Abstract
Diamond-Blackfan anaemia (DBA) is a red blood cell aplasia that in the majority of cases is associated with ribosomal protein (RP) aberrations. However, the mechanism by which this disorder leads to such a specific phenotype remains unclear. Even more elusive is the reason why non-specific agents such as glucocorticosteroids (GCs), also known as glucocorticoids, are an effective therapy for DBA. In this review, we (1) explore why GCs are successful in DBA treatment, (2) discuss the effect of GCs on erythropoiesis, and (3) summarise the GC impact on crucial pathways deregulated in DBA. Furthermore, we show that GCs do not regulate DBA erythropoiesis via a single mechanism but more likely via several interdependent pathways.
Keywords:
Diamond-Blackfan anaemia; glucocorticosteroid; erythropoiesis; GATA1; c-myc; mTOR; autophagy 1. Introduction
Human erythrocytes differ from other cells because of their particular shape, lack of organelles, and transportation of oxygen via haemoglobin. Erythropoiesis is a complex differentiation process in which cells discard several organelles, from mitochondria to the nucleus, and dramatically increase haemoglobin production. These multifaceted changes need to be tightly regulated by numerous factors, and any disruption in signalling during erythropoiesis may lead to anaemia.
Diamond-Blackfan anaemia (DBA) [1] is a rare congenital disease usually diagnosed shortly after birth. Despite normal platelet and neutrophil counts, patients present with macrocytic or normocytic anaemia, reticulocytopenia, and a shortage of erythrocyte precursors [2]. Patients with DBA also manifest physical malformations, mainly craniofacial and upper limb, and growth retardation in approximately 35–50% of cases [3].
The first gene associated with DBA was RPS19 [4]. Interestingly, RPS19 variants account for approximately 25% of all DBA cases [5]. Over the last years, the number of RP genes associated with DBA increased; currently, there are 16 ribosomal protein aberrations linked to the disease, involving both large and small RP genes [6]. Rarely, non-ribosomal protein gene abnormalities were also associated with DBA, for instance in transcription factor GATA1 [7], iron metabolism gene SLC49A1 [8], and ribosome maturation factor TSR2 [9]. The molecular pathology of approximately 20–50% of cases remains unknown; nonetheless, a number of candidate DBA genes is continually increasing with higher penetration of high-throughput DNA sequencing technologies into the clinic.
The aberration of ribosomal proteins (RPs) results in a decreased number of available ribosomes with a consequent drop in translational activity [10]. A reduced number of ribosomes leads to an imbalance between translated and non-translated mRNA [11]. Furthermore, RP defects may result in ribosomal stress [12], p53 activation [12], altered mTOR signalling [13], c-Myc [14] deregulation, and other stress-related processes (Figure 1). Deregulation of any of these signal and metabolic pathways may result in anaemia.
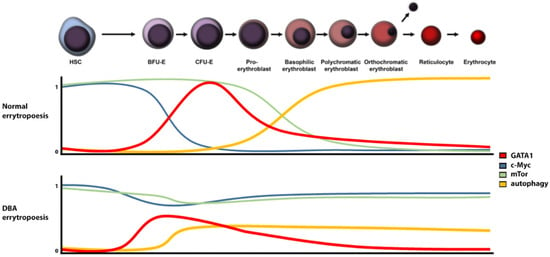
Figure 1.
Regulation of relevant signalling proteins and pathways levels in DBA versus normal erythropoiesis (HSC: hematopoietic stem cells, BFU-E: burst-forming unit-erythroid, CFU-E: Colony-forming unit-erythroid).
As for many other rare diseases, therapeutic opportunities in DBA are inadequate. The first-line non-specific therapy usually includes glucocorticosteroids (GCs). However, treatment response is limited, and long-term use is associated with drug resistance and significant side effects [2]. Blood transfusion and iron chelation are usually reserved for GC non-responder [15]. Although some alternative therapies such as L-leucine supplementation to stimulate deficient protein synthesis have been used, the only permanent treatment of DBA is bone marrow transplantation [15]. However, bone marrow replacement is a high-risk procedure reserved for transfusion-dependent individuals. Recently, the approved gene therapy for betta thalassemia is also hope for many other congenital anaemias [16]. For DBA, gene therapy was already successfully tested in mice [17], and its further development is ongoing. Nevertheless, in contrast to thalassemia, where only hematopoietic cells are affected, DBA causes complex signalling and metabolic abnormalities in many other tissues, which will be difficult to correct by currently available gene therapy technologies.
Glucocorticosteroids (GCs) [18] generally improve erythropoiesis [19] and are frequently used for DBA treatment [3]. However, the mechanism by which GCs ameliorate DBA is elusive [19,20,21]. The present review aims to analyse the possible pathways that may be affected by GCs and improve DBA pathology. This is particularly important in the development of more efficient and targeted DBA therapies without glucocorticoid side effects.
2. Glucocorticosteroids
GCs are stress hormones produced in the adrenal cortex. They regulate diverse cellular functions, including development, homeostasis, metabolism, cognition, and inflammation [22]. GCs are widely used as treatments for a broad range of pathologies, from autoimmune syndromes and allergy to cancer [23]. Although GCs have a wide range of applications, their use is limited by severe adverse effects, especially in long-term or high-dose therapies. Possible side effects include osteoporosis, skin atrophy, diabetes, abdominal obesity, glaucoma, cataracts, avascular necrosis and infection, growth retardation, and hypertension [24].
Approximately 80% of DBA patients respond to initial GC treatment with a starting dose of 2 mg/kg per day for a maximum of 4 weeks to avoid GC toxicity in children [2,25]. The most serious GC adverse effects in infants include immunosuppression and susceptibility to infection, weight gain, growth retardation, osteonecrosis, and Cushingoid features [25].
The majority of GC effects are mediated by the glucocorticoid receptor (GCR). GCR is encoded by the NR3C1 gene located on chromosome 5 and is a ligand-activated transcription factor. In an inactive form, GCR is predominantly located in the cytoplasm in complex with chaperone proteins and immunophilins [26]. However, its conformation changes upon ligand binding, whereupon it disassociates from the complex and translocates to the nucleolus, where it binds to GCR sequences and initiates the expression of target genes. Interestingly, GCR-mediated expression is tissue specific, and only a small number of genes are activated in all tissues [27]. This is probably caused by tissue-specific DNA methylation [28]. Furthermore, in addition to expression initiation, activated GCR can interact with other proteins. For example, it can modulate the activity of several kinases [29] and regulate GATA1 during erythropoiesis [30].
