
Emerging Epigenetic Therapeutics and Diagnostics for Autism Spectrum Disorder


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Autism Spectrum Disorder
1.2. Current Therapeutics
1.3. Emerging Therapeutics
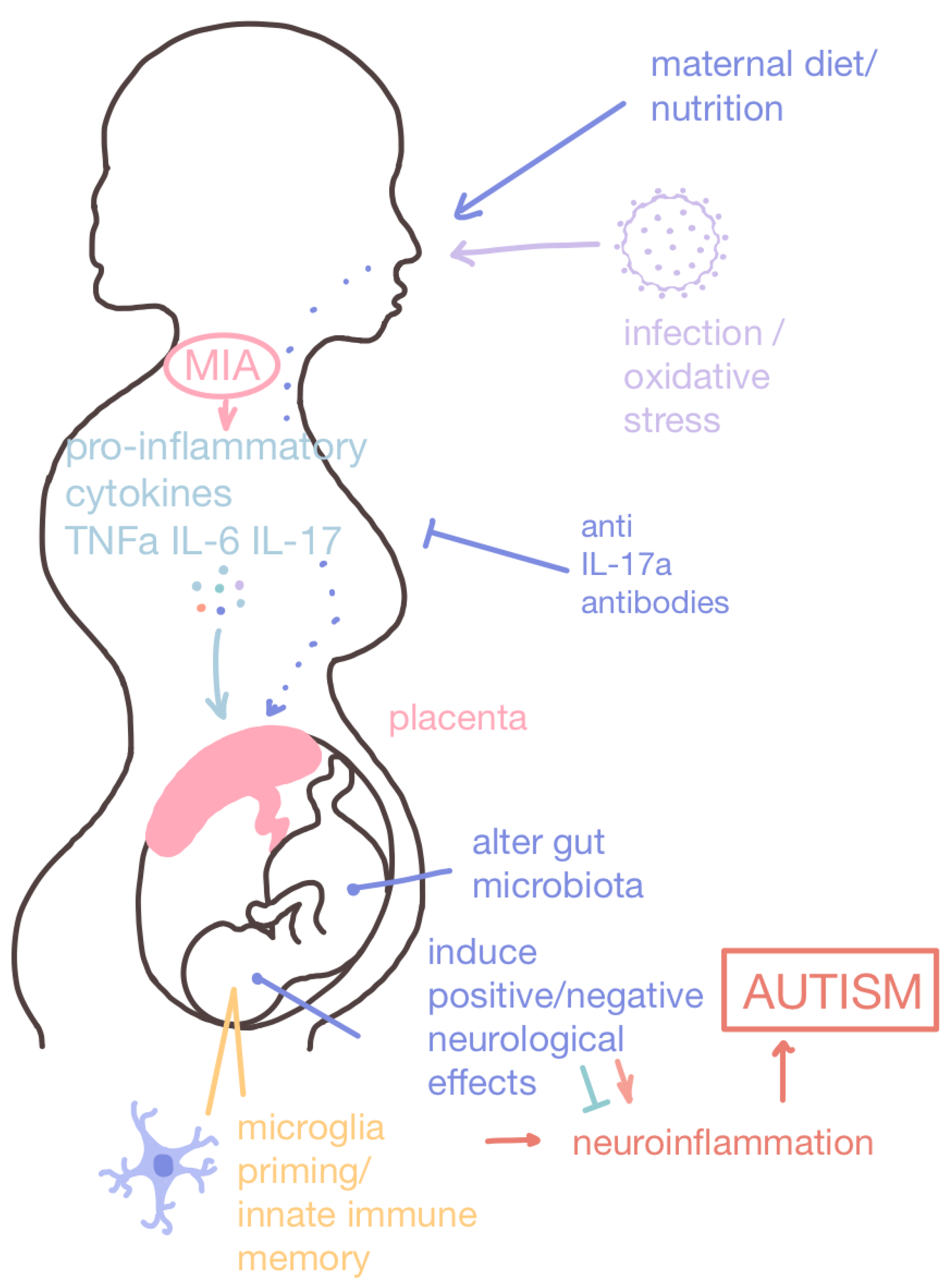
2. Maternal Interventions
2.1. The Gut Microbiome and the Gut–Brain Axis
2.2. Maternal Nutrition
2.2.1. Fatty Acid Intake
2.2.2. Folic Acid Supplementation
2.2.3. Choline Supplementation
3. Interventions on ASD Patients
3.1. Pharmacological Intervention
3.1.1. Maternal Immune Activation Therapeutics
Histamine Receptor Antagonists
IL-17a Antibodies
3.1.2. CBD-Rich Cannabis
3.1.3. Oxytocin
3.1.4. Histone Modifiers
3.1.5. DNA Methylation and Genomic Imprinting
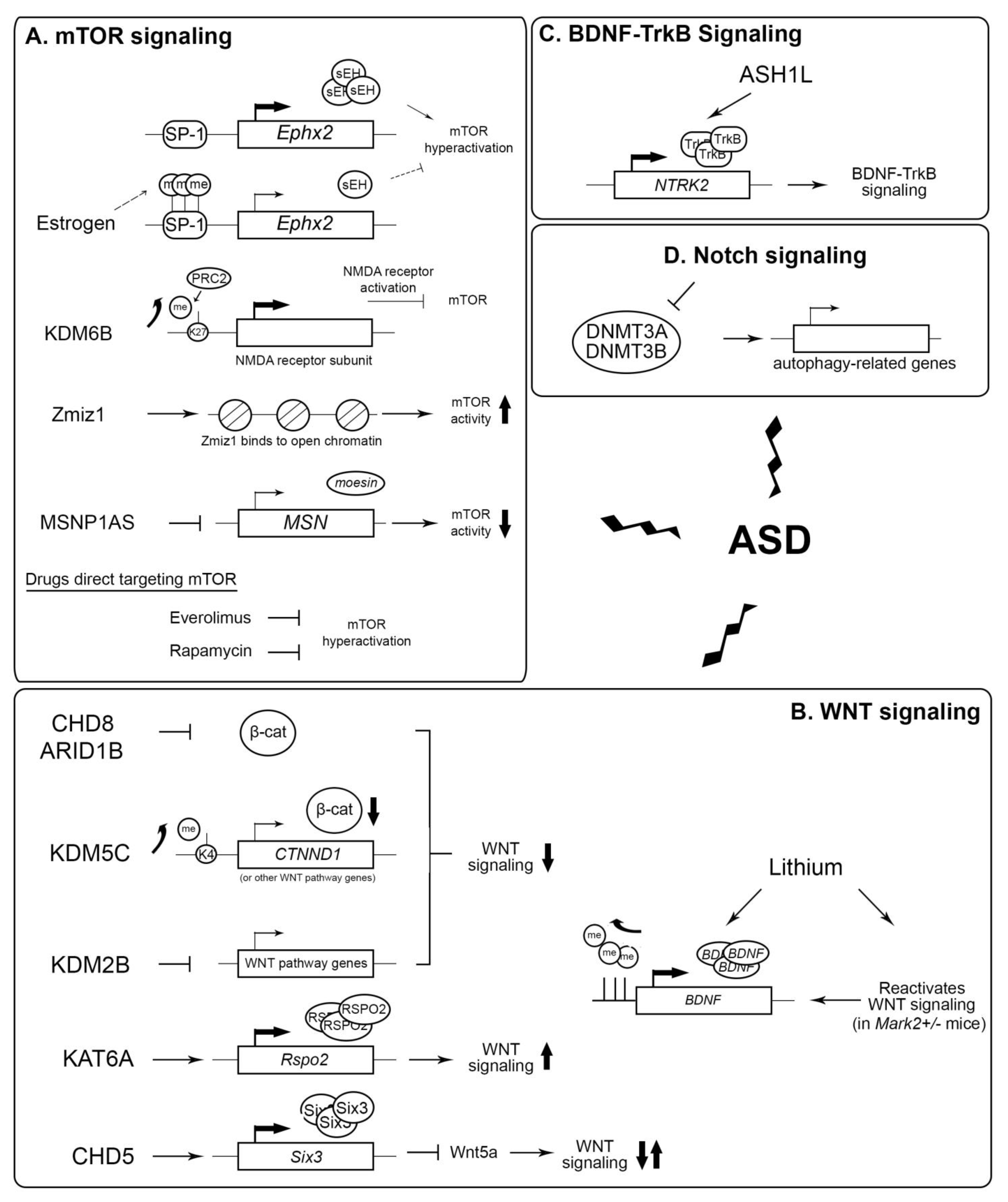
3.1.6. Epigenetic Interplay of Signal Transduction Regulation
mTOR
WNT
Other Signaling Regulation
3.2. Patient Nutrition
3.2.1. Ketogenic Diet
3.2.2. GF/CF Diet
3.2.3. Probiotics
4. Diagnostic Biomarkers for ASD
4.1. MicroRNA
4.2. Perinatal Epigenetic Signatures
4.3. Glutathione
4.4. Gut Microbiome Composition
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychological Association. Autism Spectrum Disorder. Available online: https://www.apa.org/topics/autism-spectrum-disorder (accessed on 31 July 2024).
