Abstract
Men have limited options for contraception, despite the widely accepted public health benefits of it, placing the contraceptive burden solely on women. The current study focuses on inhibiting the PP1γ2 enzyme, which plays a role in sperm maturation and motility. The study considered three top compounds based on the findings of molecular docking. The three compounds exhibited a good interaction profile with a binding affinity score of D751-0223 (−8.7 kcal/mol), D751-014 (−8.1 kcal/mol), and N117-0087 (−8 kcal/mol) measured in kcal/mol. Molecular dynamics simulation (MDS) were performed on the PP1γ2–ligand complexes along with the Apo form. The results suggested that all the complexes were stable with no major deviations observed compared to Apo. The average RMSDs for PP1γ2-D751-0223, D751-014, and Apo were 1.27 Å, 1.73 Å, 1.39 Å, and 1.69 Å, respectively. The PP1γ2–ligand complexes were observed with unique salt bridge interactions such as Glu133-Arg137, Asp4-Lys107, Asp188-Arg116, and Glu120-Arg90. The principal component analysis (PCA) findings indicated that every complex had a distinctive motion state. Furthermore, the net MM/PBSA scores for D751-0223, D751-0143, and N117-0087 were −80.01 kcal/mol, −72.18 kcal/mol, and −64.26 kcal/mol, respectively, while the MM/GBSA and MM/PBSA values were −82, −73.07,−67.26 and −80.01, −72.18, −64.26, measured in kcal/mol, respectively. The WaterSwap energy estimation was performed to validate the former technique, and the findings demonstrated that PP1γ2-D751-0223 is a stable complex, with a value of −51.05 kcal/mol. This work provides a baseline to researchers for the identification of novel therapeutic approaches for non-hormonal male contraceptives.
1. Introduction
Due to unplanned pregnancies and growing populations worldwide, nations face economic, health, environmental, and social challenges [1]. One of the most crucial elements of family planning and reproductive health involves deciding when to have children. Contraceptives and family planning reached a new phase in 1960 following the advent of Enovid, the very first female hormonal birth control pill that contained mestranol and norethynodrel [2]. Women generally carry the majority of contraceptive responsibility because they have access to a range of hormone-based contraceptive substitutes in different forms [3]. However, more than forty percent (or 100 million) of pregnancies worldwide are unwanted. Approximately 40% of these result in unplanned births, and half end in abortions [4]. According to estimations, a number of women may inevitably die annually as a result of unsafe abortion practices brought on by unplanned pregnancies [5]. To tackle these global issues, there is a dire need to broaden the range of contraceptive alternatives, including male options [6]. For male methods, the unmet demand for contraceptives is significantly higher. There are presently only two types of male contraception: vasectomy, which is safe but not readily reversible, and condoms, which are cheap and efficient for preventing sexually transmitted diseases (STDs), but have a high failure rate of 15–20% in avoiding pregnancies [7]. Nearly 30% of individuals utilize condoms and vasectomy as their primary forms of birth control, considering the lack of other options. Thus, novel male contraceptive methods might decrease unexpected births, meet individuals’ contraceptive needs, and encourage gender equality in terms of the cost of using contraceptives [8].
All eukaryotic cells express Ser/Thr protein phosphatase 1 (PP1), one of the members of the phosphoprotein phosphatase (PPP) class [9]. The primary PP1 isoform in spermatozoa, the sperm-specific gamma 2 variant (PP1c2), is an essential regulator of sperm formation and the initiation and activation of sperm mobility [10]. Several PP1 isoforms (PP1a, PP1b, PP1c1, and PP1c2), having a significant level of sequence similarity (90%), are encoded by three genes [11]. The testis-enriched or sperm-specific PP1 is referred to as PP1γ2, and it is believed to be the primary isoform within mammals that induces PP1 function in spermatozoa [12]. Protein phosphatase inhibitor 2 (PPP1R2, also referred to as I2), protein phosphatase 1 regulating unit 7 (PPP1R7, also called SDS22), and protein phosphatase 1 regulating subunit 11 (PPP1R11, referred to as I3) have been the primary inhibitors that regulate PP1γ2 activity in sperm [10]. Their relationship with PP1γ2 changes as epididymis sperm grow. In short, PP1γ2 has been linked to all three inhibitors in caudal spermatozoa, yet only with I3 in immotile spermatozoa of the caput epididymis. Because of their ability to regulate PP1γ2 activity, these alterations are crucial for the growth of motility [10]. Thus, PP1γ2 has emerged as an attractive drug target for reversible male contraception due to its pivotal role in sperm motility and maturation. Inhibition of this enzyme will stop the biochemical pathways from maturation of sperm by making them non-functional without impairing long-term reproductive health or creating hormonal disorders [13].
Considering the broad field of drug discovery, where chemistry and biology come together, finding and developing an innovative treatment can be a laborious and expensive task [14]. A crucial aspect of modern drug discovery is the powerful and multidisciplinary order of computer-aided drug design, also known as CADD [15]. It discovers and enhances potential drug candidates by fusing biological knowledge with computer systems techniques. The amalgamation of several approaches contributes to CADD’s adaptability and efficacy in the pharmaceutical sector [16]. The broad spectrum of techniques and methods that encourage CADD adds to its scope and adaptability. The efficacy of this field comes from its broad spectrum of methods, such as drug metabolism prediction and structural modeling [17]. For CADD to achieve excellent oral drug-likeness, compounds should ideally minimize violations of criteria such as molecular weight, lipophilicity, hydrogen bond donors, and acceptors. This is achieved by following Lipinski’s rule [18]. Drug development is expedited and enhanced through this systematic integration of CADD methods and adherence to drug-likeness criteria, showing the flexibility and important impact of CADD in a range of areas and the pharmaceutical sector [19]. The present study was conducted to explore novel therapeutic drug molecules against PP1γ2 by using various cheminformatics and bioinformatics techniques. The study utilized different in silico approaches as molecular docking, molecular dynamics simulation, pharmacokinetics profiling, and post-simulation analysis (Figure 1). The study identified three potent inhibitors, but extensive in vivo and in vitro investigation is needed to validate the potential efficacy of the compounds.
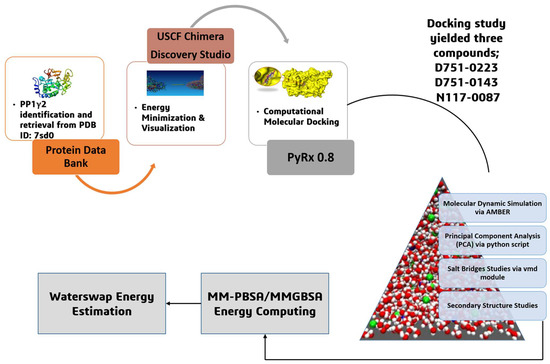
Figure 1.
The overall systematic computational study initiated with the retrieval of protein, followed by energy minimization and visualization, molecular docking and molecular dynamic simulation, principal component analysis, salt bridges studies, secondary structure analysis, MM-PBSA/GBSA energy computing, and WaterSwap energy estimation.
2. Materials and Methods
2.1. Identification, Retrieval, and Preparation of Target Receptor
The three-dimensional (3D) structure of protein phosphatase 1 gamma 2 (PP1γ2) was fetched from the Protein Data Bank (PDB) (https://www.rcsb.org/?ref=nav_home, accessed on 10 February 2025) using ID 7SD0 in UCSF Chimera v1.17. At the time of research work, PDB ID:7SD0 was the latest available structure of PP1γ2. The chain C (sequence length of 292) represents the catalytic subunit of PP1γ2 and was selected for docking investigations despite the structure having multiple chains that form a trimeric complex. All water molecule heteroatoms (apart from any necessary cofactors) and non-target chains were deleted. Following this, polar hydrogens were added to the structure, and Kollman charges were allocated using AutoDock Tools to prepare it for docking. The protein structure was visualized in Discovery Studio Visualizer v2024.
2.2. Identification of Binding Pockets
Identifying or predicting the protein’s binding regions is essential for better interaction analysis [20]. The identified active pockets were Arg132, Lys141, Lys147, and Lys150. The active site residues were obtained from [21], who characterized the binding interface using crystallographic data.