Moreover, GCR has several isoforms [31] and is highly polymorphic [32]. The genetic variability of GCR can affect body mass index, bone density or coronary artery diseases, probability of developing type II diabetes and GC treatment outcome [33,34]. This also applies in DBA. Surprisingly, the variability of GCR seems to have no or minor impact on GC treatment outcome in DBA [35]. However, it seems to modulate the onset of disease [35]. It has been shown that particular SNPs (rs6196 and rs860457) in GCR result in early onset of DBA, which can be caused by a modified GC response during embryogenesis [35].
3. The Primary Defect in DBA Cells
It is not fully understood how RP aberration causes red blood cell aplasia. A plausible mechanism involves a gap between transcription and translation [11]. The common feature of DBA is a decreased number of available ribosomes, resulting in decreased translational activity [10]. Several mRNAs are particularly affected by DBA ribosome impairment. Among the critically affected complexes are IRES-mediated mRNA and mRNAs with complex 5′ UTR regions [36].
GATA1, the specific erythropoiesis master regulator, is affected in DBA [37]. GATA1 is expressed during the final steps of erythrocyte development. It is responsible for triggering haemoglobin transcription, eliminating organelles and inducing other genes related to the terminal steps of erythropoiesis [38]. Although GATA1 mRNA levels are slightly increased or similar in RP depleted cells or DBA patients compared to controls, protein levels are downregulated [38]. The origin of this phenomenon is related to the complex 5′ UTR region of the gene [38]. GATA1 protein levels are restored and overall erythroid differentiation is improved when GATA1 5′ UTR region is substituted with less complex one [38].
It has been postulated that DBA red blood cells phenotype is induced by GATA1 translation inhibition [37]. GATA1 is expressed when committed cells are in the colony-forming unit-erythroid (CFU-E) stage of red blood cell (RBC) development [39]. At this stage, downregulation of GATA1 protein leads to apoptosis [40], which is consistent with the DBA phenotype. Furthermore, if RBC precursors escape apoptosis at the CFU-E stage, GATA1 decreased signalling may lead to impaired cell responses in later RBC developmental stages. Interestingly, mutations in the GATA1 gene have been described in several DBA-like patients [7,41]. However, although these patients showed impaired erythropoiesis similar to DBA, other typical features, such as physical malformations, were missing [7,41]. Furthermore, arsenic-induced disruption of GATA1 also leads to DBA-like erythropoiesis disruption [42].
Recent findings have suggested that erythropoiesis failure in DBA is more complicated than previously thought. More specifically, it seems that GATA1 impairment plays a crucial role in the DBA phenotype but only in ribosomal proteins of small-(RPS) and not large (RPL) subunit-related disorders [43].
4. GCs and GATA1
Impaired GATA1 protein levels seem to be the primary defect in DBA RBC precursor cells [44]. Therefore, it could be suggested that GCs restore GATA1 translation and related signalling pathways, and consequently increase erythrocyte precursor counts. However, GCs do not increase GATA1 but deplete it. GCs inhibit GATA1 by transcriptional repression and direct interaction of GCRs with GATA1 [25]. GCs also decrease protein synthesis in cells through mTOR and subsequent S6 kinase inhibition [45]. This reduction affects the levels of HSP70 [46], a chaperone that protects GATA1 from caspase-3 degradation during erythropoiesis [40]. Therefore, GCs do not seem to increase GATA1 levels but instead deplete them.
GCs can promote erythropoiesis by two mechanisms to overcome GATA1 impairment. Firstly, GCs can act before the CFU-E stage and increase the number of CFU-E precursors. Secondly, even though GCs do not rescue GATA1, they can regulate GATA1-related steps in erythropoiesis, as discussed further below.
5. Role of GCs in CFU-E Precursors
Stress erythropoiesis may be caused by blood loss, oxygen deprivation, haemolytic anaemia and long-term stress [47] (Figure 2). This type of erythropoiesis leads to the rapid proliferation of RBC precursors driven by erythropoietin (Epo) and GCs [21]. Because DBA fulfils several criteria, we can hypothesise that stress erythropoiesis plays a crucial role in RBC development in DBA patients.
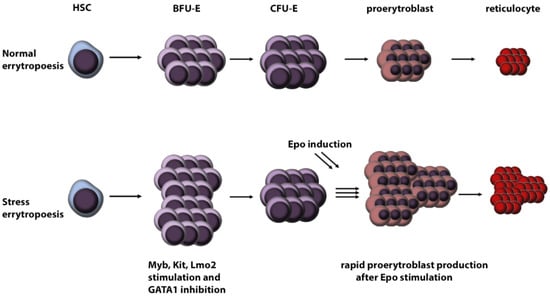
Figure 2.
Schematic overview of stress versus normal erythropoiesis. (HSC: hematopoietic stem cells, BFU-E: burst-forming unit-erythroid, CFU-E: colony-forming unit-erythroid, Epo: erythropoietin).
When the body is under stress, cortisol, the intrinsic master GC, induces many physiological responses. One of them is to stimulate the production of Epo [48] in the kidneys [49]. Epo in turn stimulates CFU-E, and its primary target is GATA1 [50]. Due to GATA1 impairment, DBA is one of very few Epo insensitive anaemias [51]. Therefore, we can speculate that Epo induction via stress erythropoiesis probably does not lead to DBA phenotype improvement.
GCs induce stress erythropoiesis also directly by activating GCR [19]. Furthermore, activation of GCR is conditional for stress erythropoiesis [19] and stimulates several transcription factors necessary for burst forming unit-erythroid (BFU-E), CFU-E precursor, cell proliferation, specifically Myb, Kit and Lmo2 [52]. Together with GATA1 inhibition, these transcription factors stimulate the rapid proliferation of BFU-E cells. This induction pathway is specific to BFU-E cells and increases the number of CFU-E precursor cells by up to 10-fold [53]. Thus, an increased number of CFU-E cells are generated, which, after Epo stimulation, undergo rapid differentiation [47]. Hence, stress erythropoiesis results in a significant increase in RBC number originating from one precursor cell.