- Will, M.N.; Currans, K.; Smith, J.; Weber, S.; Duncan, A.; Burton, J.; Kroeger-Geoppinger, K.; Miller, V.; Stone, M.; Mays, L.; et al. Evidenced-Based Interventions for Children With Autism Spectrum Disorder. Curr. Probl. Pediatr. Adolesc. Health Care 2018, 48, 234–249. [Google Scholar] [CrossRef]
- Baer, D.M.; Wolf, M.M.; Risley, T.R. Some current dimensions of applied behavior analysis. J. Appl. Behav. Anal. 1968, 1, 91–97. [Google Scholar] [CrossRef]
- McEachin, J.J.; Smith, T.; Lovaas, O.I. Long-term outcome for children with autism who received early intensive behavioral treatment. Am. J. Ment. Retard. 1993, 97, 359–372; discussion 373–391. [Google Scholar]
- Kasari, C.; Paparella, T.; Freeman, S.; Jahromi, L.B. Language outcome in autism: Randomized comparison of joint attention and play interventions. J. Consult. Clin. Psychol. 2008, 76, 125–137. [Google Scholar] [CrossRef]
- McCracken, J.T.; McGough, J.; Shah, B.; Cronin, P.; Hong, D.; Aman, M.G.; Arnold, L.E.; Lindsay, R.; Nash, P.; Hollway, J.; et al. Risperidone in children with autism and serious behavioral problems. N. Engl. J. Med. 2002, 347, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Wink, S.; Hiemstra, S.; Huppelschoten, S.; Danen, E.; Niemeijer, M.; Hendriks, G.; Vrieling, H.; Herpers, B.; van de Water, B. Quantitative high content imaging of cellular adaptive stress response pathways in toxicity for chemical safety assessment. Chem. Res. Toxicol. 2014, 27, 338–355. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, M. The clinical utility of the ADI-R and ADOS in diagnosing autism. Br. J. Psychiatry 2017, 211, 117. [Google Scholar] [CrossRef]
- Ventola, P.E.; Kleinman, J.; Pandey, J.; Barton, M.; Allen, S.; Green, J.; Robins, D.; Fein, D. Agreement among four diagnostic instruments for autism spectrum disorders in toddlers. J. Autism Dev. Disord. 2006, 36, 839–847. [Google Scholar] [CrossRef] [PubMed]
- McCrossin, R. Finding the True Number of Females with Autistic Spectrum Disorder by Estimating the Biases in Initial Recognition and Clinical Diagnosis. Children 2022, 9, 272. [Google Scholar] [CrossRef]
- Track, N.S. The gastrointestinal endocrine system. Can. Med. Assoc. J. 1980, 122, 287–292. [Google Scholar]
- Mulak, A.; Bonaz, B. Irritable bowel syndrome: A model of the brain-gut interactions. Med. Sci. Monit. 2004, 10, RA55–RA62. [Google Scholar] [PubMed]
- Porreca, F.; Burks, T.F.; Koslo, R.J. Centrally-mediated bombesin effects on gastrointestinal motility. Life Sci. 1985, 37, 125–134. [Google Scholar] [CrossRef]
- Lagod, P.P.; Naser, S.A. The Role of Short-Chain Fatty Acids and Altered Microbiota Composition in Autism Spectrum Disorder: A Comprehensive Literature Review. Int. J. Mol. Sci. 2023, 24, 17432. [Google Scholar] [CrossRef] [PubMed]
- Bettelheim, K.A.; Breadon, A.; Faiers, M.C.; O’Farrell, S.M.; Shooter, R.A. The origin of O serotypes of Escherichia coli in babies after normal delivery. J. Hyg. 1974, 72, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Sivamaruthi, B.S.; Suganthy, N.; Kesika, P.; Chaiyasut, C. The Role of Microbiome, Dietary Supplements, and Probiotics in Autism Spectrum Disorder. Int. J. Environ. Res. Public Health 2020, 17, 2647. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, B.; Kokhaei, P.; Mehranfar, F.; Bahar, A.; Abdolshahi, A.; Emadi, A.; Eslami, M. The role of the host microbiome in autism and neurodegenerative disorders and effect of epigenetic procedures in the brain functions. Neurosci. Biobehav. Rev. 2022, 132, 998–1009. [Google Scholar] [CrossRef]
- Li, Y.M.; Shen, Y.D.; Li, Y.J.; Xun, G.L.; Liu, H.; Wu, R.R.; Xia, K.; Zhao, J.P.; Ou, J.J. Maternal dietary patterns, supplements intake and autism spectrum disorders: A preliminary case-control study. Medicine 2018, 97, e13902. [Google Scholar] [CrossRef]
- Doenyas, C. Potential Role of Epigenetics and Redox Signaling in the Gut-Brain Communication and the Case of Autism Spectrum Disorder. Cell. Mol. Neurobiol. 2022, 42, 483–487. [Google Scholar] [CrossRef]
- Lewandowska-Pietruszka, Z.; Figlerowicz, M.; Mazur-Melewska, K. Microbiota in Autism Spectrum Disorder: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 16660. [Google Scholar] [CrossRef]
- Lim, J.M.; Letchumanan, V.; Tan, L.T.; Hong, K.W.; Wong, S.H.; Ab Mutalib, N.S.; Lee, L.H.; Law, J.W. Ketogenic Diet: A Dietary Intervention via Gut Microbiome Modulation for the Treatment of Neurological and Nutritional Disorders (a Narrative Review). Nutrients 2022, 14, 3566. [Google Scholar] [CrossRef]
- Niu, M.; Li, Q.; Zhang, J.; Wen, F.; Dang, W.; Duan, G.; Li, H.; Ruan, W.; Yang, P.; Guan, C.; et al. Characterization of Intestinal Microbiota and Probiotics Treatment in Children With Autism Spectrum Disorders in China. Front. Neurol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Hang, S.; Fang, Y.; Bae, S.; Zhang, Y.; Zhang, M.; Wang, G.; McCurry, M.D.; Bae, M.; Paik, D.; et al. A bacterial bile acid metabolite modulates T(reg) activity through the nuclear hormone receptor NR4A1. Cell Host Microbe 2021, 29, 1366–1377.e9. [Google Scholar] [CrossRef]
- Shen, C.; Zhu, X.; Chang, H.; Li, C.; Hou, M.; Chen, L.; Lu, C.; Zhou, Z.; Ji, M.; Xu, Z. The rebalancing of the immune system at the maternal-fetal interface ameliorates autism-like behavior in adult offspring. Cell Rep. 2024, 43, 114787. [Google Scholar] [CrossRef]
- Madore, C.; Leyrolle, Q.; Lacabanne, C.; Benmamar-Badel, A.; Joffre, C.; Nadjar, A.; Laye, S. Neuroinflammation in Autism: Plausible Role of Maternal Inflammation, Dietary Omega 3, and Microbiota. Neural Plast. 2016, 2016, 3597209. [Google Scholar] [CrossRef] [PubMed]
- American Heart Association. Polyunsaturated Fats. Available online: https://www.heart.org/en/healthy-living/healthy-eating/eat-smart/fats/polyunsaturated-fats (accessed on 31 July 2024).