2.3. Identification and Preparation of Compound Library
A natural product-based compound library consisting of 4561 molecules was retrieved from the ChemDiv database (https://www.chemdiv.com/catalog/focused-and-targeted-libraries/natural-product-based-library/, accessed on 15 February 2025). This readily accessible library consists of small molecules from diverse sources with drug-like characteristics and is inspired by natural products. The library was downloaded in a structure Data file (SDF) format. Ligand parameterization was performed using UCSF Chimera v1.17 before using them in structure-based screening [22]. The ligands were prepared using Dock Prep and the AutoDock Vina plugin. The topology and parameter files were generated via the internally run Parmck tool in UCSF Chimera v.17. The ligand library was then imported to PyRx 0.8, where energy minimization was carried out using Open Babel [23]. Partial charge assignment was performed on the ligands, which were converted into AutoDock Ligand format (PDBQT) [24].
2.4. Screening of Compound Library Against the Receptor
A key technique in computer-assisted drug design and structural molecular biology is molecular docking [25]. Ligand–protein docking aims to figure out the most prevalent modes of binding between a ligand and a known protein 3D structure [26]. By using PyRx 0.8, all 4561 compounds were screened against the target receptor [23]. Docking of each ligand with the receptor was performed 50 times. The Vina search space during the screening process was set as follows: Center (X: 100.307 Å, 115.900 Å, and Z: 116.925 Å) and dimensions of 30 Å on the XYZ axes. Thousands of candidates were filtered against the targeted protein based on the lowest binding energy in kcal/mol [27]. To remove undesirable compounds and perform extensive optimization, the compounds with the lowest binding affinity were chosen. In parallel, docking validation was performed by extracting co-crystalized ligands from a PDB ID: 4QDI and re-docked with the mentioned PDB MurF protein using the same procedure discussed above [28]. The validation procedure reported an RMSD value of 0.12 Å, thus illustrating that the protocol is valid. A DSV-formatted Excel document with the binding affinities score and interaction information was retrieved.
2.5. Interpretation and Interaction Analysis of Docking Findings
Using AutoDock Vina v4.1 through PyRx 0.8, the docking affinity of compounds was predicted, and only the best binders to the targeted receptor were selected for intermolecular binding conformation and interactions network analysis [29]. Each selected ligand was then subjected to a short-term evaluation where the most optimal binding mode of the compound at the protein active pocket was selected based on the lowest binding energy (in kcal/mol) [30]. Another parameter of evaluation was the enrichment of the ligand interactions network at the active pocket [31]. The best docked complexes, by considering the criteria discussed above, were imported into Discovery Studio Visualizer v2024 for interaction analysis and evaluation [32]. It offered details regarding the complex’s atom type, interactions, forces, and bond lengths [33].
2.6. Validating Lipinski Rule of 5 and Pharmacokinetics Assessment
The drug-like characteristics of the best active molecule were studied using the Swiss ADME http://www.swissadme.ch/ (accessed on 10 February 2025) online server accessed on 18 February 2025 [34]. The compound’s standard simplified Molecular Input Line Entry System (SMILES) was given as input [35]. The molecular structure and bioactivity of the drugs with high affinities were predicted using the Swiss ADME. This online server estimates the number of Lipinski’s rule violations about the following parameters: logP, polar surface area, weight, variety of atoms, length of OH, length of rotatable bonds, quantity, enzymes, and nuclear receptors [36]. Moreover, the compounds were evaluated for physicochemical characteristics, medicinal chemistry, lipophilicity, and pharmacokinetic profile.
2.7. Molecular Dynamics Simulation
The docked complexes were subjected to molecular dynamics simulation using the AMBER v20 software. It offers information about the accuracy of the intermolecular interactions and predicts how proteins will respond when the compounds interact with them in dynamic environments [36]. The docked complex was provided as an input in PDB format for this analysis [31]. The MD simulations were performed in three steps: (1) system preparation, (2) preprocessing step, and (3) production run [37]. The AMBERv20 software suite was employed to perform MD simulations using the ff19SB force field for the protein and the GAFF2 [38] force field for ligand parameters, which were produced using Antechamber [39]. An octahedral box of TIP3P water molecules was used to solvate the protein–ligand complexes, keeping a padding distance of 12 Å. To neutralize the system, Na+ counter ions were added [40]. The energy minimization was carried out in two stages: steepest descent and conjugate gradient algorithms for a total of 3000 rounds. During the process, the restraining force of 100 kcal/mol on hydrogen atoms, water, and sodium ions was applied, followed by a 5 kcal/mol energy constraint in the second round. Firstly, the minimization of ions and water with protein and ligands was carried out, followed by the minimization of the whole system [41]. Each system was then heated up to 310 K with a restraint of 5 kcal/mol on the carbon alpha atoms. The canonical ensemble was applied during the heating step. Afterward, equilibration was performed for 1 ns using the NPT ensemble with no restraints. The MD simulation was run for 200 ns, and the trajectories were analyzed by the xmgrace v5.1 module [42]. The SHAKE algorithm was used to remove stretching of fast bond motions [43]. The particle mesh Ewald method was employed to approximate long-range interactions [44]. The CPPTRAJ module of AMBER was used for MD trajectory analyses [45].
2.8. Salt Bridges (SS) Analysis
Protein–ligand complexes can be stabilized by non-covalent salt bridges (SS), which are effective bonds in drug development [46]. For instance, PP1γ2 can be inhibited by small compounds, where SS can play a crucial role. The SS analysis was performed by the Visual Molecular Dynamics (VMD) software v1.93, developed by University of Illinois at Urbana, CA, USA [47]. The distance cut-off set during this analysis was 3.0.
2.9. Principal Component Analysis (PCA)
PCA is an approach that transforms coordinated observations into orthogonal vectors called principal components. PCs can be explained as variance in data. PCA helps reduce the dataset’s complexity, promoting understanding while minimizing information loss [48]. PCA is widely used in downstream MD analysis for identifying dominant motions and reducing dimensionality. The collective motions of atoms are represented by the PC1, PC2, PC3, and PC4 [36]. The AMBER CPPTRAJ program was utilized for plotting PCA.
2.10. Secondary Structure Analysis
A secondary structure analysis was performed to assess the structural changes and patterns in the protein structure upon binding to the ligand [49]. The proportion of secondary structure content in a protein is a crucial statistic in the investigation of changes in structural behavior [50]. The secondary structure analysis was carried out using the AMBER CPPTRAJ module.
2.11. MMPB/GBSA Calculations
The most efficient post-MD technique to calculate the binding free energy of a system (ligand–receptor) is the Molecular Mechanics/Generalized Born Surface Area (MMGB/PBSA) [51]. The analysis was performed using the AMBER MMPBSA.py module. The MMPB/GBSA uses the following equations to carry out the calculations [52]:
where binding net energy is denoted with ∆Gbind (Equation (1)), calculated by utilizing Equation (2). T∆S represents the variation in conformational entropy. ∆Ggas denotes the sum of the bond angle, Van der Waals, and electrostatic part of the internal energy (Equation (3)), while the ∆Gsol, called salvation energy, is the combination of polar solvation energy (∆EGB) and non-polar solvation energy (∆ESURF). The MMPB/GBSA analysis was performed on 5000 frames picked at regular intervals from simulation trajectories.
∆Gbind = G complex − Gprotein − Gligand
∆Gbind = ∆Ggas + ∆Gsol − T∆S
∆Ggas = Bond + Angle + Dihed + EEL + VDWAAL
∆Gsol = ∆EGB + ∆ESURF
2.12. WaterSwap Energy Estimation
The complicated nature of enzyme–water and drug–water interactions is usually not taken into consideration by the MM/GBPBSA approach, which uses an implicit water system and takes snapshots at regular times of MD simulations [53]. On the other hand, WaterSwap addressed the limitations of MM/GBPBSA by employing an explicit water model. A computational tool called WaterSwap can calculate the absolute binding free energy by swapping equal volume water in the protein binding pocket with a ligand [54]. Three algorithms were used for this calculation: free-energy perturbation (FEP), Bennett’s acceptance ratio (BAR), and thermodynamic integration (TI) [55]. An energy difference of 1 kcal/mol among the mentioned three algorithms indicates that the system’s energy converged well.
