a,b |
| C18:1t | 94.951 ± 0.269 | 103.939 ± 0.239 a | 137.438 ± 0.607 a,b |
| C18:1c | 84.793 ± 0.239 | 97.285 ± 0.223 a | 123.254 ± 0.544 a,b |
| C18:2c+t | 164.795 ± 0.466 | 190.118 ± 0.438 a | 211.336 ± 0.933 a,b |
| C18:3n3c+t | 33.525 ± 0.095 | 37.895 ± 0.084 a | 43.130 ± 0.190 a,b |
| C20 | 114.894 ± 0.325 | 124.500 ± 0.285 a | 145.548 ± 0.642 a,b |
| C20:1 | 22.631 ± 0.064 | 28.770 ± 0.061 a | 34.097 ± 0.151 a,b |
| C20:2 | 25.598 ± 0.073 | 30.836 ± 0.064 a | 37.139 ± 0.164 a,b |
| C20:3n3 | 16.541 ± 0.047 | 21.163 ± 0.049 a | 20.906 ± 0.106 a |
| C20:3n4 | 9.804 ± 0.028 | 14.784 ± 0.033 a | 15.770 ± 0.069 a |
| C20:3n6 | 15.310 ± 0.043 | 19.079 ± 0.044 a | 26.214 ± 0.116 a,b |
| C22:6 | 101.980 ± 0.288 | 122.416 ± 0.281 a | 158.623 ± 0.700 a,b |
| Heart | C group | S group | M+S group |
| L carnitine | 0.884 ± 0.003 | 0.983 ± 0.004 a | 1.017 ± 0.004 a,b |
| TGAs | 0.467 ± 0.006 | 0.651 ± 0.005 a | 0.233 ± 0.003 a,b |
| Glycerol | 2.076 ± 0.007 | 1.952 ± 0.007 a | 1.274 ± 0.005 a,b |
| FFAs | 1512.182 ± 3.536 | 1402.85 ± 6.708 a | 1371.662 ± 7.284 a,b |
| Glucose | 0.792 ± 0.001 | 1.850 ± 0.002 a | 0.737 ± 0.003 a,b |
| Lactate | 4.398 ± 0.005 | 4.870 ± 0.019 a | 4.706 ± 0.010 a,b |
| Inositol | 0.467 ± 0.074 | 0.921 ± 0.003 a | 0.738 ± 0.001 a,b |
| C14:0 | 43.859 ± 0.010 | 35.237 ± 0.157 a | 28.021 ± 0.148 a,b |
| C15:0 | 4.610 ± 0.011 | 5.741 ± 0.0253 a | 4.559 ± 0.0265 b |
| C16:0 | 127.17 ± 0.291 | 120.423 ± 0.538 a | 121.225 ± 0.655 a |
| C16:1 | 40.896 ± 0.0925 | 33.153 ± 0.148 a | 24.578 ± 0.130 a,b |
| C17:0 | 14.158 ± 0.028 | 10.379 ± 0.045 a | 8.072 ± 0.048 a,b |
| C17:1 | 6.604 ± 0.013 | 4.431 ± 0.020 a | 3.299 ± 0.017 a,b |
| C18:0 | 196.863 ± 0.448 | 178.384 ± 0.841 a | 165.965 ± 0.961 a,b |
| C18:1t | 162.939 ± 0.348 | 149.855 ± 0.669 a | 134.488 ± 0.768 a,b |
| C18:1c | 103.710 ± 0.236 | 130.540 ± 0.583 a | 119.179 ± 0.632 a,b |
| C18:2c+t | 194.688 ± 0.421 | 180.918 ± 0.807 a | 166.481 ± 0.884 a,b |
| C18:3n3c+t | 49.210 ± 0.112 | 42.275 ± 0.189 a | 35.112 ± 0.186 a,b |
| C20 | 233.621 ± 0.509 | 207.710 ± 0.927 a | 197.160 ± 1.047 a,b |
| C20:1 | 53.837 ± 0.123 | 36.157 ± 0.161 a | 28.730 ± 0.153 a,b |
| C20:2 | 51.895 ± 0.119 | 50.704 ± 0.227 | 44.225 ± 0.235 |
| C20:3n3 | 42.273 ± 0.096 | 33.790 ± 0.160 a | 25.986 ± 0.137 a,b |
| C20:3n4 | 38.309 ± 0.088 | 29.691 ± 0.132 a | 20.218 ± 0.108 a,b |
| C20:3n6 | 33.590 ± 0.077 | 27.084 ± 0.130 a | 14.846 ± 0.078 a,b |
| C22:6 | 186.953 ± 0.425 | 212.379 ± 0.948 a | 211.119 ± 1.069 a |