- Lyall, K.; Munger, K.L.; O’Reilly, E.J.; Santangelo, S.L.; Ascherio, A. Maternal dietary fat intake in association with autism spectrum disorders. Am. J. Epidemiol. 2013, 178, 209–220. [Google Scholar] [CrossRef]
- Julvez, J.; Mendez, M.; Fernandez-Barres, S.; Romaguera, D.; Vioque, J.; Llop, S.; Ibarluzea, J.; Guxens, M.; Avella-Garcia, C.; Tardon, A.; et al. Maternal Consumption of Seafood in Pregnancy and Child Neuropsychological Development: A Longitudinal Study Based on a Population with High Consumption Levels. Am. J. Epidemiol. 2016, 183, 169–182. [Google Scholar] [CrossRef]
- Hibbeln, J.R.; Davis, J.M.; Steer, C.; Emmett, P.; Rogers, I.; Williams, C.; Golding, J. Maternal seafood consumption in pregnancy and neurodevelopmental outcomes in childhood (ALSPAC study): An observational cohort study. Lancet 2007, 369, 578–585. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services; U.S. Department of Agriculture. 2015–2020 Dietary Guidelines for Americans. 8th Edition. December 2015. Available online: https://odphp.health.gov/sites/default/files/2019-09/2015-2020_Dietary_Guidelines.pdf (accessed on 19 June 2025).
- Buffington, S.A.; Di Prisco, G.V.; Auchtung, T.A.; Ajami, N.J.; Petrosino, J.F.; Costa-Mattioli, M. Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 2016, 165, 1762–1775. [Google Scholar] [CrossRef] [PubMed]
- van Otterdijk, S.D.; Klett, H.; Boerries, M.; Michels, K.B. The impact of pre-pregnancy folic acid intake on placental DNA methylation in a fortified cohort. FASEB J. 2023, 37, e22698. [Google Scholar] [CrossRef]
- Mikkelsen, K.; Apostolopoulos, V. Vitamin B12, folic acid and the immune system. In Nutrition and Immunity; Mahmoudi, M., Rezaei, N., Eds.; Springer Nature: Cham, Switzerland, 2019; pp. 103–114. [Google Scholar]
- Bansal, M.; Singh, N.; Pal, S.; Dev, I.; Ansari, K.M. Chemopreventive Role Dietary Phytochemicals in Colorectal Cancer. In Advances in Molecular Toxicology; Fishbein, J.C., Heilman, J.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 12, pp. 69–121. [Google Scholar]
- National Institutes of Health. Folate—Fact Sheet for Health Professionals. Available online: https://ods.od.nih.gov/factsheets/Folate-HealthProfessional/ (accessed on 31 July 2024).
- World Health Organization. Periconceptional Folic Acid Supplementation to Prevent Neural Tube Defects. Available online: https://www.who.int/tools/elena/interventions/folate-periconceptional (accessed on 31 July 2024).
- Coffey-Vega, K. Folate Deficiency. Available online: https://emedicine.medscape.com/article/200184-overview#a1 (accessed on 15 August 2024).
- Liu, X.; Zou, M.; Sun, C.; Wu, L.; Chen, W.X. Prenatal Folic Acid Supplements and Offspring’s Autism Spectrum Disorder: A Meta-analysis and Meta-regression. J. Autism Dev. Disord. 2022, 52, 522–539. [Google Scholar] [CrossRef]
- Tan, M.; Yang, T.; Zhu, J.; Li, Q.; Lai, X.; Li, Y.; Tang, T.; Chen, J.; Li, T. Maternal folic acid and micronutrient supplementation is associated with vitamin levels and symptoms in children with autism spectrum disorders. Reprod. Toxicol. 2020, 91, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Kumaravel, T.S.; Lohani, A.; Pedersen, W.A.; Cutler, R.G.; Kruman, Y.; Haughey, N.; Lee, J.; Evans, M.; Mattson, M.P. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J. Neurosci. 2002, 22, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.A.; Nguyen, M.L.; Barrett, A.N.; Tan, Y.Y.; Choolani, M.A.; Chen, E.S. Synthetic combinations of missense polymorphic genetic changes underlying Down syndrome susceptibility. Cell. Mol. Life Sci. 2016, 73, 4001–4017. [Google Scholar] [CrossRef] [PubMed]
- Davison, J.M.; Mellott, T.J.; Kovacheva, V.P.; Blusztajn, J.K. Gestational choline supply regulates methylation of histone H3, expression of histone methyltransferases G9a (Kmt1c) and Suv39h1 (Kmt1a), and DNA methylation of their genes in rat fetal liver and brain. J. Biol. Chem. 2009, 284, 1982–1989. [Google Scholar] [CrossRef]
- Liu, S.X.; Barks, A.K.; Lunos, S.; Gewirtz, J.C.; Georgieff, M.K.; Tran, P.V. Prenatal Iron Deficiency and Choline Supplementation Interact to Epigenetically Regulate Jarid1b and Bdnf in the Rat Hippocampus into Adulthood. Nutrients 2021, 13, 4527. [Google Scholar] [CrossRef]
- Irvine, N.; England-Mason, G.; Field, C.J.; Dewey, D.; Aghajafari, F. Prenatal Folate and Choline Levels and Brain and Cognitive Development in Children: A Critical Narrative Review. Nutrients 2022, 14, 364. [Google Scholar] [CrossRef]
- Tartaglione, A.M.; Villani, A.; Ajmone-Cat, M.A.; Minghetti, L.; Ricceri, L.; Pazienza, V.; De Simone, R.; Calamandrei, G. Maternal immune activation induces autism-like changes in behavior, neuroinflammatory profile and gut microbiota in mouse offspring of both sexes. Transl. Psychiatry 2022, 12, 384. [Google Scholar] [CrossRef]
- Ellul, P.; Maruani, A.; Vantalon, V.; Humeau, E.; Amestoy, A.; Anchordoqui, A.; Atzori, P.; Baleyte, J.M.; Benmansour, S.; Bonnot, O.; et al. Maternal immune activation during pregnancy is associated with more difficulties in socio-adaptive behaviors in autism spectrum disorder. Sci. Rep. 2023, 13, 17687. [Google Scholar] [CrossRef]
- Zawadzka, A.; Cieslik, M.; Adamczyk, A. The Role of Maternal Immune Activation in the Pathogenesis of Autism: A Review of the Evidence, Proposed Mechanisms and Implications for Treatment. Int. J. Mol. Sci. 2021, 22, 11516. [Google Scholar] [CrossRef]
- Loewen, S.M.; Chavesa, A.M.; Murray, C.J.; Traetta, M.E.; Burns, S.E.; Pekarik, K.H.; Tremblay, M.E. The Outcomes of Maternal Immune Activation Induced with the Viral Mimetic Poly I:C on Microglia in Exposed Rodent Offspring. Dev. Neurosci. 2023, 45, 191–209. [Google Scholar] [CrossRef]
- Stelzer, I.A.; Arck, P.C. Immunity and the Endocrine System. Encycl. Immunobiol. 2016, 9, 73–85. [Google Scholar]
- Eissa, N.; Sadeq, A.; Sasse, A.; Sadek, B. Role of Neuroinflammation in Autism Spectrum Disorder and the Emergence of Brain Histaminergic System. Lessons Also for BPSD? Front. Pharmacol. 2020, 11, 886. [Google Scholar] [CrossRef] [PubMed]
- Mastenbroek, L.J.M.; Kooistra, S.M.; Eggen, B.J.L.; Prins, J.R. The role of microglia in early neurodevelopment and the effects of maternal immune activation. Semin. Immunopathol. 2024, 46, 1. [Google Scholar] [CrossRef]
- Netea, M.G.; Dominguez-Andres, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Bertocchi, A.; Mancinelli, S.; Bellini, M.; Erreni, M.; Borreca, A.; Braga, D.; Giugliano, S.; Mozzarelli, A.M.; Manganaro, D.; et al. Identification of a choroid plexus vascular barrier closing during intestinal inflammation. Science 2021, 374, 439–448. [Google Scholar] [CrossRef]