3. Results
3.1. Identification, Retrieval, and Preparation of Male Contraceptives with the Main PP1γ2
PP1γ2 is testis-specific and helps in sperm maturation. Inhibiting this is a promising strategy for non-hormonal male contraceptives [56]. The crustal structure of the target enzyme was downloaded from PDB via ID: 7sd0. The target protein is accessible at 2.95 Å by the electron microscopy method. The protein has Global Symmetry: Asymmetric-C1 and Global Stoichiometry: Hetero 3-mer—A1B1C1. The structure was energy-minimized and subsequently visualized by Discovery Studio [32]. The crystal structure, along with the active pockets labeled, is depicted in Figure 2.
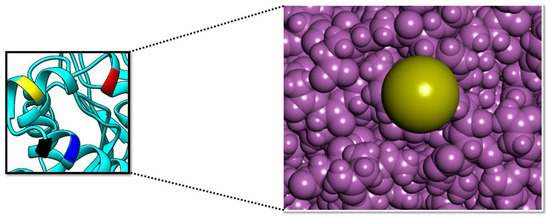
Figure 2.
The surface of the PP1γ2 active pockets (right side); the surface of the Arg132 (yellow), Lys141 (red), Lys147 (black), and Lys150 (blue) active pockets (left side). The purple color shows the active site surface in close view.
3.2. Molecular Docking Analysis
In silico molecular docking was carried out using AutoDock Vina to analyze the molecular interactions and the binding affinity of the protein with the Natural Product-Based Library consisting of 4561 compounds. To identify the best binding from the drug library, target-based virtual screening was performed [57]. The docking findings yielded the top 10 compounds based on their docking score, ranging from −8.7 to −7 kcal/mol. Binding mode and interaction analysis were performed on the first three hits that were considered to be the best binding compounds based on their lower binding affinity value. The top 10 compounds selected are given in Table 1, presenting detailed information about the compound’s chemical name, binding score, chemical structure, and H-bond interactions involved.

Table 1.
The top 10 hits with their IDs, chemical name and structure, binding affinities score, and associated hydrogen bond interactions.
3.3. Docking Analysis and Interpretation of Selected Hits
Each of the three compounds is strongly attached to the binding pocket of PP1γ2. The top-1 hit compound (D751-0223) is 2-methyl-5-(3-(14-methyl-5-oxo-7,8,13b,14-tetrahydroindolo[2″,3′:3,4]pyrido[2,1-b]quinazolin-13(5H)-yl)propanamido)-1,3,4-thiadiazole-3,4-diium, where the compound is strongly bound to three hydrogen atoms as VAL 223, ASP197, and GLU218. Besides h-bonding, several van der Waals interactions were noted: TRP 216, CYS202, GLY222, GLY199, and GLN198 (Figure 3A). The top hit (D751-0143) is N-(3-(3-methyl-1,2,4-oxadiazolidin-5-yl)propyl)-2-(14-methyl-5-oxo-7,8,13b,14-tetrahydroindolo[2′,3′:3,4]pyrido[2,1-b]quinazolin-13(5H)-yl)acetamide, showing three h-bonds, Glu218, Asp197, and Val223, while the van der Waals interactions formed were Trp216, Gly217, Ser224, Gly222, and Pro178 (Figure 3B). (N117-0087) is 3-(4-(4-(4-fluorophenyl) piperazine-1-carbonyl) piperidine-1-carbonyl) octahydro-1H-1, 5-methanopyrido [1, 2-a] [1, 5] diazocin-8(2H), with one h-bond observed with residue GLU218. On the other hand, the compound formed van der Waals interactions with Phe235, Lys237, Val231, Leu180, Ser177, and Gln181, along with other alkyl and pi-alkyl interactions (Figure 3C). The top three hits were found to possess good binding affinity and appropriate interactions with the residues of the enzyme’s active site. Hence, the three hits seem to be novel inhibitors of PP1γ2. The intermolecular binding mode of the selected compounds with the PP1γ2 enzyme can be seen in Figure 3D.
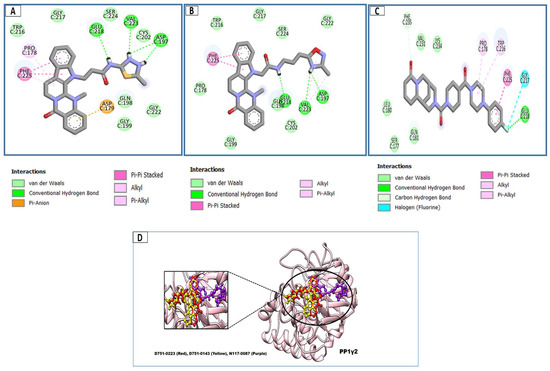
Figure 3.
The two-dimensional (2D) and three-dimensional (3D) images of the top three hits: D751-0223 (A), D751-0143 (B), and N117-0087 (C), with a close-up view of the binding poses and interactions (D).
3.4. Lipinski Rule of 5 and Pharmacokinetics Characteristics
Swiss ADME assessed the criteria of Lipinski’s rule and other parameters of drug-likeness, such as mass; logP; polar surface area; range of atoms; range of O, N, or NH; hydrogen bond donors; total number of hydrogen bond recipients; and range of rotatable bonds, as shown in Table S1. The top three hits met the Lipinski Rule 5 criteria and were deemed drug-like. Moreover, the physiochemical features, pharmacokinetics, and medicinal chemistry were within the acceptable range. The compounds were processed for further computational screening through MD simulation.
3.5. Molecular Dynamic Simulation of PP1γ2–Ligand Complexes and Apo
The stability and dynamics of PP1γ2-compound complexes were studied using a 200 ns molecular simulation. To determine the intermolecular strength of interaction and steady dynamics of complexes, multiple statistical analyses were carried out from the simulation trajectories, which include the radius of gyration (RoG), RMSD, root mean square fluctuation (RMSF), beta factor, and solvent accessible surface area (SASA) [58]. The lower RMSD shows the greater stability of the receptor protein upon ligand binding and vice versa [59]. The RMSD (Figure 4A) of all three complexes showed that they attained stability at the start of the simulation period, with the ligand-bound systems exhibiting decreased deviations compared to Apo, indicating better stability upon ligand binding. The average RMSD scores for D751-0223, D751-0143, N117-0087, and Apo are 1.27 Å, 1.73 Å, 1.39 Å, and 1.69 Å, respectively. The RMSF of the complexes was evaluated afterward to identify residue-specific fluctuations in the compound’s presence at the enzyme’s active region. The majority of the receptor residues are in a good stable range, with a mean RMSF < 2 Å, as shown in Table 2. Certain variations that lead to a higher RMSF are driven by specific residues with notable fluctuations around 200 in the Apo. The fluctuations then decreased upon ligand binding (Figure 4B). The analysis of the beta factor validated the flexibility seen in the RMSF plot, representing greater values of Apo at 200 residues. The average beta factor scores were D751-0223 (14.75 Å), D751-0143 (20.29 Å), N117-0087 (15.56 Å), and Apo (17.29 Å). The PP1γ2 rigidity and compactness during the 200 ns simulation period were then assessed by the radius of the gyration plot. A higher ROG score represents reduced rigidity, while a lower value suggests a more compact structure of PP1γ2 (Figure 4C). The D751-0223, D751-0143, and N117-0087 showed lower RoG compared to Apo, suggesting more compactness upon ligand binding. The RoG values for the top three hits are mentioned in Table 2. Similarly, the beta factor analysis was performed, which confirms the RMSF analysis and validates that there are no major residue-wise fluctuations (Figure 4D).
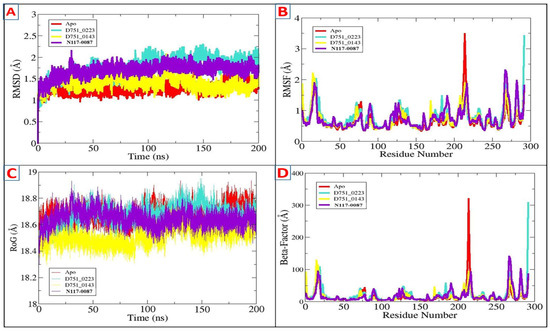
Figure 4.