Furthermore, it seems that stress erythropoiesis impairment plays a crucial role mainly in RPL-related DBA, and its external stimulation by GC treatment may result in restoration of this pathway [43].
6. Regulation of Ribosomal Stress and p53
It has been suggested that apoptosis in DBA RBC precursors is caused by p53 induction via ribosomal stress [54]. However, this hypothesis has been partially refuted since the haploinsufficiency of most RPs does not lead to p53 stimulation via ribosomal stress mechanisms [55]. Even though ribosomal stress and p53 signalling are critical for normal erythropoiesis [56], the activation of p53 in DBA is most likely mediated by impaired metabolism and DNA damage [6,57].
Unfortunately, it is difficult to elucidate the interplay between p53 and GCs. On the one hand, GCs decrease p53 levels [58], whereas on the other hand, increased p53, common in DBA cells, limits proper GCs receptor activation [59]. Moreover, GCs do not only regulate p53 directly but also indirectly by reducing p53 activation signals.
As mentioned above, in addition to ribosomal stress, p53 could be activated by HEM-mediated ROS production [60,61]. Although it has been reported that GCs reduce ROS species [62,63,64], their overall influence on ROS production is ambiguous [65]. More specifically, it seems that short-term GC usage leads to a reduction in ROS generation, whereas long-term exposure leads to ROS induction [65]. Furthermore, increased ROS levels have been detected in DBA patients regardless of treatment [60]. Therefore, GC-mediated p53 regulation does not seem to reflect ROS levels. Thus, the precise mechanism by which GCs modulate p53 levels in DBA remains mysterious. Nevertheless, we can conclude that GC treatment improves p53 status in DBA [66].
7. Regulation of Enucleation through c-Myc
Enucleation is a critical step in erythropoiesis, in which the cells lose a major part of the nucleus to generate reticulocytes. Inhibition of this process may explain reticulocytopenia in DBA patients [67]. Before enucleation, the nucleolus must be condensed and histones deacetylated.
Protein c-Myc is a master transcription factor that promotes and regulates proliferation, transcription, translation and cellular metabolism with an important role in several cell transduction pathways [68]. GATA1 silences c-Myc expression in RBC precursors [37], which highlights the importance of c-Myc during erythropoiesis. Only a slight increase in c-Myc levels disables enucleation and reticulocyte development by impairing histone deacetylases [68]. Activated c-Myc also induces proerythroblast apoptosis during RBC development [68].
c-Myc is deregulated in DBA in multiple ways. Downregulated GATA1 is probably the central factor leading to upregulation of c-Myc in all DBA erythroid progenitors. Interestingly, some RPs affected in DBA regulate c-Myc on their own, e.g., RPL11 and RPL5. Both these RPs regulate ribosomal stress induction [69] and c-Myc regulation [70] at mRNA and protein levels [14]. Impairment of RPL5 and RPL11 has also been associated with a more severe DBA phenotype [71]. In addition, c-Myc driven lymphomagenesis has been observed in a DBA mouse model [14]. Interestingly, similar or identical mutations of those RPs reported in DBA have also been detected in some cancers [72,73], characterized by p53 deactivation and activation of c-Myc signalling.
GCs transcriptionally repress c-Myc expression [74], leading to cell cycle arrest without apoptosis, which is necessary for proper RBC differentiation [75]. In DBA, GCs may support enucleation during erythropoiesis, and thus prevent reticulocytopenia by downregulating c-Myc.
8. mTOR Pathway and Autophagy
Autophagy is crucial in RBC development [76]. During erythrocyte differentiation, cells discard and/or recycle most subcellular structures, starting from mitochondria to the nucleus. The nucleus is removed through the enucleation step, whereas other organelles are typically removed via autophagy [77]. Autophagy is activated by GATA1 [78] and mTOR [79] mediated induction of autophagy genes transcription. Notably, increased mTOR leads to macrocytic anaemia [80], consistent with the DBA phenotype [81].
mTOR has a dual role in RBC development [82]. Its activity is necessary for proper BFU-E and CFU-E cell proliferation in the primary steps of RBC development. But in later steps of erythropoiesis, it needs to be silenced for autophagy induction. mTOR is known to be activated in DBA cells [13]. Activated mTOR in DBA probably facilitates the increased proliferation of BFU-E and CFU-E cells and in a back-loop manner stimulates deficient protein translation S6-kinase activation and in turn RPS6 phosphorylation [83].
Furthermore, L-leucine, an inductor of mTOR activation, is routinely used as a DBA treatment but with a variable response rate [84,85]. Thus, we suggest induction of mTOR is beneficial for DBA cells, especially during RBC development until the CFU-E stage. On the other hand, constitutive mTOR induction during terminal differentiation may lead to autophagy failure, followed by aberration of the final erythropoiesis steps. We can speculate that this may be the underlying mechanism in DBA because DBA erythroid precursors treated by rapamycin, an mTOR inhibitor, have been shown to increase the proliferation of late erythroid cells in DBA [86].
In DBA, autophagy is increased despite mTOR activation [13]. It seems to be induced by ROS [60], hypoxia [87] or some mTOR-independent pathways [88]. However, mTOR-independent autophagy may not be sufficient for proper RBC development [89]. Therefore, autophagy induction may offer a possible target for future treatment of DBA patients [86].
Inhibition of mTOR by GCs followed by autophagy has been described in several cell types [90,91,92]. Therefore, a plausible hypothesis is that GCs may also improve erythropoiesis by modulating mTOR, and subsequently autophagy. The other important pathway is inactivation of Akt signalling by GCs in DBA cells [13,93]. Even though GC-induced mTOR inactivation may lead to decreased proliferation of BFU-E and CFU-E cells [82], stress-induced erythropoiesis may compensate for this [39] (Figure 3).