- Santana-Coelho, D.; Layne-Colon, D.; Valdespino, R.; Ross, C.C.; Tardif, S.D.; O’Connor, J.C. Advancing Autism Research From Mice to Marmosets: Behavioral Development of Offspring Following Prenatal Maternal Immune Activation. Front. Psychiatry 2021, 12, 705554. [Google Scholar] [CrossRef] [PubMed]
- Mueller, F.S.; Scarborough, J.; Schalbetter, S.M.; Richetto, J.; Kim, E.; Couch, A.; Yee, Y.; Lerch, J.P.; Vernon, A.C.; Weber-Stadlbauer, U.; et al. Behavioral, neuroanatomical, and molecular correlates of resilience and susceptibility to maternal immune activation. Mol. Psychiatry 2021, 26, 396–410. [Google Scholar] [CrossRef]
- Sadek, B.; Saad, A.; Sadeq, A.; Jalal, F.; Stark, H. Histamine H3 receptor as a potential target for cognitive symptoms in neuropsychiatric diseases. Behav. Brain Res. 2016, 312, 415–430. [Google Scholar] [CrossRef]
- Rani, B.; Silva-Marques, B.; Leurs, R.; Passani, M.B.; Blandina, P.; Provensi, G. Short- and Long-Term Social Recognition Memory Are Differentially Modulated by Neuronal Histamine. Biomolecules 2021, 11, 555. [Google Scholar] [CrossRef]
- Barata-Antunes, S.; Cristovao, A.C.; Pires, J.; Rocha, S.M.; Bernardino, L. Dual role of histamine on microglia-induced neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 764–769. [Google Scholar] [CrossRef]
- Sang, Z.; Huang, L. Alzheimer’s disease therapeutics: Current strategies and future directions. In Privileged Scaffolds in Drug Discovery; Yu, B., Li, N., Fu, C., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 405–473. [Google Scholar]
- Zhou, Z.; An, Q.; Zhang, W.; Li, Y.; Zhang, Q.; Yan, H. Histamine and receptors in neuroinflammation: Their roles on neurodegenerative diseases. Behav. Brain Res. 2024, 465, 114964. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.D.; Abdalla, S.; Eissa, N.; Akour, A.; Jha, N.K.; Ojha, S.; Sadek, B. Targeting Microglia in Neuroinflammation: H3 Receptor Antagonists as a Novel Therapeutic Approach for Alzheimer’s Disease, Parkinson’s Disease, and Autism Spectrum Disorder. Pharmaceuticals 2024, 17, 831. [Google Scholar] [CrossRef]
- Taheri, F.; Esmaeilpour, K.; Sepehri, G.; Sheibani, V.; Shekari, M.A. Amelioration of cognition impairments in the valproic acid-induced animal model of autism by ciproxifan, a histamine H3-receptor antagonist. Behav. Pharmacol. 2023, 34, 179–196. [Google Scholar] [CrossRef]
- Albin, R.L.; Mink, J.W. Recent advances in Tourette syndrome research. Trends Neurosci. 2006, 29, 175–182. [Google Scholar] [CrossRef]
- Han, S.; Marquez-Gomez, R.; Woodman, M.; Ellender, T. Histaminergic Control of Corticostriatal Synaptic Plasticity during Early Postnatal Development. J. Neurosci. 2020, 40, 6557–6571. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Um, J.Y.; Cho, J.S.; Lee, S.H.; Lee, S.H.; Lee, H.M. Histamine Promotes the Release of Interleukin-6 via the H1R/p38 and NF-kappaB Pathways in Nasal Fibroblasts. Allergy Asthma Immunol. Res. 2014, 6, 567–572. [Google Scholar] [CrossRef]
- Carthy, E.; Ellender, T. Histamine, Neuroinflammation and Neurodevelopment: A Review. Front. Neurosci. 2021, 15, 680214. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Barata-Antunes, S.; Santos, T.; Ferreiro, E.; Cristovao, A.C.; Serra-Almeida, C.; Ferreira, R.; Bernardino, L. Histamine modulates hippocampal inflammation and neurogenesis in adult mice. Sci. Rep. 2019, 9, 8384. [Google Scholar] [CrossRef]
- Thangam, E.B.; Jemima, E.A.; Singh, H.; Baig, M.S.; Khan, M.; Mathias, C.B.; Church, M.K.; Saluja, R. The Role of Histamine and Histamine Receptors in Mast Cell-Mediated Allergy and Inflammation: The Hunt for New Therapeutic Targets. Front. Immunol. 2018, 9, 1873. [Google Scholar] [CrossRef]
- Al-Ayadhi, L.Y.; Mostafa, G.A. Elevated serum levels of interleukin-17A in children with autism. J. Neuroinflamm. 2012, 9, 158. [Google Scholar] [CrossRef]
- Kim, E.; Paik, D.; Ramirez, R.N.; Biggs, D.G.; Park, Y.; Kwon, H.K.; Choi, G.B.; Huh, J.R. Maternal gut bacteria drive intestinal inflammation in offspring with neurodevelopmental disorders by altering the chromatin landscape of CD4(+) T cells. Immunity 2022, 55, 145–158.e7. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef]
- Fala, L. Cosentyx (Secukinumab): First IL-17A Antagonist Receives FDA Approval for Moderate-to-Severe Plaque Psoriasis. Am. Health Drug Benefits 2016, 9, 60–63. [Google Scholar] [PubMed]
- U.S. Food and Drug Administration. FDA Approves New Psoriasis Drug Taltz. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-new-psoriasis-drug-taltz (accessed on 19 June 2025).
- Fujitani, M.; Miyajima, H.; Otani, Y.; Liu, X. Maternal and Adult Interleukin-17A Exposure and Autism Spectrum Disorder. Front. Psychiatry 2022, 13, 836181. [Google Scholar] [CrossRef]
- Ellison, G.L.; Helzlsouer, K.J.; Rosenfield, S.M.; Kim, Y.; Ashare, R.L.; Blaes, A.H.; Cullen, J.; Doran, N.; Ebbert, J.O.; Egan, K.M.; et al. Perceptions, prevalence, and patterns of cannabis use among cancer patients treated at 12 NCI-Designated Cancer Centers. J. Natl. Cancer Inst. Monogr. 2024, 2024, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Montagner, P.S.S.; Medeiros, W.; da Silva, L.C.R.; Borges, C.N.; Brasil-Neto, J.; de Deus Silva Barbosa, V.; Caixeta, F.V.; Malcher-Lopes, R. Individually tailored dosage regimen of full-spectrum Cannabis extracts for autistic core and comorbid symptoms: A real-life report of multi-symptomatic benefits. Front. Psychiatry 2023, 14, 1210155. [Google Scholar] [CrossRef]
- Aran, A.; Harel, M.; Cassuto, H.; Polyansky, L.; Schnapp, A.; Wattad, N.; Shmueli, D.; Golan, D.; Castellanos, F.X. Cannabinoid treatment for autism: A proof-of-concept randomized trial. Mol. Autism 2021, 12, 6. [Google Scholar] [CrossRef]
- Bilge, S.; Ekici, B. CBD-enriched cannabis for autism spectrum disorder: An experience of a single center in Turkey and reviews of the literature. J. Cannabis Res. 2021, 3, 53. [Google Scholar] [CrossRef]
- Hacohen, M.; Stolar, O.E.; Berkovitch, M.; Elkana, O.; Kohn, E.; Hazan, A.; Heyman, E.; Sobol, Y.; Waissengreen, D.; Gal, E.; et al. Children and adolescents with ASD treated with CBD-rich cannabis exhibit significant improvements particularly in social symptoms: An open label study. Transl. Psychiatry 2022, 12, 375. [Google Scholar] [CrossRef]
- Ma, L.; Platnick, S.; Platnick, H. Cannabidiol in Treatment of Autism Spectrum Disorder: A Case Study. Cureus 2022, 14, e28442. [Google Scholar] [CrossRef]
- Leas, E.C.; Hendrickson, E.M.; Nobles, A.L.; Todd, R.; Smith, D.M.; Dredze, M.; Ayers, J.W. Self-reported Cannabidiol (CBD) Use for Conditions With Proven Therapies. JAMA Netw. Open 2020, 3, e2020977. [Google Scholar] [CrossRef] [PubMed]