Findings of MDs through AMBER xmgrace module. (A) RMSD, (B) RMSF, (C) RoG, and (D) beta factor for D751-0223 (cyan), D751-0143 (yellow), and N117-0087 (purple) in comparison with Apo (Red).
Table 2.
The simulation values for the top three complexes, D751-0223, D751-0143, and N117-0087, in comparison to Apo.
Further, ligand RMSD analysis was performed in order to investigate ligand dynamics stability over the course of simulation time (Figure 5). As can be seen in the figure, the RMSD plot of each ligand shows constant stability with no observed major fluctuations. The minor fluctuations noticed were due to flexible loops in the protein structure, which naturally allow the protein to accommodate the bounded ligand. The mean RMSDs of D751-0223 (cyan), D751-0143 (yellow), and N117-0087 (purple) are 1.73 Å, 1.39 Å, and 1.69 Å, respectively. The analysis of solvent-accessible surface area was conducted to assess the surface region of PP1γ2 exposed to solvents such as water [58]. D751-0223-PP1γ2 complex showed a good SASA score with higher exposure to a solvent, indicating better stability in its structure. The maximum SASAs for the top three PP1γ2 complexes, along with Apo, were 140.39 Å2, 14,014.4 Å2, 136,462.7 Å2, and 13,850.9 Å2. The SASAs indicate no major water molecule concentration shift, indicating the stable equilibrium of the protein–ligand complexes (Figure 6).
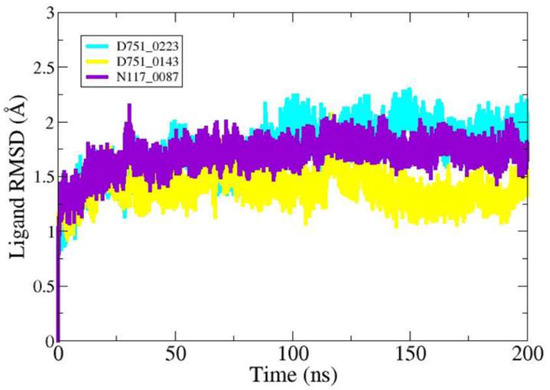
Figure 5.
Compounds’ RMSD over simulation time.
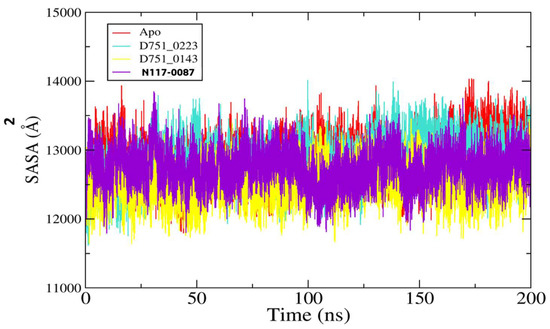
Figure 6.
The analysis of SASA for D751-0223 (cyan), D751-0143 (yellow), and N117-0087 (purple) in comparison to Apo (red).
3.6. Salt Bridge Studies: (SB)
Salt bridges are formed when proteins containing oppositely charged residues are sufficiently connected to attract each other via electrostatic attraction [60]. While they are unlikely to naturally increase a protein’s free energy of unfolding, they can contribute to the protein structure and the unique characteristics of interactions. Table 3 offers a detailed overview of the SB interactions of complexes and the Apo form. SB non-covalent interactions that combine electrostatic interactions between charged residues are typically positive and negative [61]. The greatest number of salt bridges in proteins is found between the negatively charged acidic Asp and Glu residues and the positively charged amino acid residues, such as Lys or Arg [62]. The permissible distance for SB is <4 Å [46]. The complexes and the Apo form that shared common SB interactions are Glu96-Arg137, Glu178-Arg182, Glu28-Arg9, and Asp132-Arg136. These interactions were redundant and repetitive, suggesting their role in protein stability and functionality. The unique SBs observed are Glu133-Arg137 (D751-0223), Asp4-Lys107 (D751-0223 and Apo), Asp188-Arg116 (D751-0143), and Glu120-Arg90 (N117-0087 and Apo), as shown in Figures S1 and S2. The unique SBs formed as a result of conformational variations upon ligand binding to the protein, supporting the stability of complexes in specified functional conditions. The complexes remained stable during the 200ns simulation since the SB interactions length was under the permissible range (<4 Å).
Table 3.
The salt bridge interactions for D751-0223, D751-0143, N117-0087, and Apo. SB (Salt bridges).
3.7. Principal Component Analysis (PCA)
The structural and energy data from MD simulation on protein–ligand complexes and Apo proteins are evaluated utilizing principal component analysis (PCA) [63]. The coordinate covariance matrix generated from the 200 ns MD simulation of hit compound complexes was subjected to PCA to capture the most noticeable motions over the MD simulation [64]. The PCA was plotted for the first two components, such as PC1 and PC2, capturing the majority of the data. Here, the PCA for D751-0223-PP1γ2, D751-0143-PP1γ2, and N117-0087-PP1γ2 complexes was performed along with Apo (unbound state). The periodic changes were noticed by the color gradient changing from purple to yellow. Every complex had a distinctive motion style, according to PCA. With blue dots at the start and purple at the end of the spectrum, the motion of the D751-0143, N117-0087, and Apo forms was relatively scattered (Figure 7). In contrast, the D751-0223-PP1γ2 exhibited a compact and clustered form of motions covering the majority of conformational data in PC1 compared to other complexes. On the contrary, the N117-0087-PP1γ2 (C) and Apo form (D) exhibited two distinct clusters, which may show transitions between multiple conformational states, suggesting less stable binding. The PCA for PP1γ2–ligand complexes, along with Apo, is depicted in Figure 7.
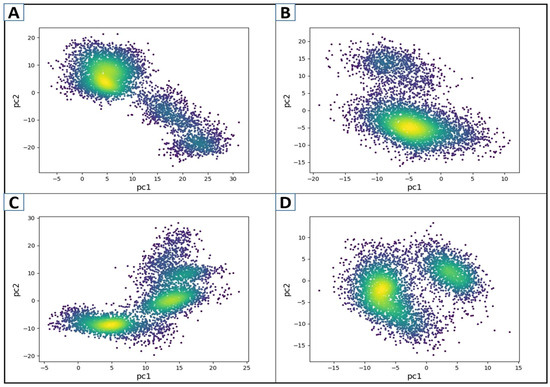
Figure 7.
The PCA plots of the first two components, PC1 and PC2, for D751-0223-PP1γ2 (A), D751-0143-PP1γ2 (B), N117-0087-PP1γ2 (C), and Apo form (D). Color gradient represents simulation frames (from blue to yellow to red).
3.8. Secondary Structure Studies (SS)
To determine the changes in secondary structures induced by the binding of the specified compound, an analysis of secondary structure was carried out on the complexes and Apo form [65]. The percentage of secondary structure concentration in a protein is an important variable in the study of structural behavior changes [50]. Figure 8 depicts the SS changes across their different residues. The y-axis in the figure represents the frequency of various structural conditions, while the x-axis shows the residue numbers. The three rows in each figure of complexes show different significances. The row named “Extended” designates extended structures and beta-sheets, while the “percentage” indicates the alpha-helical sites of the protein. Furthermore, the “other” sections in the figure specify coils and turns. The novel identified compounds’ secondary structure percentage stayed mostly unchanged. However, it seems that N117-0087 showed a consistent SS. With extended sites and fewer unstructured residues. The portion of the alpha helix and beta sheets was largely unchanged, suggesting that ligand binding did not cause significant changes in the structure. This shows a non-disruptive interaction of the protein and ligand.
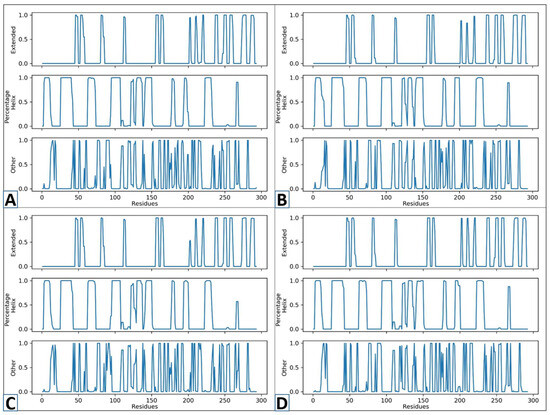
Figure 8.