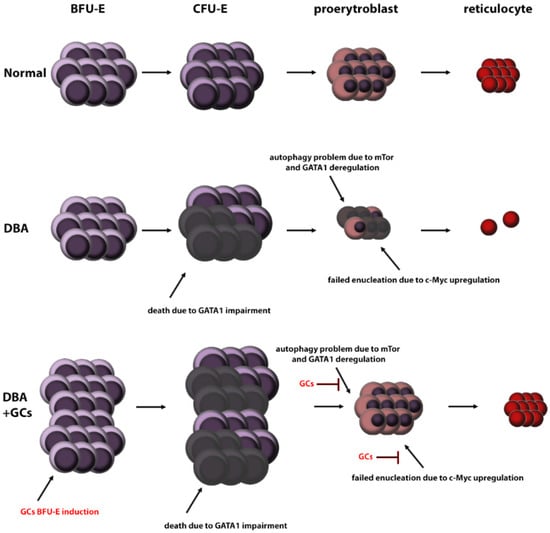
Figure 3.
Possible mechanism involved in GC-mediated improvement of DBA erythropoiesis. (HSC: hematopoietic stem cells, BFU-E: burst-forming unit-erythroid, CFU-E: colony-forming unit-erythroid, DBA: Diamond Blackfan anemia, GC: glucocorticosteroids).
9. GC Resistance
GC resistance is common in almost all GC-based therapies [94,95]; DBA is no exception [96]. Multiple mechanisms may induce GC resistance, and these mechanisms may vary among individual patients [97]. GCR polymorphism, alternative splicing and downregulation of GCR expression may also lead to GCR resistance [97], expression of pharmacological multidrug transporters may also be involved [94]. GC resistance in DBA has been shown to be associated with p57 cyclin dependent kinase inhibitor dysregulation [96]. Expression of p57 is directly induced by the GCR responsive element promoter [98]. Therefore, more than one mechanism may play a role to effect GC resistance.
10. Conclusions
GCs have been used to treat DBA for decades, but their mechanism of action remains elusive, and it is difficult to understand them fully due to their pleiotropic effects. We suggest that GCs improve the DBA pathology via at least four relatively independent ways: (i) induction of BFU-E cell proliferation via stress erythropoiesis, (ii) (de)regulation of P53 signalling, (iii) deactivation of c-Myc, and (iv) inhibition of mTOR signalling. Altogether, these four steps result in the induction of BFU-E cell proliferation and terminal differentiation to RBCs.
Although all four pathways seem to be relevant in DBA treatment, to date, only L-leucine modulation of mTOR activity is routinely used as a therapy [84]. Stress erythropoiesis seems to be tightly connected with GCs as stress hormones [19], and there is currently no evidence for other biologically active compounds. Protein p53 [99] and c-Myc [100] inhibitors are tested in cancer clinical trials, but their efficacy in DBA patients or DBA preclinical models is unknown. Thus, further studies are needed to ascertain the role of the above-mentioned GC-dependent pathways in the regulation and pathology of DBA, with the ultimate goal of developing new, more effective, and less toxic therapies for children suffering from this disease.
Author Contributions
Z.M., A.K.—review preparation, J.B.D.S., M.H.—review outline, revision and proofreading. All authors have read and agreed to the published version of the manuscript.
Funding
Supported by grants from the Czech Ministry of Education, Youth and Sports (8F20005, EATRIS-CZ LM2018133, ACGT-CZ.02.1.01/0.0/0.0/16_026/0008448 and NCLG LM2018132), Technological Agency of the Czech Republic (PerMed TACR TN 01000013) and Palacky University Olomouc (IGA UP LF_2021_019, IGA UP LF_2021_038, and IGA_LF_2021_004).
Conflicts of Interest
The authors declare that there is no conflict of interest.
References
- Bleiber, R.; Eggert, W.; Reichmann, G.; Muhlack, D.; Andres, J. An early childhood erythrocyte phospholipid distribution as further indication of persistence of neonatal erythrocyte characteristics in Diamond-Blackfan anemia. Folia Haematol. 1983, 110, 71–80. [Google Scholar]
- Vlachos, A.; Muir, E. How I treat Diamond-Blackfan anemia. Blood 2010, 116, 3715–3723. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, A.; Ball, S.; Dahl, N.; Alter, B.P.; Sheth, S.; Ramenghi, U.; Meerpohl, J.; Karlsson, S.; Liu, J.M.; Leblanc, T.; et al. Diagnosing and treating Diamond Blackfan anaemia: Results of an international clinical consensus conference. Br. J. Haematol. 2008, 142, 859–876. [Google Scholar] [CrossRef] [PubMed]
- Draptchinskaia, N.; Gustavsson, P.; Andersson, B.; Pettersson, M.; Willig, T.N.; Dianzani, I.; Ball, S.; Tchernia, G.; Klar, J.; Matsson, H.; et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999, 21, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Pospisilova, D.; Cmejlova, J.; Ludikova, B.; Stary, J.; Cerna, Z.; Hak, J.; Timr, P.; Petrtylova, K.; Blatny, J.; Vokurka, S.; et al. The Czech National Diamond-Blackfan Anemia Registry: Clinical data and ribosomal protein mutations update. Blood Cells Mol. Dis. 2012, 48, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Gazda, H.T. Ribosomopathies: How a common root can cause a tree of pathologies. Dis. Model. Mech. 2015, 8, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Klar, J.; Khalfallah, A.; Arzoo, P.S.; Gazda, H.T.; Dahl, N. Recurrent GATA1 mutations in Diamond-Blackfan anaemia. Br. J. Haematol. 2014, 166, 949–951. [Google Scholar] [CrossRef] [PubMed]
- Ulirsch, J.C.; Verboon, J.M.; Kazerounian, S.; Guo, M.H.; Yuan, D.; Ludwig, L.S.; Handsaker, R.E.; Abdulhay, N.J.; Fiorini, C.; Genovese, G.; et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am. J. Hum. Genet. 2018, 103, 930–947. [Google Scholar] [CrossRef]
- Gripp, K.W.; Curry, C.; Olney, A.H.; Sandoval, C.; Fisher, J.; Chong, J.X.; Pilchman, L.; Sahraoui, R.; Stabley, D.L.; Sol-Church, K. Diamond-Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am. J. Med. Genet. Part A 2014, 164, 2240–2249. [Google Scholar] [CrossRef]