- Aran, A.; Cassuto, H.; Lubotzky, A.; Wattad, N.; Hazan, E. Brief Report: Cannabidiol-Rich Cannabis in Children with Autism Spectrum Disorder and Severe Behavioral Problems-A Retrospective Feasibility Study. J. Autism Dev. Disord. 2019, 49, 1284–1288. [Google Scholar] [CrossRef]
- Straiker, A.; Dvorakova, M.; Zimmowitch, A.; Mackie, K. Cannabidiol Inhibits Endocannabinoid Signaling in Autaptic Hippocampal Neurons. Mol. Pharmacol. 2018, 94, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S.; Subbanna, S. Molecular Insights into Epigenetics and Cannabinoid Receptors. Biomolecules 2022, 12, 1560. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A. Oxytocin and human social behavior. Personal. Soc. Psychol. Rev. 2010, 14, 281–295. [Google Scholar] [CrossRef]
- Gregory, S.G.; Connelly, J.J.; Towers, A.J.; Johnson, J.; Biscocho, D.; Markunas, C.A.; Lintas, C.; Abramson, R.K.; Wright, H.H.; Ellis, P.; et al. Genomic and epigenetic evidence for oxytocin receptor deficiency in autism. BMC Med. 2009, 7, 62. [Google Scholar] [CrossRef]
- Evenepoel, M.; Moerkerke, M.; Daniels, N.; Chubar, V.; Claes, S.; Turner, J.; Vanaudenaerde, B.; Willems, L.; Verhaeghe, J.; Prinsen, J.; et al. Endogenous oxytocin levels in children with autism: Associations with cortisol levels and oxytocin receptor gene methylation. Transl. Psychiatry 2023, 13, 235. [Google Scholar] [CrossRef]
- Szabo, J.; Mlynar, M.; Fejes, A.; Renczes, E.; Borbelyova, V.; Ostatnikova, D.; Celec, P. Intranasal oxytocin in a genetic animal model of autism. Mol. Psychiatry 2024, 29, 342–347. [Google Scholar] [CrossRef]
- Moerkerke, M.; Daniels, N.; Tibermont, L.; Tang, T.; Evenepoel, M.; Van der Donck, S.; Debbaut, E.; Prinsen, J.; Chubar, V.; Claes, S.; et al. Chronic oxytocin administration stimulates the oxytocinergic system in children with autism. Nat. Commun. 2024, 15, 58. [Google Scholar] [CrossRef]
- Le, J.; Zhang, L.; Zhao, W.; Zhu, S.; Lan, C.; Kou, J.; Zhang, Q.; Zhang, Y.; Li, Q.; Chen, Z.; et al. Infrequent Intranasal Oxytocin Followed by Positive Social Interaction Improves Symptoms in Autistic Children: A Pilot Randomized Clinical Trial. Psychother. Psychosom. 2022, 91, 335–347. [Google Scholar] [CrossRef]
- Rapanelli, M.; Williams, J.B.; Ma, K.; Yang, F.; Zhong, P.; Patel, R.; Kumar, M.; Qin, L.; Rein, B.; Wang, Z.J.; et al. Targeting histone demethylase LSD1 for treatment of deficits in autism mouse models. Mol. Psychiatry 2022, 27, 3355–3366. [Google Scholar] [CrossRef]
- Kratsman, N.; Getselter, D.; Elliott, E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology 2016, 102, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Rein, B.; Zhong, P.; Shwani, T.; Conrow-Graham, M.; Wang, Z.J.; Yan, Z. Synergistic inhibition of histone modifiers produces therapeutic effects in adult Shank3-deficient mice. Transl. Psychiatry 2021, 11, 99. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Ma, K.; Wang, Z.J.; Hu, Z.; Matas, E.; Wei, J.; Yan, Z. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat. Neurosci. 2018, 21, 564–575. [Google Scholar] [CrossRef]
- Barlow, D.P.; Bartolomei, M.S. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a018382. [Google Scholar] [CrossRef]
- Liao, J.; Szabo, P.E. Role of transcription in imprint establishment in the male and female germ lines. Epigenomics 2024, 16, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Prabha, P.K.; Singla, R.; Kaur, G.; Sharma, A.R.; Joshi, R.; Suroy, B.; Medhi, B. Epigenetic Interface of Autism Spectrum Disorders (ASDs): Implications of Chromosome 15q11-q13 Segment. ACS Chem. Neurosci. 2022, 13, 1684–1696. [Google Scholar] [CrossRef]
- Lu, Z.; Li, J.; Lu, Y.; Li, L.; Wang, W.; Zhang, C.; Xu, L.; Nie, Y.; Gao, C.; Bian, X.; et al. Cynomolgus-rhesus hybrid macaques serve as a platform for imprinting studies. Innovation 2023, 4, 100436. [Google Scholar] [CrossRef]
- Strong, E.; Butcher, D.T.; Singhania, R.; Mervis, C.B.; Morris, C.A.; De Carvalho, D.; Weksberg, R.; Osborne, L.R. Symmetrical Dose-Dependent DNA-Methylation Profiles in Children with Deletion or Duplication of 7q11.23. Am. J. Hum. Genet. 2015, 97, 216–227. [Google Scholar] [CrossRef]
- Wong, C.C.Y.; Smith, R.G.; Hannon, E.; Ramaswami, G.; Parikshak, N.N.; Assary, E.; Troakes, C.; Poschmann, J.; Schalkwyk, L.C.; Sun, W.; et al. Genome-wide DNA methylation profiling identifies convergent molecular signatures associated with idiopathic and syndromic autism in post-mortem human brain tissue. Hum. Mol. Genet. 2019, 28, 2201–2211. [Google Scholar] [CrossRef]
- Aspra, Q.; Cabrera-Mendoza, B.; Morales-Marin, M.E.; Marquez, C.; Chicalote, C.; Ballesteros, A.; Aguilar, M.; Castro, X.; Gomez-Cotero, A.; Balboa-Verduzco, A.M.; et al. Epigenome-Wide Analysis Reveals DNA Methylation Alteration in ZFP57 and Its Target RASGFR2 in a Mexican Population Cohort with Autism. Children 2022, 9, 462. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, J.I.; Bakulski, K.M.; Jaffe, A.E.; Tryggvadottir, R.; Brown, S.C.; Goldman, L.R.; Croen, L.A.; Hertz-Picciotto, I.; Newschaffer, C.J.; Fallin, M.D.; et al. Paternal sperm DNA methylation associated with early signs of autism risk in an autism-enriched cohort. Int. J. Epidemiol. 2015, 44, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Schrott, R.; Greeson, K.W.; King, D.; Symosko Crow, K.M.; Easley, C.A., IV; Murphy, S.K. Cannabis alters DNA methylation at maternally imprinted and autism candidate genes in spermatogenic cells. Syst. Biol. Reprod. Med. 2022, 68, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Schanen, N.C. Epigenetics of autism spectrum disorders. Hum. Mol. Genet. 2006, 15 (Suppl. S2), R138–R150. [Google Scholar] [CrossRef]
- Hogart, A.; Nagarajan, R.P.; Patzel, K.A.; Yasui, D.H.; Lasalle, J.M. 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum. Mol. Genet. 2007, 16, 691–703. [Google Scholar] [CrossRef]
- Ben-David, E.; Shohat, S.; Shifman, S. Allelic expression analysis in the brain suggests a role for heterogeneous insults affecting epigenetic processes in autism spectrum disorders. Hum. Mol. Genet. 2014, 23, 4111–4124. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Delgado, M.; Camprubi, C.; Tumer, Z.; Martinez, F.; Mila, M.; Monk, D. Screening individuals with intellectual disability, autism and Tourette’s syndrome for KCNK9 mutations and aberrant DNA methylation within the 8q24 imprinted cluster. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165B, 472–478. [Google Scholar] [CrossRef]
- Schaafsma, S.M.; Pfaff, D.W. Etiologies underlying sex differences in Autism Spectrum Disorders. Front. Neuroendocrinol. 2014, 35, 255–271. [Google Scholar] [CrossRef]
- Baranova, J.; Dragunas, G.; Botellho, M.C.S.; Ayub, A.L.P.; Bueno-Alves, R.; Alencar, R.R.; Papaiz, D.D.; Sogayar, M.C.; Ulrich, H.; Correa, R.G. Autism Spectrum Disorder: Signaling Pathways and Prospective Therapeutic Targets. Cell. Mol. Neurobiol. 2021, 41, 619–649. [Google Scholar] [CrossRef]