The secondary structure studies of D751-0223 (A), D751-0143 (B), N117-0087 (C), and Apo (D) from.
3.9. MM/GBSA and MM/PBSA Calculations
In computer-assisted drug design, the MM/GBSA and MM/PBSA techniques are frequently utilized to evaluate a drug’s binding affinity towards a specific biological macromolecule [66]. One of the benefits of these techniques is that they require fewer computing resources than chemical binding free-energy techniques [67]. The strategies additionally act as endpoint ways of evaluating the various binding energies resulting from the drugs’ interaction with the target. All three virtually identified hit compounds exhibited strong Van der Waals and electrostatic energy contributions. The van der Waals contributed significantly to all compounds, followed by electrostatic energy. These two types of energy combine to form the gas-phase energy of the compounds. The MMGBSA/PBSA values are given in detail in Table 4. The primary field of difference between the two methods is how they handle solvation effects, yet both calculate binding free energy using molecular mechanics energies [36]. While MM/PBSA utilizes the Poisson–Boltzmann (PB) model, which solves the PB equation to produce more precise electrostatic solvation energy values, MM/GBSA uses the Generalized Born (GB) model, which corresponds to the solvent environment more quickly but less correctly. While MM/GBSA delivers far quicker processing, MM/PBSA is typically thought to be more physically demanding. Combining the two enables a complementary assessment of binding energetics [68]. The net MM/GBSA energy for D751-0223 (−82 kcal/mol), D751-0143 (−73.07 kcal/mol), and N117-0087 (−67.26 kcal/mol). Furthermore, the net MP/GBSA scores for D751-0223, D751-0143, and N117-0087 were −80.01 kcal/mol, −72.18 kcal/mol, and −64.26 kcal/mol, respectively. According to the findings, each of the three compounds achieved a stable phase within the enzyme’s region and was bound by a strong system of chemical bonds.
Table 4.
The top compounds’ (D751-0223, D751-0143, and N117-0087) MM/GBPBSA binding free-energy investigation. The units of measurement are kcal/mol.
3.10. WaterSwap Energy Estimation
Protein–water, ligand–water, and protein–water–ligand interaction characteristics are considered by the precise solvation mechanism utilized by WaterSwap [69]. Since the MMGB/PBSA does not have this data, it cannot be used to precisely predict where water molecules might work in interactions between proteins and ligands. This is particularly important in cases where water molecules are employed to link the ligand to the receptor. The WaterSwap method is vital to figuring out total binding free energy and has been successfully utilized in a variety of biological settings [54]. After 1000 frames, the WaterSwap energies for each PL complex exhibited a considerable convergence. Furthermore, every number suggested that the intermolecular docked configuration was stable. Three algorithms, such as Bennet’s, free-energy perturbation (FEP), and thermodynamic integration (TI), were used to calculate the net mean (Table 5). The findings suggest that PP1γ2-D751-0223 ranked first, with a value of −51.05, followed by PP1γ2-D751-0143 (−47.21) and PP1γ2-N117-0087 (−37.2).
Table 5.
The WaterSwap energies for each complex.
4. Discussion
The world’s population, which has been increasing at an alarming pace, is estimated to exceed 9.8 billion people by 2050 [12]. Despite the provision of contraceptive methods, nearly 40% of pregnancies globally were unplanned between 2010 and 2014 [12]. Male contraceptive options are more limited and less frequently used than female options [70]. Hormonal control has remained the main objective of modern male contraceptive technology so far; however, the pharmaceutical sector stopped much of its efforts in this area after receiving reports of serious adverse effects [71].
The current study was conducted to develop non-hormonal male contraceptives by inhibiting PP1γ2 to avoid systemic adverse effects. In this research, a systematic in silico investigation was performed to target the enzyme PP1γ2, which plays a key role in sperm maturation. A compound library called the Natural Product-Based Library, consisting of 4561 compounds, was used against the target receptor PP1γ2. Extensive virtual screening was carried out to identify potential inhibitors of PP1γ2. The study selected the top 10 hits based on their binding affinities, indicating strong interactions with the target in a static environment. For further analysis, the three most promising compounds were chosen as lead compounds. These compounds—D751-0223, D751-0143, and N117-0087—exhibited binding affinities of −8.7 kcal/mol, −8.1 kcal/mol, and −8.0 kcal/mol, respectively. Molecular dynamics simulation was then performed in three phases: (i) system preparation, (ii) pre-processing, and (iii) the production run of these three compound-PP1γ2 complexes, along with the Apo form. A detailed analysis of the MD simulation trajectories included the RMSF, RMSD, RoG, beta factor, and SASA.
The in silico study suggested that the PP1γ2–ligand complexes showed improved overall stability and compactness over the simulation run compared to the Apo form (the unbound state). This suggested that the protein achieved stability with less deviation upon binding to ligands. To further proceed with the study, the salt bridge analysis of three complexes showed unique interactions. such as Glu133-Arg137 (D751-0223), Asp4-Lys107 (D751-0223 and Apo), Asp188-Arg116 (D751-0143), and Glu120-Arg90 (N117-0087 and Apo). Unique SBs were created due to conformational variations upon ligand binding to protein, aiding in the stability of the PP1γ2 complexes identified. The PCA demonstrated that each PP1γ2-complex had a distinct motion style, according to PCA. The MM/PBSA and MM/GBSA binding free energy classified the compounds as follows: D751-0223 > D751-0143 > N117-0087, with free-energy scores of −93.05 kcal/mol, −88 kcal/mol, and −80.75 kcal/mol.
A study was conducted by [72] to disrupt sperm motility by inhibiting an enzyme called sperm hyaluronidase. This study involved both experimental and computational analysis and utilized bioactive compounds from natural sources with contraceptive properties. Natural bioactive compounds with contraceptive potential were great substitutes for current hormonal methods of contraception. Reversible and non-hormonal contraceptives for men can be developed from natural non-hormonal compounds that inhibit the enzyme sperm hyaluronidase. The male contraceptive characteristics of the plant “Aegle marmelos Linn” leaf extracts were evaluated using the in vitro inhibition method along with computer-assisted methods. In silico approaches such as molecular docking, molecular dynamics, non-covalent interaction evaluation, molecular mechanics, and Poisson–Boltzmann surface area were used to identify the interaction pattern of marmin, aegeline, and marmenol on hyaluronidases. The findings from the use of computer-assisted sperm analysis and the in vitro hyaluronidase inhibition assay suggest that the usage of leaf extracts limits the enzymatic activity of hyaluronidase and decreases sperm counts. The findings from the in silico approach suggest that phytocompounds such as aegeline and marmin have the potential to inhibit the enzyme sperm hyaluronidase.
Compared to standard combinatorial chemistry, CADD employs a considerably more focused search, which may boost the hit rate for novel therapeutic molecules [73]. Apart from elucidating the molecular foundations of therapeutic actions, it also seeks to predict potential compounds that could enhance efficiency [74]. Three main objectives are frequently achieved by CADD in drug discovery: (1) filtering massive compound libraries into reduced sets of suggested active compounds that can be evaluated through experiments; (2) contributing to the optimization of hits, whether the objective is to enhance their affinity or just their features of drug metabolism and pharmacokinetics (DMPK), including absorption, distribution, metabolism, excretion, and possibility of toxicity (ADMET); and (3) developing novel drugs by either putting together pieces into distinct chemotypes or “developing” and setting up molecules one functional group at a time [75]. The study identified three novel drugs targeting thePP1γ2 with promising computational findings, but further experimental validation needs to be conducted to improve their efficiency. Though the CADD results presented in this work are interesting, they have several limitations. CADD relies on computational models, which are usually simplifications of the complex biological reality and thus cannot represent all the microscopic events in real time. Thus, the models may result in inaccurate binding affinity predictions, off-target effects, or pharmacokinetics. Also, the findings of the study might not be able to fully capture the complexity of system biology, polypharmacology, and multi-target effects.