- Cmejlova, J.; Dolezalova, L.; Pospisilova, D.; Petrtylova, K.; Petrak, J.; Cmejla, R. Translational efficiency in patients with Diamond-Blackfan anemia. Haematologica 2006, 91, 1456–1464. [Google Scholar]
- Gazda, H.T.; Kho, A.T.; Sanoudou, D.; Zaucha, J.M.; Kohane, I.S.; Sieff, C.A.; Beggs, A.H. Defective ribosomal protein gene expression alters transcription, translation, apoptosis, and oncogenic pathways in Diamond-Blackfan anemia. Stem Cells 2006, 24, 2034–2044. [Google Scholar] [CrossRef]
- Aspesi, A.; Monteleone, V.; Betti, M.; Actis, C.; Morleo, G.; Sculco, M.; Guarrera, S. Author Correction: Lymphoblastoid cell lines from Diamond Blackfan anaemia patients exhibit a full ribosomal stress phenotype that is rescued by gene therapy. Sci. Rep. 2018, 8, 17227. [Google Scholar] [CrossRef] [PubMed]
- Heijnen, H.F.; van Wijk, R.; Pereboom, T.C.; Goos, Y.J.; Seinen, C.W.; van Oirschot, B.A.; van Dooren, R.; Gastou, M.; Giles, R.H.; van Solinge, W.; et al. Ribosomal protein mutations induce autophagy through S6 kinase inhibition of the insulin pathway. PLoS Genet. 2014, 10, e1004371. [Google Scholar] [CrossRef]
- Morgado-Palacin, L.; Varetti, G.; Llanos, S.; Gómez-López, G.; Martinez, D.; Serrano, M. Partial Loss of Rpl11 in Adult Mice Recapitulates Diamond-Blackfan Anemia and Promotes Lymphomagenesis. Cell Rep. 2015, 13, 712–722. [Google Scholar] [CrossRef] [PubMed]
- Bartels, M.; Bierings, M. How I manage children with Diamond-Blackfan anaemia. Br. J. Haematol. 2019, 184, 123–133. [Google Scholar] [CrossRef]
- Harrison, C. First gene therapy for β-thalassemia approved. Nat. Biotechnol. 2019, 37, 1102–1103. [Google Scholar] [CrossRef]
- Liu, Y.; Dahl, M.; Debnath, S.; Rothe, M.; Smith, E.M.; Grahn, T.H.M.; Warsi, S.; Chen, J.; Flygare, J.; Schambach, A.; et al. Successful gene therapy of Diamond-Blackfan anemia in a mouse model and human CD34+ cord blood hematopoietic stem cells using a clinically applicable lentiviral vector. Haematologica 2021. [Google Scholar] [CrossRef] [PubMed]
- Barczyk, K.; Ehrchen, J.; Tenbrock, K.; Ahlmann, M.; Kneidl, J.; Viemann, D.; Roth, J. Glucocorticoids promote survival of anti-inflammatory macrophages via stimulation of adenosine receptor A3. Blood 2010, 116, 446–455. [Google Scholar] [CrossRef]
- Bauer, A.; Tronche, F.; Wessely, O.; Kellendonk, C.; Reichardt, H.M.; Steinlein, P.; Schütz, G.; Beug, H. The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev. 1999, 13, 2996–3002. [Google Scholar] [CrossRef]
- Narla, A.; Vlachos, A.; Nathan, D.G. Diamond Blackfan anemia treatment: Past, present, and future. Semin. Hematol. 2011, 48, 117–123. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Narla, A. Perspective on Diamond-Blackfan anemia: Lessons from a rare congenital bone marrow failure syndrome. Leukemia 2018, 32, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed]
- Lossignol, D. A little help from steroids in oncology. J. Transl. Intern. Med. 2016, 4, 52–54. [Google Scholar] [CrossRef]
- Schäcke, H.; Döcke, W.D.; Asadullah, K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol. Ther. 2002, 96, 23–43. [Google Scholar] [CrossRef]
- Aljebab, F.; Choonara, I.; Conroy, S. Systematic Review of the Toxicity of Long-Course Oral Corticosteroids in Children. PLoS ONE 2017, 12, e0170259. [Google Scholar] [CrossRef]
- Grad, I.; Picard, D. The glucocorticoid responses are shaped by molecular chaperones. Mol. Cell. Endocrinol. 2007, 275, 2–12. [Google Scholar] [CrossRef]
- Lamberts, S.W.; Huizenga, A.T.; de Lange, P.; de Jong, F.H.; Koper, J.W. Clinical aspects of glucocorticoid sensitivity. Steroids 1996, 61, 157–160. [Google Scholar] [CrossRef][Green Version]
- John, S.; Sabo, P.J.; Thurman, R.E.; Sung, M.H.; Biddie, S.C.; Johnson, T.A.; Hager, G.L.; Stamatoyannopoulos, J.A. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 2011, 43, 264–268. [Google Scholar] [CrossRef]
- Samarasinghe, R.A.; Witchell, S.F.; DeFranco, D.B. Cooperativity and complementarity: Synergies in non-classical and classical glucocorticoid signaling. Cell Cycle 2012, 11, 2819–2827. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.J.; Scher, B.M.; Waxman, S.; Scher, W. Inhibition of mouse GATA-1 function by the glucocorticoid receptor: Possible mechanism of steroid inhibition of erythroleukemia cell differentiation. Mol. Endocrinol. 1993, 7, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.Z.; Cidlowski, J.A. The origin and functions of multiple human glucocorticoid receptor isoforms. Ann. N. Y. Acad. Sci. 2004, 1024, 102–123. [Google Scholar] [CrossRef]
- Koper, J.W.; van Rossum, E.F.; van den Akker, E.L. Glucocorticoid receptor polymorphisms and haplotypes and their expression in health and disease. Steroids 2014, 92, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Jewell, C.M.; Cidlowski, J.A. Molecular evidence for a link between the N363S glucocorticoid receptor polymorphism and altered gene expression. J. Clin. Endocrinol. Metab. 2007, 92, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- van Rossum, E.F.; Lamberts, S.W. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog. Horm. Res. 2004, 59, 333–357. [Google Scholar] [CrossRef]