- Caracci, M.O.; Avila, M.E.; Espinoza-Cavieres, F.A.; Lopez, H.R.; Ugarte, G.D.; De Ferrari, G.V. Wnt/beta-Catenin-Dependent Transcription in Autism Spectrum Disorders. Front. Mol. Neurosci. 2021, 14, 764756. [Google Scholar] [CrossRef]
- Chen, R.; Davis, L.K.; Guter, S.; Wei, Q.; Jacob, S.; Potter, M.H.; Cox, N.J.; Cook, E.H.; Sutcliffe, J.S.; Li, B. Leveraging blood serotonin as an endophenotype to identify de novo and rare variants involved in autism. Mol. Autism 2017, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.R.; Zhang, Q.; Miao, J.K.; Yang, T.; Chen, J.; Chen, H.Y.; Mou, Q.H.; Xiang, X.L.; Long, D.; Wei, Q.H.; et al. Association of the retinol to all-trans retinoic acid pathway with autism spectrum disorder. World J. Pediatr. 2024, 20, 1043–1058. [Google Scholar] [CrossRef]
- Ilchibaeva, T.; Tsybko, A.; Lipnitskaya, M.; Eremin, D.; Milutinovich, K.; Naumenko, V.; Popova, N. Brain-Derived Neurotrophic Factor (BDNF) in Mechanisms of Autistic-like Behavior in BTBR Mice: Crosstalk with the Dopaminergic Brain System. Biomedicines 2023, 11, 1482. [Google Scholar] [CrossRef]
- Pagani, M.; Barsotti, N.; Bertero, A.; Trakoshis, S.; Ulysse, L.; Locarno, A.; Miseviciute, I.; De Felice, A.; Canella, C.; Supekar, K.; et al. mTOR-related synaptic pathology causes autism spectrum disorder-associated functional hyperconnectivity. Nat. Commun. 2021, 12, 6084. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Larsen, R.S.; Bjorklund, G.R.; Li, X.; Wu, Y.; Philpot, B.D.; Snider, W.D.; Newbern, J.M. Layer specific and general requirements for ERK/MAPK signaling in the developing neocortex. Elife 2016, 5, e11123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.H.; Wang, T.; Li, Y.F.; Deng, Y.N.; Shen, F.G. Roles of the Notch signaling pathway and microglia in autism. Behav. Brain Res. 2023, 437, 114131. [Google Scholar] [CrossRef]
- Trifonova, E.A.; Klimenko, A.I.; Mustafin, Z.S.; Lashin, S.A.; Kochetov, A.V. The mTOR Signaling Pathway Activity and Vitamin D Availability Control the Expression of Most Autism Predisposition Genes. Int. J. Mol. Sci. 2019, 20, 6332. [Google Scholar] [CrossRef]
- Brauer, B.; Ancaten-Gonzalez, C.; Ahumada-Marchant, C.; Meza, R.C.; Merino-Veliz, N.; Nardocci, G.; Varela-Nallar, L.; Arriagada, G.; Chavez, A.E.; Bustos, F.J. Impact of KDM6B mosaic brain knockout on synaptic function and behavior. Sci. Rep. 2024, 14, 20416. [Google Scholar] [CrossRef]
- Sen, G.L.; Webster, D.E.; Barragan, D.I.; Chang, H.Y.; Khavari, P.A. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes. Dev. 2008, 22, 1865–1870. [Google Scholar] [CrossRef]
- Burket, J.A.; Benson, A.D.; Tang, A.H.; Deutsch, S.I. NMDA receptor activation regulates sociability by its effect on mTOR signaling activity. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 60, 60–65. [Google Scholar] [CrossRef]
- Rajan, K.C.; Tiemroth, A.S.; Thurmon, A.N.; Meadows, S.M.; Galazo, M.J. Zmiz1 is a novel regulator of brain development associated with autism and intellectual disability. Front. Psychiatry 2024, 15, 1375492. [Google Scholar]
- Lu, W.; Chen, Z.; Xu, H.; Shen, Z.; Wu, Z.; Li, M. Decreased ZMIZ1 suppresses melanogenesis in vitiligo by regulating mTOR/AKT/GSK-3beta-mediated glucose uptake. In Vitro Cell. Dev. Biol. Anim. 2024, 60, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Noroozi, R.; Brand, S.; Hussen, B.M.; Eghtedarian, R.; Taheri, M.; Ebrahimzadeh, K. Emerging Role of Non-coding RNAs in Autism Spectrum Disorder. J. Mol. Neurosci. 2022, 72, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.; Ou, J.N.; Cao, L.F.; Peng, X.Q.; Li, Y.M.; Tian, Y.Q. The Autism-Related lncRNA MSNP1AS Regulates Moesin Protein to Influence the RhoA, Rac1, and PI3K/Akt Pathways and Regulate the Structure and Survival of Neurons. Autism Res. 2020, 13, 2073–2082. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Sawada, H.; Kihara, T.; Urushitani, M.; Nakamizo, T.; Akaike, A.; Shimohama, S. Phosphatidylinositol 3-kinase mediates neuroprotection by estrogen in cultured cortical neurons. J. Neurosci. Res. 2000, 60, 321–327. [Google Scholar] [CrossRef]
- Yang, Y.M.; Sun, D.; Kandhi, S.; Froogh, G.; Zhuge, J.; Huang, W.; Hammock, B.D.; Huang, A. Estrogen-dependent epigenetic regulation of soluble epoxide hydrolase via DNA methylation. Proc. Natl. Acad. Sci. USA 2018, 115, 613–618. [Google Scholar] [CrossRef]
- Iyer, M.R.; Kundu, B.; Wood, C.M. Soluble epoxide hydrolase inhibitors: An overview and patent review from the last decade. Expert. Opin. Ther. Pat. 2022, 32, 629–647. [Google Scholar] [CrossRef]
- Ma, M.; Ren, Q.; Yang, J.; Zhang, K.; Xiong, Z.; Ishima, T.; Pu, Y.; Hwang, S.H.; Toyoshima, M.; Iwayama, Y.; et al. Key role of soluble epoxide hydrolase in the neurodevelopmental disorders of offspring after maternal immune activation. Proc. Natl. Acad. Sci. USA 2019, 116, 7083–7088. [Google Scholar] [CrossRef]
- Chu, M.C.; Wu, H.F.; Lee, C.W.; Wu, C.C.; Chi, H.; Ko, C.Y.; Lee, Y.C.; Tang, C.W.; Chen, P.S.; Lin, H.C. Soluble epoxide hydrolase deletion rescues behavioral and synaptic deficits by AMPK-mTOR pathway in autism animals. Prog. Neuropsychopharmacol. Biol. Psychiatry 2025, 136, 111190. [Google Scholar] [CrossRef]
- Imataka, G.; Mori, S.; Yui, K.; Igawa, K.; Shiraishi, H.; Yoshihara, S. The Therapeutic Potential of Oral Everolimus for Facial Angiofibromas in Pediatric Tuberous Sclerosis Complex: A Case-Based Analysis of Efficacy. Diseases 2024, 12, 334. [Google Scholar] [CrossRef]
- Iezzi, D.; Curti, L.; Ranieri, G.; Gerace, E.; Costa, A.; Ilari, A.; La Rocca, A.; Luceri, C.; D’Ambrosio, M.; Silvestri, L.; et al. Acute rapamycin rescues the hyperexcitable phenotype of accumbal medium spiny neurons in the valproic acid rat model of autism spectrum disorder. Pharmacol. Res. 2022, 183, 106401. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.D.; Jha, N.K.; Ojha, S.; Sadek, B. mTOR Signaling Disruption and Its Association with the Development of Autism Spectrum Disorder. Molecules 2023, 28, 1889. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bhalla, S.; Mehan, S. PI3K/AKT/mTOR signalling inhibitor chrysophanol ameliorates neurobehavioural and neurochemical defects in propionic acid-induced experimental model of autism in adult rats. Metab. Brain Dis. 2022, 37, 1909–1929. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, S.B.; Valenzuela-Bezanilla, D.; Mardones, M.D.; Varela-Nallar, L. Role of Wnt Signaling in Adult Hippocampal Neurogenesis in Health and Disease. Front. Cell Dev. Biol. 2020, 8, 860. [Google Scholar] [CrossRef]
- McLeod, F.; Salinas, P.C. Wnt proteins as modulators of synaptic plasticity. Curr. Opin. Neurobiol. 2018, 53, 90–95. [Google Scholar] [CrossRef]