5. Conclusions
To sum up, this in silico study employed molecular docking and molecular dynamics modeling to investigate how PP1γ2 binds to multiple ligands. The top three compounds with the best binding energy score were selected as drug candidates. The protein–ligand (PL) complexes’ flexibility and stability at the molecular level in a dynamic environment were shown by a 200 ns molecular dynamics simulation. The best compounds identified (D751-0223, D751-0143, and N117-0087) could potentially be the most effective against the target protein based on the latest research. However, further extensive investigation (including both in vitro and in vivo testing) is required to confirm their accuracy and performance as successful therapeutic options.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/cimb47080658/s1.
Author Contributions
Conceptualization, H.M.A. and A.A.E.; methodology, B.T.B.; software, A.M.S.; validation, A.Y.A., R.M.A. and M.M.J.; formal analysis, M.A.A.; investigation, A.A.E. and H.M.A.; resources, A.A.E.; data curation, B.T.B.; writing—original draft preparation, A.M.S., A.Y.A., R.M.A. and M.M.J.; writing—review and editing, M.A.A.; visualization, A.A.E.; supervision, A.A.E.; project administration, H.M.A.; funding acquisition, A.A.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no specific grants from any funding agency.
Institutional Review Board Statement
Not required.
Informed Consent Statement
Not required.
Data Availability Statement
The data generated in the work is presented in the manuscript. All the raw data is also uploaded to the journal along with Supplementary Files.
Acknowledgments
The authors would like to acknowledge Taibah University, Medina, Saudi Arabia, for providing all the necessary funds.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Coulson, J.; Sharma, V.; Wen, H. Understanding the global dynamics of continuing unmet need for family planning and unintended pregnancy. China Popul. Dev. Stud. 2023, 7, 1–14. [Google Scholar] [CrossRef]
- Barton, B.E.; Erickson, J.A.; Allred, S.I.; Jeffries, J.M.; Stephens, K.K.; Hunter, M.I.; Woodall, K.A.; Winuthayanon, W. Reversible female contraceptives: Historical, current, and future perspectives. Biol. Reprod. 2024, 110, 14–32. [Google Scholar] [CrossRef]
- Le Guen, M.; Schantz, C.; Régnier-Loilier, A.; de La Rochebrochard, E. Reasons for rejecting hormonal contraception in Western countries: A systematic review. Soc. Sci. Med. 2021, 284, 114247. [Google Scholar] [CrossRef]
- Page, S.T.; Amory, J.K. The world needs better male contraceptives: What is taking so long? FASEB J. 2022, 36, e22658. [Google Scholar] [CrossRef] [PubMed]
- Darko Okyere, C. The Conundrum of Unsafe Abortion Among the Youth in Ghana: A Case of Awutu Senya East Municipal Assembly. Master’s Thesis, OsloMet-Storbyuniversitetet, Oslo, Norway, 2022. [Google Scholar]
- Cleland, J. The Contraceptive Revolution. In International Handbook of Population Policies; Springer: Berlin/Heidelberg, Germany, 2022; pp. 595–615. [Google Scholar]
- Pyo, Y.; Kwon, K.H. A review of various types of male contraception. J. Mens. Health 2024, 20, 1–8. [Google Scholar]
- Jacobstein, R.; Radloff, S.; Khan, F.; Mimno, K.; Pal, M.; Snell, J.; Stafford, R.; Touré, C.; Tripathi, V. Down but not out: Vasectomy is faring poorly almost everywhere—we can do better to make it a true method option. Glob. Heal. Sci. Pract. 2023, 11, e2200369. [Google Scholar] [CrossRef] [PubMed]
- Kerk, D.; White-Gloria, C.; Johnson, J.J.; Moorhead, G.B. Eukaryotic-like phosphoprotein phosphatase (PPP) enzyme evolution: Interactions with environmental toxins and regulatory proteins. Biosci. Rep. 2023, 43, BSR20230378. [Google Scholar] [CrossRef]
- Ferreira, A.F.; Santiago, J.; Silva, J.V.; Oliveira, P.F.; Fardilha, M. PP1, PP2A and PP2B interplay in the regulation of sperm motility: Lessons from protein phosphatase inhibitors. Int. J. Mol. Sci. 2022, 23, 15235. [Google Scholar] [CrossRef]
- Mehta, V.; Chamousset, D.; Law, J.; Ooi, S.; Campuzano, D.; Nguyen, V.; Boisvert, F.-M.; Moorhead, G.B.; Trinkle-Mulcahy, L. Subcellular distribution of PP1 isoforms in holoenzyme complexes. bioRxiv 2022. [Google Scholar] [CrossRef]
- Silva, J.V.; Freitas, M.J.; Santiago, J.; Jones, S.; Guimarães, S.; Vijayaraghavan, S.; Publicover, S.; Colombo, G.; Howl, J.; Fardilha, M. Disruption of protein phosphatase 1 complexes with the use of bioportides as a novel approach to target sperm motility. Fertil. Steril. 2021, 115, 348–362. [Google Scholar] [CrossRef]
- Marques, L.; Costa, B.; Pereira, M.; Silva, A.; Santos, J.; Saldanha, L.; Silva, I.; Magalhães, P.; Schmidt, S.; Vale, N. Advancing precision medicine: A review of innovative In Silico approaches for drug development, clinical pharmacology and personalized healthcare. Pharmaceutics 2024, 16, 332. [Google Scholar] [CrossRef]
- Nagarajan, K.; Sundaram, D.P.; Marimuthu, S.K. Innovations In Pharmaceutical Biotechnology; Academic Guru Publishing House: Bhopal, India, 2024; ISBN 8197059136. [Google Scholar]
- Rajaei, F.; Minoccheri, C.; Wittrup, E.; Wilson, R.C.; Athey, B.D.; Omenn, G.S.; Najarian, K. AI-based Computational Methods in Early Drug Discovery and Post Market Drug Assessment: A Survey. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2024, 22, 97–115. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Jain, M. Limitations and future challenges of computer-aided drug design methods. In Computer Aided Drug Design (CADD): From Ligand-Based Methods to Structure-Based Approaches; Elsevier: Amsterdam, The Netherlands, 2022; pp. 283–297. [Google Scholar]
- Bouribab, A.; Errougui, A.; Chtita, S. CADD Methods for Developing Novel Compounds Synthesized to Inhibit Tyrosine Kinase Receptors. Curr. Top. Med. Chem. 2024, 25, 1141–1164. [Google Scholar] [CrossRef] [PubMed]
- Giménez, B.G.; Santos, M.S.; Ferrarini, M.; Dos Santos Fernandes, J.P. Evaluation of blockbuster drugs under the rule-of-five. Pharmazie 2010, 65, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Niazi, S.K.; Mariam, Z. Computer-aided drug design and drug discovery: A prospective analysis. Pharmaceuticals 2023, 17, 22. [Google Scholar] [CrossRef]
- Nisius, B.; Sha, F.; Gohlke, H. Structure-based computational analysis of protein binding sites for function and druggability prediction. J. Biotechnol. 2012, 159, 123–134. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Johnson, M.C.; Izadi, S.; Gerosa, L.; Hammel, M.; Bruning, J.M.; Wendorff, T.J.; Phung, W.; Hymowitz, S.G.; Sudhamsu, J. Structural basis for SHOC2 modulation of RAS signalling. Nature 2022, 609, 400–407. [Google Scholar] [CrossRef]
- Aloliqi, A.A. Towards identification of therapeutics against multi-infections and cancers causing Propionibacterium acnes: Molecular modeling and dynamics simulation investigation. J. Mol. Liq. 2024, 126373. [Google Scholar] [CrossRef]