- Lonetti, A.; Indio, V.; Dianzani, I.; Ramenghi, U.; Da Costa, L.; Pospíšilová, D.; Migliaccio, A.R. The Glucocorticoid Receptor Polymorphism Landscape in Patients With Diamond Blackfan Anemia Reveals an Association Between Two Clinically Relevant Single Nucleotide Polymorphisms and Time to Diagnosis. Front. Physiol. 2021, 12, 745032. [Google Scholar] [CrossRef]
- Horos, R.; Ijspeert, H.; Pospisilova, D.; Sendtner, R.; Andrieu-Soler, C.; Taskesen, E.; Nieradka, A.; Cmejla, R.; Sendtner, M.; Touw, I.P.; et al. Ribosomal deficiencies in Diamond-Blackfan anemia impair translation of transcripts essential for differentiation of murine and human erythroblasts. Blood 2012, 119, 262–272. [Google Scholar] [CrossRef]
- Boultwood, J.; Pellagatti, A. Reduced translation of GATA1 in Diamond-Blackfan anemia. Nat. Med. 2014, 20, 703–704. [Google Scholar] [CrossRef]
- Gutiérrez, L.; Caballero, N.; Fernández-Calleja, L.; Karkoulia, E.; Strouboulis, J. Regulation of GATA1 levels in erythropoiesis. IUBMB Life 2020, 72, 89–105. [Google Scholar] [CrossRef]
- Akiyama, M.; Yanagisawa, T.; Yuza, Y.; Yokoi, K.; Ariga, M.; Fujisawa, K.; Hoshi, Y.; Eto, Y. Successful treatment of Diamond-Blackfan anemia with metoclopramide. Am. J. Hematol. 2005, 78, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.A.; Zermati, Y.; Vandekerckhove, J.; Cathelin, S.; Kersual, J.; Dussiot, M.; Coulon, S.; Moura, I.C.; Zeuner, A.; Kirkegaard-Sørensen, T.; et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007, 445, 102–105. [Google Scholar] [CrossRef]
- Sankaran, V.G.; Ghazvinian, R.; Do, R.; Thiru, P.; Vergilio, J.A.; Beggs, A.H.; Sieff, C.A.; Orkin, S.H.; Nathan, D.G.; Lander, E.S.; et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J. Clin. Investig. 2012, 122, 2439–2443. [Google Scholar] [CrossRef]
- Zhou, X.; Medina, S.; Bolt, A.M.; Zhang, H.; Wan, G.; Xu, H.; Lauer, F.T.; Wang, S.C.; Burchiel, S.W.; Liu, K.J. Inhibition of red blood cell development by arsenic-induced disruption of GATA-1. Sci. Rep. 2020, 10, 19055. [Google Scholar] [CrossRef] [PubMed]
- Iskander, D.; Wang, G. Single-cell profiling of human bone marrow progenitors reveals mechanisms of failing erythropoiesis in Diamond-Blackfan anemia. Sci. Transl. Med. 2021, 13, eabf0113. [Google Scholar] [CrossRef]
- Ludwig, L.S.; Gazda, H.T.; Eng, J.C.; Eichhorn, S.W.; Thiru, P.; Ghazvinian, R.; George, T.I.; Gotlib, J.R.; Beggs, A.H.; Sieff, C.A.; et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat. Med. 2014, 20, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Wei, L.; Barrett, E.J. Dexamethasone inhibits the stimulation of muscle protein synthesis and PHAS-I and p70 S6-kinase phosphorylation. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E570–E575. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Roh, S.; Lee, J.S.; Yang, B.H.; Choi, M.R.; Chai, Y.G.; Kim, S.H. The Effects of Venlafaxine and Dexamethasone on the Expression of HSP70 in Rat C6 Glioma Cells. Psychiatry Investig. 2010, 7, 43–48. [Google Scholar] [CrossRef]
- Paulson, R.F.; Hariharan, S.; Little, J.A. Stress erythropoiesis: Definitions and models for its study. Exp. Hematol. 2020, 89, 43–54.e42. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.H.; Amato, D.; Saunders, E.F. Haem synthesis in the Diamond-Blackfan syndrome. Br. J. Haematol. 1975, 31, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Malgor, L.A.; Torales, P.R.; Klainer, T.E.; Barrios, L.; Blanc, C.C. Effects of dexamethasone on bone marrow erythropoiesis. Hormones 1974, 5, 269–277. [Google Scholar] [CrossRef]
- Zhao, W.; Kitidis, C.; Fleming, M.D.; Lodish, H.F.; Ghaffari, S. Erythropoietin stimulates phosphorylation and activation of GATA-1 via the PI3-kinase/AKT signaling pathway. Blood 2006, 107, 907–915. [Google Scholar] [CrossRef]
- Sjögren, S.E.; Flygare, J. Progress towards mechanism-based treatment for Diamond-Blackfan anemia. Sci. World J. 2012, 2012, 184362. [Google Scholar] [CrossRef] [PubMed]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From stem cell to red cell: Regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef]
- Flygare, J.; Rayon Estrada, V.; Shin, C.; Gupta, S.; Lodish, H.F. HIF1alpha synergizes with glucocorticoids to promote BFU-E progenitor self-renewal. Blood 2011, 117, 3435–3444. [Google Scholar] [CrossRef]
- Russo, A.; Russo, G. Ribosomal Proteins Control or Bypass p53 during Nucleolar Stress. Int. J. Mol. Sci. 2017, 18, 140. [Google Scholar] [CrossRef]
- Nicolas, E.; Parisot, P.; Pinto-Monteiro, C.; de Walque, R.; De Vleeschouwer, C.; Lafontaine, D.L. Involvement of human ribosomal proteins in nucleolar structure and p53-dependent nucleolar stress. Nat. Commun. 2016, 7, 11390. [Google Scholar] [CrossRef]