- Speese, S.D.; Budnik, V. Wnts: Up-and-coming at the synapse. Trends Neurosci. 2007, 30, 268–275. [Google Scholar] [CrossRef]
- Karwacki-Neisius, V.; Jang, A.; Cukuroglu, E.; Tai, A.; Jiao, A.; Predes, D.; Yoon, J.; Brookes, E.; Chen, J.; Iberg, A.; et al. WNT signalling control by KDM5C during development affects cognition. Nature 2024, 627, 594–603. [Google Scholar] [CrossRef]
- Thompson, B.A.; Tremblay, V.; Lin, G.; Bochar, D.A. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol. Cell. Biol. 2008, 28, 3894–3904. [Google Scholar] [CrossRef]
- Zhang, B.; Zhao, C.; Shen, W.; Li, W.; Zheng, Y.; Kong, X.; Wang, J.; Wu, X.; Zeng, T.; Liu, Y.; et al. KDM2B regulates hippocampal morphogenesis by transcriptionally silencing Wnt signaling in neural progenitors. Nat. Commun. 2023, 14, 6489. [Google Scholar] [CrossRef]
- Liu, Y.; Fan, M.; Yang, J.; Mihaljevic, L.; Chen, K.H.; Ye, Y.; Sun, S.; Qiu, Z. KAT6A deficiency impairs cognitive functions through suppressing RSPO2/Wnt signaling in hippocampal CA3. Sci. Adv. 2024, 10, eadm9326. [Google Scholar] [CrossRef]
- Shrestha, P.; Jaganathan, A.; Huilgol, D.; Ballon, C.; Hwangbo, Y.; Mills, A.A. Chd5 Regulates the Transcription Factor Six3 to Promote Neuronal Differentiation. Stem Cells 2023, 41, 242–251. [Google Scholar] [CrossRef] [PubMed]
- van Amerongen, R.; Fuerer, C.; Mizutani, M.; Nusse, R. Wnt5a can both activate and repress Wnt/beta-catenin signaling during mouse embryonic development. Dev. Biol. 2012, 369, 101–114. [Google Scholar] [CrossRef]
- Gong, M.; Li, J.; Qin, Z.; Machado Bressan Wilke, M.V.; Liu, Y.; Li, Q.; Liu, H.; Liang, C.; Morales-Rosado, J.A.; Cohen, A.S.A.; et al. MARK2 variants cause autism spectrum disorder via the downregulation of WNT/beta-catenin signaling pathway. Am. J. Hum. Genet. 2024, 111, 2392–2410. [Google Scholar] [CrossRef]
- Rybakowski, J.K. Lithium in neuropsychiatry: A 2010 update. World J. Biol. Psychiatry 2011, 12, 340–348. [Google Scholar] [CrossRef]
- Dwivedi, T.; Zhang, H. Lithium-induced neuroprotection is associated with epigenetic modification of specific BDNF gene promoter and altered expression of apoptotic-regulatory proteins. Front. Neurosci. 2014, 8, 457. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.G.; Kuddo, T.; Song, E.Y.; Dambrosia, J.M.; Kohler, S.; Satyanarayana, G.; Vandunk, C.; Grether, J.K.; Nelson, K.B. Selected neurotrophins, neuropeptides, and cytokines: Developmental trajectory and concentrations in neonatal blood of children with autism or Down syndrome. Int. J. Dev. Neurosci. 2006, 24, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Ricci, S.; Businaro, R.; Ippoliti, F.; Lo Vasco, V.R.; Massoni, F.; Onofri, E.; Troili, G.M.; Pontecorvi, V.; Morelli, M.; Rapp Ricciardi, M.; et al. Altered cytokine and BDNF levels in autism spectrum disorder. Neurotox. Res. 2013, 24, 491–501. [Google Scholar] [CrossRef]
- Zhang, W.; Shi, Y.; Peng, Y.; Zhong, L.; Zhu, S.; Zhang, W.; Tang, S.J. Neuron activity-induced Wnt signaling up-regulates expression of brain-derived neurotrophic factor in the pain neural circuit. J. Biol. Chem. 2018, 293, 15641–15651. [Google Scholar] [CrossRef]
- Cheon, S.; Culver, A.M.; Bagnell, A.M.; Ritchie, F.D.; Vacharasin, J.M.; McCord, M.M.; Papendorp, C.M.; Chukwurah, E.; Smith, A.J.; Cowen, M.H.; et al. Counteracting epigenetic mechanisms regulate the structural development of neuronal circuitry in human neurons. Mol. Psychiatry 2022, 27, 2291–2303. [Google Scholar] [CrossRef]
- Li, Y.; Ma, L.; Deng, Y.; Du, Z.; Guo, B.; Yue, J.; Liu, X.; Zhang, Y. The Notch1/Hes1 signaling pathway affects autophagy by adjusting DNA methyltransferases expression in a valproic acid-induced autism spectrum disorder model. Neuropharmacology 2023, 239, 109682. [Google Scholar] [CrossRef]
- Sgritta, M.; Dooling, S.W.; Buffington, S.A.; Momin, E.N.; Francis, M.B.; Britton, R.A.; Costa-Mattioli, M. Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron 2019, 101, 246–259.e6. [Google Scholar] [CrossRef]
- Herbert, B.M.; Blechert, J.; Hautzinger, M.; Matthias, E.; Herbert, C. Intuitive eating is associated with interoceptive sensitivity. Effects on body mass index. Appetite 2013, 70, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Jurecka, A.; Zikanova, M.; Jurkiewicz, E.; Tylki-Szymanska, A. Attenuated adenylosuccinate lyase deficiency: A report of one case and a review of the literature. Neuropediatrics 2014, 45, 50–55. [Google Scholar] [PubMed]
- Saad, K.; Shabaan, I.; Hassan, A.M.; Ezzat, M.; Abouzed, M.A.; Hamed, Y.; Ibrahim, M.F.M.; Gad, E.F. Gluten-Free, Casein-Free Diet for Children with Autism Spectrum Disorder: A Case-Controlled Study. J. Pharm. Bioallied Sci. 2024, 16 (Suppl. S1), S905–S908. [Google Scholar] [CrossRef]
- Elder, J.H.; Kreider, C.M.; Schaefer, N.M.; de Laosa, M.B. A review of gluten- and casein-free diets for treatment of autism: 2005–2015. Nutr. Diet. Suppl. 2015, 7, 87–101. [Google Scholar] [CrossRef] [PubMed]
- de Magistris, L.; Picardi, A.; Siniscalco, D.; Riccio, M.P.; Sapone, A.; Cariello, R.; Abbadessa, S.; Medici, N.; Lammers, K.M.; Schiraldi, C.; et al. Antibodies against food antigens in patients with autistic spectrum disorders. Biomed. Res. Int. 2013, 2013, 729349. [Google Scholar] [CrossRef]
- Karhu, E.; Zukerman, R.; Eshraghi, R.S.; Mittal, J.; Deth, R.C.; Castejon, A.M.; Trivedi, M.; Mittal, R.; Eshraghi, A.A. Nutritional interventions for autism spectrum disorder. Nutr. Rev. 2020, 78, 515–531. [Google Scholar] [CrossRef]
- Macfabe, D.F. Short-chain fatty acid fermentation products of the gut microbiome: Implications in autism spectrum disorders. Microb. Ecol. Health Dis. 2012, 23, 19260. [Google Scholar] [CrossRef]
- Mazzone, L.; Dooling, S.W.; Volpe, E.; Uljarevic, M.; Waters, J.L.; Sabatini, A.; Arturi, L.; Abate, R.; Riccioni, A.; Siracusano, M.; et al. Precision microbial intervention improves social behavior but not autism severity: A pilot double-blind randomized placebo-controlled trial. Cell Host Microbe 2024, 32, 106–116.e6. [Google Scholar] [CrossRef]
- Phan, J.; Calvo, D.C.; Nair, D.; Jain, S.; Montagne, T.; Dietsche, S.; Blanchard, K.; Treadwell, S.; Adams, J.; Krajmalnik-Brown, R. Precision synbiotics increase gut microbiome diversity and improve gastrointestinal symptoms in a pilot open-label study for autism spectrum disorder. mSystems 2024, 9, e0050324. [Google Scholar] [CrossRef]
- Autism Research Institute. Screening & Assessment. Available online: https://autism.org/screening-assessment/ (accessed on 15 August 2024).
- Empower Behavioral Health. What Are the 4 Main Tests for Autism? Available online: https://www.empowerbh.com/blog/what-are-the-4-main-tests-for-autism/ (accessed on 15 August 2024).