- Abdullahi, M.; Adeniji, S.E. In-silico Molecular Docking and ADME/Pharmacokinetic Prediction Studies of Some Novel Carboxamide Derivatives as Anti-tubercular Agents. Chem. Afr. 2020, 3, 989–1000. [Google Scholar] [CrossRef]
- Kondapuram, S.K.; Sarvagalla, S.; Coumar, M.S. Docking-based virtual screening using PyRx Tool: Autophagy target Vps34 as a case study. In Molecular Docking for Computer-Aided Drug Design; Elsevier: Amsterdam, The Netherlands, 2021; pp. 463–477. [Google Scholar]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Crampon, K.; Giorkallos, A.; Deldossi, M.; Baud, S.; Steffenel, L.A. Machine-learning methods for ligand-protein molecular docking. Drug Discov. Today 2022, 27, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lin, S.; Yang, F.; Chen, Z.; Guo, L.; Yang, J.; Lin, X.; Wang, L.; Duan, Y.; Wen, A. Structural and functional basis of low-affinity SAM/SAH-binding in the conserved MTase of the multi-segmented Alongshan virus distantly related to canonical unsegmented flaviviruses. PLoS Pathog. 2023, 19, e1011694. [Google Scholar] [CrossRef]
- Wong, F.; Krishnan, A.; Zheng, E.J.; Stärk, H.; Manson, A.L.; Earl, A.M.; Jaakkola, T.; Collins, J.J. Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery. Mol. Syst. Biol. 2022, 18, e11081. [Google Scholar] [CrossRef]
- Acharya, A.; Agarwal, R.; Baker, M.B.; Baudry, J.; Bhowmik, D.; Boehm, S.; Byler, K.G.; Chen, S.Y.; Coates, L.; Cooper, C.J.; et al. Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to COVID-19. J. Chem. Inf. Model. 2020, 60, 5832–5852. [Google Scholar] [CrossRef]
- Alamri, M.A.; Mirza, M.U.; Adeel, M.M.; Ashfaq, U.A.; Tahir Ul Qamar, M.; Shahid, F.; Ahmad, S.; Alatawi, E.A.; Albalawi, G.M.; Allemailem, K.S.; et al. Structural Elucidation of Rift Valley Fever Virus L Protein towards the Discovery of Its Potential Inhibitors. Pharmaceuticals 2022, 15, 659. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832, Erratum in Nat. Protoc. 2022, 17, 177. [Google Scholar] [CrossRef]
- Jejurikar, B.L.; Rohane, S.H. Drug designing in discovery studio. Asian J. Res. Chem. 2021, 14, 135–138. [Google Scholar]
- Makhlouf, J.; El Bakri, Y.; Valkonen, A.; Saravanan, K.; Ahmad, S.; Smirani, W. Growth, single crystal investigations, hirshfeld surface analysis, DFT studies, molecular dynamics simulations, molecular docking, physico-chemical characterization and biological activity of novel thiocyanic complex with zinc transition metal precursor. Polyhedron 2022, 222, 115937. [Google Scholar] [CrossRef]
- Kamble, A.N.S.; Mitkar, A.A. Swiss ADME predictions of pharmacokinetics and drug-likeness properties of secondary metabolites present in trigonella foenum-graecum. J. Pharmacogn. Phytochem. 2023, 12, 341–349. [Google Scholar] [CrossRef]
- Šegota, S.B.; Anđelić, N.; Lorencin, I.; Musulin, J.; Štifanić, D.; Car, Z. Preparation of simplified molecular input line entry system notation datasets for use in convolutional neural networks. In Proceedings of the 2021 IEEE 21st International Conference on Bioinformatics and Bioengineering (BIBE), Kragujevac, Serbia, 25–27 October 2021; IEEE: New York, NY, USA, 2021; pp. 1–6. [Google Scholar]
- Akash, S.; Bayıl, I.; Rahman, M.A.; Mukerjee, N.; Maitra, S.; Islam, M.R.; Rajkhowa, S.; Ghosh, A.; Al-Hussain, S.A.; Zaki, M.E.A.; et al. Target specific inhibition of West Nile virus envelope glycoprotein and methyltransferase using phytocompounds: An in silico strategy leveraging molecular docking and dynamics simulation. Front. Microbiol. 2023, 14, 1189786. [Google Scholar] [CrossRef]
- Bhrdwaj, A.; Abdalla, M.; Pande, A.; Madhavi, M.; Chopra, I.; Soni, L.; Vijayakumar, N.; Panwar, U.; Khan, M.A.; Prajapati, L. Structure-based virtual screening, molecular docking, molecular dynamics simulation of EGFR for the clinical treatment of glioblastoma. Appl. Biochem. Biotechnol. 2023, 195, 5094–5119. [Google Scholar] [CrossRef] [PubMed]
- Guterres, H.; Park, S.; Zhang, H.; Perone, T.; Kim, J.; Im, W. CHARMM-GUI high-throughput simulator for efficient evaluation of protein–ligand interactions with different force fields. Protein Sci. 2022, 31, e4413. [Google Scholar] [CrossRef]
- Raguette, L.E.; Cuomo, A.E.; Belfon, K.A.A.; Tian, C.; Hazoglou, V.; Witek, G.; Telehany, S.M.; Wu, Q.; Simmerling, C. phosaa14SB and phosaa19SB: Updated Amber Force Field Parameters for Phosphorylated Amino Acids. J. Chem. Theory Comput. 2024, 20, 7199–7209. [Google Scholar] [CrossRef]
- Liao, J.; Wang, Q.; Wu, F.; Huang, Z. In silico methods for identification of potential active sites of therapeutic targets. Molecules 2022, 27, 7103. [Google Scholar] [CrossRef]
- Szél, V.; Zsidó, B.Z.; Jeszenői, N.; Hetényi, C. Target–ligand binding affinity from single point enthalpy calculation and elemental composition. Phys. Chem. Chem. Phys. 2023, 25, 31714–31725. [Google Scholar] [CrossRef]
- Rawat, R.; Kant, K.; Kumar, A.; Bhati, K.; Verma, S.M. HeroMDAnalysis: An automagical tool for GROMACS-based molecular dynamics simulation analysis. Future Med. Chem. 2021, 13, 447–456. [Google Scholar] [CrossRef]
- Pan, X.; Van, R.; Epifanovsky, E.; Liu, J.; Pu, J.; Nam, K.; Shao, Y. Accelerating ab initio quantum mechanical and molecular mechanical (QM/MM) molecular dynamics simulations with multiple time step integration and a recalibrated semiempirical QM/MM Hamiltonian. J. Phys. Chem. B 2022, 126, 4226–4235. [Google Scholar] [CrossRef] [PubMed]
- Pederson, J.P.; McDaniel, J.G. DFT-based QM/MM with particle-mesh Ewald for direct, long-range electrostatic embedding. J. Chem. Phys. 2022, 156, 174105. [Google Scholar] [CrossRef] [PubMed]
- Maszota-Zieleniak, M.; Samsonov, S.A. Molecular Dynamics Simulation-Based Prediction of Glycosaminoglycan Interactions with Drug Molecules. In Computational Drug Discovery and Design; Springer: Berlin/Heidelberg, Germany, 2023; pp. 143–153. [Google Scholar]
- Spassov, D.S.; Atanasova, M.; Doytchinova, I. A role of salt bridges in mediating drug potency: A lesson from the N-myristoyltransferase inhibitors. Front. Mol. Biosci. 2023, 9, 1066029. [Google Scholar] [CrossRef]
- Bibi, S.; Khan, M.S.; El-Kafrawy, S.A.; Alandijany, T.A.; El-Daly, M.M.; Yousafi, Q.; Fatima, D.; Faizo, A.A.; Bajrai, L.H.; Azhar, E.I. Virtual screening and molecular dynamics simulation analysis of Forsythoside A as a plant-derived inhibitor of SARS-CoV-2 3CLpro. Saudi Pharm. J. 2022, 30, 979–1002. [Google Scholar] [CrossRef]
- Hasan, B.M.S.; Abdulazeez, A.M. A review of principal component analysis algorithm for dimensionality reduction. J. Soft Comput. Data Min. 2021, 2, 20–30. [Google Scholar] [CrossRef]
- Montgomerie, S.; Sundararaj, S.; Gallin, W.J.; Wishart, D.S. Improving the accuracy of protein secondary structure prediction using structural alignment. BMC Bioinform. 2006, 7, 301. [Google Scholar] [CrossRef]