- Le Goff, S.; Boussaid, I.; Floquet, C.; Raimbault, A.; Hatin, I.; Andrieu-Soler, C.; Salma, M.; Leduc, M.; Gautier, E.F.; Guyot, B.; et al. p53 activation during ribosome biogenesis regulates normal erythroid differentiation. Blood 2021, 137, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Danilova, N.; Bibikova, E.; Covey, T.M.; Nathanson, D.; Dimitrova, E.; Konto, Y.; Lindgren, A.; Glader, B.; Radu, C.G.; Sakamoto, K.M.; et al. The role of the DNA damage response in zebrafish and cellular models of Diamond Blackfan anemia. Dis. Model. Mech. 2014, 7, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qian, W.; Weng, X.; Wu, Z.; Li, H.; Zhuang, Q.; Feng, B.; Bian, Y. Glucocorticoid receptor and sequential P53 activation by dexamethasone mediates apoptosis and cell cycle arrest of osteoblastic MC3T3-E1 cells. PLoS ONE 2012, 7, e37030. [Google Scholar] [CrossRef]
- Sengupta, S.; Vonesch, J.L.; Waltzinger, C.; Zheng, H.; Wasylyk, B. Negative cross-talk between p53 and the glucocorticoid receptor and its role in neuroblastoma cells. EMBO J. 2000, 19, 6051–6064. [Google Scholar] [CrossRef]
- Kapralova, K.; Jahoda, O.; Koralkova, P.; Gursky, J.; Lanikova, L. Oxidative DNA Damage, Inflammatory Signature, and Altered Erythrocytes Properties in Diamond-Blackfan Anemia. Int. J. Mol. Sci. 2020, 21, 9652. [Google Scholar] [CrossRef]
- Rio, S.; Gastou, M.; Karboul, N.; Derman, R.; Suriyun, T.; Manceau, H.; Leblanc, T.; El Benna, J.; Schmitt, C.; Azouzi, S.; et al. Regulation of globin-heme balance in Diamond-Blackfan anemia by HSP70/GATA1. Blood 2019, 133, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Sanner, B.M.; Meder, U.; Zidek, W.; Tepel, M. Effects of glucocorticoids on generation of reactive oxygen species in platelets. Steroids 2002, 67, 715–719. [Google Scholar] [CrossRef]
- Gerö, D.; Szabo, C. Glucocorticoids Suppress Mitochondrial Oxidant Production via Upregulation of Uncoupling Protein 2 in Hyperglycemic Endothelial Cells. PLoS ONE 2016, 11, e0154813. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Mohanty, P.; Hamouda, W.; Aljada, A.; Kumbkarni, Y.; Garg, R. Effect of dexamethasone on reactive oxygen species generation by leukocytes and plasma interleukin-10 concentrations: A pharmacodynamic study. Clin. Pharmacol. Ther. 1999, 66, 58–65. [Google Scholar] [CrossRef]
- Chen, L.; Hu, S.L.; Xie, J.; Yan, D.Y.; Weng, S.J.; Tang, J.H.; Wang, B.Z.; Xie, Z.J.; Wu, Z.Y.; Yang, L. Proanthocyanidins-Mediated Nrf2 Activation Ameliorates Glucocorticoid-Induced Oxidative Stress and Mitochondrial Dysfunction in Osteoblasts. Oxidative Med. Cell. Longev. 2020, 2020, 9102012. [Google Scholar] [CrossRef] [PubMed]
- Sjögren, S.E.; Siva, K.; Soneji, S.; George, A.J.; Winkler, M.; Jaako, P.; Wlodarski, M.; Karlsson, S.; Hannan, R.D.; Flygare, J. Glucocorticoids improve erythroid progenitor maintenance and dampen Trp53 response in a mouse model of Diamond-Blackfan anaemia. Br. J. Haematol. 2015, 171, 517–529. [Google Scholar] [CrossRef]
- Naithani, R.; Chandra, J.; Narayan, S.; Singh, V.; Dutta, A.K. Diamond-Blackfan anemia: Clinical features and treatment results in 4 cases. Hematology 2006, 11, 193–195. [Google Scholar] [CrossRef]
- Jayapal, S.R.; Lee, K.L.; Ji, P.; Kaldis, P.; Lim, B.; Lodish, H.F. Down-regulation of Myc is essential for terminal erythroid maturation. J. Biol. Chem. 2010, 285, 40252–40265. [Google Scholar] [CrossRef] [PubMed]
- Sloan, K.E.; Bohnsack, M.T.; Watkins, N.J. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013, 5, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.M.; Zhou, X.; Gatignol, A.; Lu, H. Ribosomal proteins L5 and L11 co-operatively inactivate c-Myc via RNA-induced silencing complex. Oncogene 2014, 33, 4916–4923. [Google Scholar] [CrossRef] [PubMed]
- Quarello, P.; Garelli, E.; Carando, A.; Brusco, A.; Calabrese, R.; Dufour, C.; Longoni, D.; Misuraca, A.; Vinti, L.; Aspesi, A.; et al. Diamond-Blackfan anemia: Genotype-phenotype correlations in Italian patients with RPL5 and RPL11 mutations. Haematologica 2010, 95, 206–213. [Google Scholar] [CrossRef]
- Oršolić, I.; Bursać, S.; Jurada, D.; Drmić Hofman, I.; Dembić, Z. Cancer-associated mutations in the ribosomal protein L5 gene dysregulate the HDM2/p53-mediated ribosome biogenesis checkpoint. Oncogene 2020, 39, 3443–3457. [Google Scholar] [CrossRef]
- Ajore, R.; Raiser, D.; McConkey, M.; Jöud, M.; Boidol, B.; Mar, B.; Saksena, G.; Weinstock, D.M.; Armstrong, S.; Ellis, S.R.; et al. Deletion of ribosomal protein genes is a common vulnerability in human cancer, especially in concert with TP53 mutations. EMBO Mol. Med. 2017, 9, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Medh, R.D.; Thompson, E.B. Glucocorticoid mediated transcriptional repression of c-myc in apoptotic human leukemic CEM cells. J. Steroid Biochem. Mol. Biol. 2000, 73, 195–202. [Google Scholar] [CrossRef][Green Version]
- Ausserlechner, M.J.; Obexer, P.; Böck, G.; Geley, S.; Kofler, R. Cyclin D3 and c-MYC control glucocorticoid-induced cell cycle arrest but not apoptosis in lymphoblastic leukemia cells. Cell Death Differ. 2004, 11, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wu, K.; Xiao, X.; Liao, J.; Hu, Q.; Chen, H.; Liu, J.; An, X. Autophagy as a regulatory component of erythropoiesis. Int. J. Mol. Sci. 2015, 16, 4083–4094. [Google Scholar] [CrossRef]
- Grosso, R.; Fader, C.M.; Colombo, M.I. Autophagy: A necessary event during erythropoiesis. Blood Rev. 2017, 31, 300–305. [Google Scholar] [CrossRef]