| Adrenal Glands | C Group | S Group | M + S Group |
|---|---|---|---|
| Noradrenaline | 165.320 ± 4.300 | 139.705 ± 4.032 a | 129.545 ± 2.345 a |
| Adrenaline | 720.924 ± 28.162 | 484.769 ± 14.267 a | 510.224 ± 22.258 a |
| Serum | C group | S group | M + S group |
| Noradrenaline | 1764.275 ± 85.679 | 3116.625 ± 144.720 a | 1765.050 ± 98.654 a,b |
| Adrenaline | 1968.475 ± 92.005 | 4091.450 ± 229.687 a | 4159.283 ± 202.225 a |
| TGAs | 1.194 ± 0.004 | 0.860 ± 0.005 a | 0.516 ± 0.003 a,b |
| Glycerol | 1.591 ± 0.005 | 5.064 ± 0.042 a | 0.820 ± 0.001 a,b |
| FFAs | 1976.773 ± 38.599 | 1428.852 ± 2.615 a | 1399.508 ± 1.32 a |
| Glucose | 4.228 ± 0.023 | 6.473 ± 0.045 a | 2.187 ± 0.010 a,b |
| Lactate | 0.224 ± 0.003 | 0.647 ± 0.002 a | 0.705 ± 0.001 a |
| Inositol | 2.116 ± 0.020 | 5.044 ± 0.017 a | 3.654 ± 0.005 a,b |
| Groups | C Group | S Group | M + S Group |
|---|---|---|---|
| Application of meldonium in water | none | none | + |
| Intraperitoneal application of saline | + | none | none |
| Intraperitoneal application of LPS | none | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Đurašević, S.; Ružičić, A.; Lakić, I.; Tosti, T.; Đurović, S.; Glumac, S.; Pejić, S.; Todorović, A.; Drakulić, D.; Stanković, S.; et al. The Effects of a Meldonium Pre-Treatment on the Course of the LPS-Induced Sepsis in Rats. Int. J. Mol. Sci. 2022, 23, 2395. https://doi.org/10.3390/ijms23042395
Đurašević S, Ružičić A, Lakić I, Tosti T, Đurović S, Glumac S, Pejić S, Todorović A, Drakulić D, Stanković S, et al. The Effects of a Meldonium Pre-Treatment on the Course of the LPS-Induced Sepsis in Rats. International Journal of Molecular Sciences. 2022; 23(4):2395. https://doi.org/10.3390/ijms23042395
Chicago/Turabian StyleĐurašević, Siniša, Aleksandra Ružičić, Iva Lakić, Tomislav Tosti, Saša Đurović, Sofija Glumac, Snežana Pejić, Ana Todorović, Dunja Drakulić, Sanja Stanković, and et al. 2022. "The Effects of a Meldonium Pre-Treatment on the Course of the LPS-Induced Sepsis in Rats" International Journal of Molecular Sciences 23, no. 4: 2395. https://doi.org/10.3390/ijms23042395
APA StyleĐurašević, S., Ružičić, A., Lakić, I., Tosti, T., Đurović, S., Glumac, S., Pejić, S., Todorović, A., Drakulić, D., Stanković, S., Jasnić, N., Đorđević, J., & Todorović, Z. (2022). The Effects of a Meldonium Pre-Treatment on the Course of the LPS-Induced Sepsis in Rats. International Journal of Molecular Sciences, 23(4), 2395. https://doi.org/10.3390/ijms23042395