- Micsonai, A.; Moussong, E.; Wien, F.; Boros, E.; Vadászi, H.; Murvai, N.; Lee, Y.-H.; Molnár, T.; Réfrégiers, M.; Goto, Y. BeStSel: Webserver for secondary structure and fold prediction for protein CD spectroscopy. Nucleic Acids Res. 2022, 50, W90–W98. [Google Scholar] [CrossRef]
- Miandad, K.; Ullah, A.; Bashir, K.; Khan, S.; Abideen, S.A.; Shaker, B.; Alharbi, M.; Alshammari, A.; Ali, M.; Haleem, A.; et al. Virtual Screening of Artemisia annua Phytochemicals as Potential Inhibitors of SARS-CoV-2 Main Protease Enzyme. Molecules 2022, 27, 8103. [Google Scholar] [CrossRef]
- Acikgoz, A.; Demircan, G.; Yılmaz, D.; Aktas, B.; Yalcin, S.; Yorulmaz, N. Structural, mechanical, radiation shielding properties and albedo parameters of alumina borate glasses: Role of CeO2 and Er2O3. Mater. Sci. Eng. B 2022, 276, 115519. [Google Scholar] [CrossRef]
- Rani, P.; Chahal, S.; Kataria, R.; Kumar, P.; Kumar, S.; Sindhu, J. Unravelling the thermodynamics and binding interactions of bovine serum albumin (BSA) with thiazole based carbohydrazide: Multi-spectroscopic, DFT and molecular dynamics approach. J. Mol. Struct. 2022, 1270, 133939. [Google Scholar] [CrossRef]
- Karnik, K.S.; Sarkate, A.P.; Jambhorkar, V.S.; Wakte, P. WaterSwap Analysis, a Computation-based Method for the Discovery of Effective and Stable Binding Compounds for Mutant EGFR Inhibition. arXiv 2021. [Google Scholar] [CrossRef]
- Fratev, F.; Sirimulla, S. An improved free energy perturbation FEP+ sampling protocol for flexible ligand-binding domains. Sci. Rep. 2019, 9, 16829. [Google Scholar] [CrossRef] [PubMed]
- Mariani, N.A.P.; Silva, J.V.; Fardilha, M.; Silva, E.J.R. Advances in non-hormonal male contraception targeting sperm motility. Hum. Reprod. Update 2023, 29, 545–569. [Google Scholar] [CrossRef]
- Uddin, A.; Gupta, S.; Mohammad, T.; Shahi, D.; Hussain, A.; Alajmi, M.F.; El-Seedi, H.R.; Hassan, I.; Singh, S.; Abid, M. Target-based virtual screening of natural compounds identifies a potent antimalarial with selective falcipain-2 inhibitory activity. Front. Pharmacol. 2022, 13, 850176. [Google Scholar] [CrossRef]
- Li, L.; Mohammed, A.H.; Auda, N.A.; Alsallameh, S.M.S.; Albekairi, N.A.; Muhseen, Z.T.; Butch, C.J. Network Pharmacology, Molecular Docking, and Molecular Dynamics Simulation Analysis Reveal Insights into the Molecular Mechanism of Cordia myxa in the Treatment of Liver Cancer. Biology 2024, 13, 315. [Google Scholar] [CrossRef]
- Altharawi, A.; Ahmad, S.; Alamri, M.A.; ul Qamar, M.T. Structural insight into the binding pattern and interaction mechanism of chemotherapeutic agents with Sorcin by docking and molecular dynamic simulation. Colloids Surf. B Biointerfaces 2021, 208, 112098. [Google Scholar] [CrossRef]
- Roy, S.; Ghosh, P.; Bandyopadhyay, A.; Basu, S. Capturing a crucial ‘disorder-to-order transition’at the heart of the coronavirus molecular pathology—triggered by highly persistent, interchangeable salt-bridges. Vaccines 2022, 10, 301. [Google Scholar] [CrossRef]
- Reeda, V.S.J.; Sakthivel, S.; Divya, P.; Javed, S.; Jothy, V.B. Conformational stability, quantum computational (DFT), vibrational, electronic and non-covalent interactions (QTAIM, RDG and IGM) of antibacterial compound N-(1-naphthyl) ethylenediamine dihydrochloride. J. Mol. Struct. 2024, 1298, 137043. [Google Scholar] [CrossRef]
- Padilla-Bernal, G.; Vargas, R.; Martínez, A. Salt bridge: Key interaction between antipsychotics and receptors. Theor. Chem. Acc. 2023, 142, 65. [Google Scholar] [CrossRef]
- Furkan, M.; Khan, M.S.; Shahwan, M.; Hassan, N.; Yadav, D.K.; Anwar, S.; Khan, R.H.; Shamsi, A. Identifying repurposed drugs as potential inhibitors of Apolipoprotein E: A bioinformatics approach to target complex diseases associated with lipid metabolism and neurodegeneration. Int. J. Biol. Macromol. 2024, 259, 129167. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, K.K.; Ahmad, I.; Pati, S.; Ghosh, A.; Rabha, B.; Sarkar, T.; Bhattacharjya, D.; Patel, H.; Baishya, D. Screening of phytocompounds for identification of prospective histone deacetylase 1 (HDAC1) inhibitor: An in silico molecular docking, molecular dynamics simulation, and MM-GBSA approach. Appl. Biochem. Biotechnol. 2024, 196, 3747–3764. [Google Scholar] [CrossRef] [PubMed]
- Miles, A.J.; Ramalli, S.G.; Wallace, B.A. DichroWeb, a website for calculating protein secondary structure from circular dichroism spectroscopic data. Protein Sci. 2022, 31, 37–46. [Google Scholar] [CrossRef]
- Hasan, M.R.; Alsaiari, A.A.; Fakhurji, B.Z.; Molla, M.H.R.; Asseri, A.H.; Sumon, M.A.A.; Park, M.N.; Ahammad, F.; Kim, B. Application of mathematical modeling and computational tools in the modern drug design and development process. Molecules 2022, 27, 4169. [Google Scholar] [CrossRef]
- Cournia, Z.; Chipot, C.; Roux, B.; York, D.M.; Sherman, W. Free energy methods in drug discovery—introduction. In Free Energy Methods in Drug Discovery: Current State and Future Directions; ACS Publications: Washington, DC, USA, 2021; pp. 1–38. ISBN 1947-5918. [Google Scholar]
- Sheng, Y.; Yin, Y.; Ma, Y.; Ding, H. Improving the performance of MM/PBSA in protein–protein interactions via the screening electrostatic energy. J. Chem. Inf. Model. 2021, 61, 2454–2462. [Google Scholar] [CrossRef] [PubMed]
- Sobhia, M.E.; Ghosh, K.; Kumar, G.S.; Sivangula, S.; Laddha, K.; Kumari, S.; Kumar, H. The Role of Water Network Chemistry in Proteins: A Structural Bioinformatics Perspective in Drug Discovery and Development. Curr. Top. Med. Chem. 2022, 22, 1636–1653. [Google Scholar] [CrossRef]
- Reynolds-Wright, J.J.; Cameron, N.J.; Anderson, R.A. Will men use novel male contraceptive methods and will women trust them? A systematic review. J. Sex Res. 2021, 58, 838–849. [Google Scholar] [CrossRef] [PubMed]
- Dominiak, Z.; Huras, H.; Kręcisz, P.; Krzeszowski, W.; Szymański, P.; Czarnecka, K. Promising results in development of male contraception. Bioorg. Med. Chem. Lett. 2021, 41, 128005. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, P.; Velmurugan, Y.; Arputharaj, D.S.; Savaridasson, J.K.; Hemamalini, M.; Venkatachalam, R. In vitro contraceptive activities, molecular docking, molecular dynamics, MM-PBSA, non-covalent interaction and DFT studies of bioactive compounds from Aegle marmelos. Linn., leaves. Front. Chem. 2023, 11, 1096177. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Martínez, F.D.; López-López, E.; Juárez-Mercado, K.E.; Medina-Franco, J.L. Computational drug design methods—current and future perspectives. In In Silico Drug Design; Academic Press: Cambridge, MA, USA, 2019; pp. 19–44. [Google Scholar]
- Schenone, M.; Dančík, V.; Wagner, B.K.; Clemons, P.A. Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 2013, 9, 232–240. [Google Scholar] [CrossRef]
- Kiriiri, G.K.; Njogu, P.M.; Mwangi, A.N. Exploring different approaches to improve the success of drug discovery and development projects: A review. Futur. J. Pharm. Sci. 2020, 6, 27. [Google Scholar] [CrossRef]


















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).