- Kang, Y.A.; Sanalkumar, R.; O’Geen, H.; Linnemann, A.K.; Chang, C.J.; Bouhassira, E.E.; Farnham, P.J.; Keles, S.; Bresnick, E.H. Autophagy driven by a master regulator of hematopoiesis. Mol. Cell. Biol. 2012, 32, 226–239. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef]
- Knight, Z.A.; Schmidt, S.F.; Birsoy, K.; Tan, K.; Friedman, J.M. A critical role for mTORC1 in erythropoiesis and anemia. eLife 2014, 3, e01913. [Google Scholar] [CrossRef]
- Yang, Z.; Keel, S.B.; Shimamura, A.; Liu, L.; Gerds, A.T.; Li, H.Y.; Wood, B.L.; Scott, B.L.; Abkowitz, J.L. Delayed globin synthesis leads to excess heme and the macrocytic anemia of Diamond Blackfan anemia and del(5q) myelodysplastic syndrome. Sci. Transl. Med. 2016, 8, 338ra367. [Google Scholar] [CrossRef]
- Liu, Q.; Luo, L.; Ren, C.; Zou, M.; Yang, S.; Cai, B.; Wu, L.; Wang, Y.; Fu, S.; Hua, X.; et al. The opposing roles of the mTOR signaling pathway in different phases of human umbilical cord blood-derived CD34(+) cell erythropoiesis. Stem Cells 2020, 38, 1492–1505. [Google Scholar] [CrossRef]
- Chauvin, C.; Koka, V.; Nouschi, A.; Mieulet, V.; Hoareau-Aveilla, C.; Dreazen, A.; Cagnard, N.; Carpentier, W.; Kiss, T.; Meyuhas, O.; et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 2014, 33, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Pospisilova, D.; Cmejlova, J.; Hak, J.; Adam, T.; Cmejla, R. Successful treatment of a Diamond-Blackfan anemia patient with amino acid leucine. Haematologica 2007, 92, e66–e67. [Google Scholar] [CrossRef]
- Payne, E.M.; Virgilio, M.; Narla, A.; Sun, H.; Levine, M.; Paw, B.H.; Berliner, N.; Look, A.T.; Ebert, B.L.; Khanna-Gupta, A. L-Leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood 2012, 120, 2214–2224. [Google Scholar] [CrossRef]
- Doulatov, S.; Vo, L.T.; Macari, E.R.; Wahlster, L.; Kinney, M.A.; Taylor, A.M.; Barragan, J.; Gupta, M.; McGrath, K.; Lee, H.Y.; et al. Drug discovery for Diamond-Blackfan anemia using reprogrammed hematopoietic progenitors. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Brumwell, A.; Fell, L.; Obress, L.; Uniacke, J. Hypoxia influences polysome distribution of human ribosomal protein S12 and alternative splicing of ribosomal protein mRNAs. Rna 2020, 26, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: Autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef]
- Malik, N.; Dunn, K.M.; Cassels, J.; Hay, J.; Estell, C.; Sansom, O.J.; Michie, A.M. mTORC1 activity is essential for erythropoiesis and B cell lineage commitment. Sci. Rep. 2019, 9, 16917. [Google Scholar] [CrossRef]
- Shimizu, N.; Yoshikawa, N.; Ito, N.; Maruyama, T.; Suzuki, Y.; Takeda, S.; Nakae, J.; Tagata, Y.; Nishitani, S.; Takehana, K.; et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab. 2011, 13, 170–182. [Google Scholar] [CrossRef]
- Fu, L.; Wu, W.; Sun, X.; Zhang, P. Glucocorticoids Enhanced Osteoclast Autophagy Through the PI3K/Akt/mTOR Signaling Pathway. Calcif. Tissue Int. 2020, 107, 60–71. [Google Scholar] [CrossRef]
- Polman, J.A.; Hunter, R.G.; Speksnijder, N.; van den Oever, J.M.; Korobko, O.B.; McEwen, B.S.; de Kloet, E.R.; Datson, N.A. Glucocorticoids modulate the mTOR pathway in the hippocampus: Differential effects depending on stress history. Endocrinology 2012, 153, 4317–4327. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.M.; Wu, L.G.; Cai, J.W.; Wu, L.T.; Liang, M. Dexamethasone suppresses osteogenesis of osteoblast via the PI3K/Akt signaling pathway in vitro and in vivo. J. Recept. Signal Transduct. 2019, 39, 80–86. [Google Scholar] [CrossRef]
- Kopriva, F.; Dzubak, P.; Potesil, J.; Hajduch, M. The anti-inflammatory effects of inhaled corticosteroids versus anti-leukotrienes on the lymphocyte P-glycoprotein (PGP) expression in asthmatic children. J. Asthma 2009, 46, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Spenerova, M.; Dzubak, P.; Srovnal, J.; Radova, L.; Burianova, R.; Konecny, P.; Salkova, S.; Novak, Z.; Pospisilova, D.; Stary, J.; et al. Combination of prednisolone and low dosed dexamethasone exhibits greater in vitro antileukemic activity than equiactive dose of prednisolone and overcomes prednisolone drug resistance in acute childhood lymphoblastic leukemia. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc. Czech Repub. 2014, 158, 422–427. [Google Scholar] [CrossRef]
- Ashley, R.J.; Yan, H.; Wang, N.; Hale, J.; Dulmovits, B.M.; Papoin, J.; Olive, M.E.; Udeshi, N.D.; Carr, S.A.; Vlachos, A.; et al. Steroid resistance in Diamond Blackfan anemia associates with p57Kip2 dysregulation in erythroid progenitors. J. Clin. Investig. 2020, 130, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Adcock, I.M.; Barnes, P.J. Molecular mechanisms of corticosteroid resistance. Chest 2008, 134, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, M.K.; Pazirandeh, A.; Davani, B.; Okret, S. p57Kip2, a glucocorticoid-induced inhibitor of cell cycle progression in HeLa cells. Mol. Endocrinol. 1999, 13, 1811–1822. [Google Scholar] [CrossRef] [PubMed]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of p53 inhibitors: Progress, challenges and perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef]
- Madden, S.K.; de Araujo, A.D. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).