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson Rosenberg, C.; White, T.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Durkin, M.S.; Maenner, M.J.; Baio, J.; Christensen, D.; Daniels, J.; Fitzgerald, R.; Imm, P.; Lee, L.C.; Schieve, L.A.; Van Naarden Braun, K.; et al. Autism Spectrum Disorder Among US Children (2002–2010): Socioeconomic, Racial, and Ethnic Disparities. Am. J. Public Health 2017, 107, 1818–1826. [Google Scholar] [CrossRef] [PubMed]
- Tek, S.; Landa, R.J. Differences in autism symptoms between minority and non-minority toddlers. J. Autism Dev. Disord. 2012, 42, 1967–1973. [Google Scholar] [CrossRef] [PubMed]
- Gabbay-Dizdar, N.; Ilan, M.; Meiri, G.; Faroy, M.; Michaelovski, A.; Flusser, H.; Menashe, I.; Koller, J.; Zachor, D.A.; Dinstein, I. Early diagnosis of autism in the community is associated with marked improvement in social symptoms within 1–2 years. Autism 2022, 26, 1353–1363. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Miska, E.A. How microRNAs control cell division, differentiation and death. Curr. Opin. Genet. Dev. 2005, 15, 563–568. [Google Scholar] [CrossRef]
- Wu, X.; Li, W.; Zheng, Y. Recent Progress on Relevant microRNAs in Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 5904. [Google Scholar] [CrossRef]
- Hicks, S.D.; Confair, A. Infant Saliva Levels of microRNA miR-151a-3p Are Associated with Risk for Neurodevelopmental Delay. Int. J. Mol. Sci. 2023, 24, 1476. [Google Scholar] [CrossRef]
- Hicks, S.D.; Ignacio, C.; Gentile, K.; Middleton, F.A. Salivary miRNA profiles identify children with autism spectrum disorder, correlate with adaptive behavior, and implicate ASD candidate genes involved in neurodevelopment. BMC Pediatr. 2016, 16, 52. [Google Scholar] [CrossRef]
- Kalemaj, Z.; Marino, M.M.; Santini, A.C.; Tomaselli, G.; Auti, A.; Cagetti, M.G.; Borsello, T.; Costantino, A.; Inchingolo, F.; Boccellino, M.; et al. Salivary microRNA profiling dysregulation in autism spectrum disorder: A pilot study. Front. Neurosci. 2022, 16, 945278. [Google Scholar] [CrossRef]
- Che, X.; Roy, A.; Bresnahan, M.; Mjaaland, S.; Reichborn-Kjennerud, T.; Magnus, P.; Stoltenberg, C.; Shang, Y.; Zhang, K.; Susser, E.; et al. Metabolomic analysis of maternal mid-gestation plasma and cord blood in autism spectrum disorders. Mol. Psychiatry 2023, 28, 2355–2369. [Google Scholar] [CrossRef]
- Suren, P.; Roth, C.; Bresnahan, M.; Haugen, M.; Hornig, M.; Hirtz, D.; Lie, K.K.; Lipkin, W.I.; Magnus, P.; Reichborn-Kjennerud, T.; et al. Association between maternal use of folic acid supplements and risk of autism spectrum disorders in children. JAMA 2013, 309, 570–577. [Google Scholar] [CrossRef]
- Wiggs, K.K.; Rickert, M.E.; Sujan, A.C.; Quinn, P.D.; Larsson, H.; Lichtenstein, P.; Oberg, A.S.; D’Onofrio, B.M. Antiseizure medication use during pregnancy and risk of ASD and ADHD in children. Neurology 2020, 95, e3232–e3240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Zhang, S.Y.; Wen, R.; Zhang, T.N.; Yang, N. Role of histone deacetylases and their inhibitors in neurological diseases. Pharmacol. Res. 2024, 208, 107410. [Google Scholar] [CrossRef]
- Sambra, V.; Echeverria, F.; Valenzuela, A.; Chouinard-Watkins, R.; Valenzuela, R. Docosahexaenoic and Arachidonic Acids as Neuroprotective Nutrients throughout the Life Cycle. Nutrients 2021, 13, 986. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.E.; Hundert, A.S.; Goguen, D.; Weaver, I.C.; Karten, B. Presymptomatic Alterations in Amino Acid Metabolism and DNA Methylation in the Cerebellum of a Murine Model of Niemann-Pick Type C Disease. Am. J. Pathol. 2016, 186, 1582–1597. [Google Scholar] [CrossRef]
- Bahado-Singh, R.O.; Vishweswaraiah, S.; Aydas, B.; Radhakrishna, U. Placental DNA methylation changes and the early prediction of autism in full-term newborns. PLoS ONE 2021, 16, e0253340. [Google Scholar] [CrossRef] [PubMed]
- Mordaunt, C.E.; Jianu, J.M.; Laufer, B.I.; Zhu, Y.; Hwang, H.; Dunaway, K.W.; Bakulski, K.M.; Feinberg, J.I.; Volk, H.E.; Lyall, K.; et al. Cord blood DNA methylome in newborns later diagnosed with autism spectrum disorder reflects early dysregulation of neurodevelopmental and X-linked genes. Genome Med. 2020, 12, 88. [Google Scholar] [CrossRef]
- Zwaigenbaum, L.; Penner, M. Autism spectrum disorder: Advances in diagnosis and evaluation. BMJ 2018, 361, k1674. [Google Scholar] [CrossRef]
- Jia, Q.; Li, H.; Wang, M.; Wei, C.; Xu, L.; Ye, L.; Wang, C.; Ke, S.; Li, L.; Yao, P. Transcript levels of 4 genes in umbilical cord blood are predictive of later autism development: A longitudinal follow-up study. J. Psychiatry Neurosci. 2023, 48, E334–E344. [Google Scholar] [CrossRef]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shi, X.J.; Liu, H.; Mao, X.; Gui, L.N.; Wang, H.; Cheng, Y. Oxidative stress marker aberrations in children with autism spectrum disorder: A systematic review and meta-analysis of 87 studies (N = 9109). Transl. Psychiatry 2021, 11, 15. [Google Scholar] [CrossRef]
- Bjorklund, G.; Tinkov, A.A.; Hosnedlova, B.; Kizek, R.; Ajsuvakova, O.P.; Chirumbolo, S.; Skalnaya, M.G.; Peana, M.; Dadar, M.; El-Ansary, A.; et al. The role of glutathione redox imbalance in autism spectrum disorder: A review. Free Radic. Biol. Med. 2020, 160, 149–162. [Google Scholar] [CrossRef]
- Chauhan, A.; Audhya, T.; Chauhan, V. Brain region-specific glutathione redox imbalance in autism. Neurochem. Res. 2012, 37, 1681–1689. [Google Scholar] [CrossRef]
- Liu, X.; Lin, J.; Zhang, H.; Khan, N.U.; Zhang, J.; Tang, X.; Cao, X.; Shen, L. Oxidative Stress in Autism Spectrum Disorder-Current Progress of Mechanisms and Biomarkers. Front. Psychiatry 2022, 13, 813304. [Google Scholar] [CrossRef] [PubMed]
- Radwan, K.; Wu, G.; Banks-Word, K.; Rosenberger, R. An Open-Label Case Series of Glutathione Use for Symptomatic Management in Children with Autism Spectrum Disorder. Med. Sci. 2023, 11, 73. [Google Scholar] [CrossRef]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e8. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wang, Y.; Meng, F.; Chen, X.; Chang, T.; Huang, H.; He, F.; Zheng, Y. Altered Gut Microbiota as Potential Biomarkers for Autism Spectrum Disorder in Early Childhood. Neuroscience 2023, 523, 118–131. [Google Scholar] [CrossRef]
- Al-Ayadhi, L.; Zayed, N.; Bhat, R.S.; Moubayed, N.M.S.; Al-Muammar, M.N.; El-Ansary, A. The use of biomarkers associated with leaky gut as a diagnostic tool for early intervention in autism spectrum disorder: A systematic review. Gut Pathog. 2021, 13, 54. [Google Scholar] [CrossRef]
- Ding, X.; Xu, Y.; Zhang, X.; Zhang, L.; Duan, G.; Song, C.; Li, Z.; Yang, Y.; Wang, Y.; Wang, X.; et al. Gut microbiota changes in patients with autism spectrum disorders. J. Psychiatr. Res. 2020, 129, 149–159. [Google Scholar] [CrossRef]
- Iglesias-Vazquez, L.; Van Ginkel Riba, G.; Arija, V.; Canals, J. Composition of Gut Microbiota in Children with Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Nutrients 2020, 12, 792. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zou, R.; Guo, M.; Duan, M.; Li, Q.; Zheng, H. Comparison of gut microbiota between adults with autism spectrum disorder and obese adults. PeerJ 2021, 9, e10946. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Wong, O.W.H.; Lu, W.; Wan, Y.; Zhang, L.; Xu, W.; Li, M.K.T.; Liu, C.; Cheung, C.P.; Ching, J.Y.L.; et al. Multikingdom and functional gut microbiota markers for autism spectrum disorder. Nat. Microbiol. 2024, 9, 2344–2355. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, B.; Velasco, S.; Kedaigle, A.J.; Pigoni, M.; Quadrato, G.; Deo, A.J.; Adiconis, X.; Uzquiano, A.; Sartore, R.; Yang, S.M.; et al. Autism genes converge on asynchronous development of shared neuron classes. Nature 2022, 602, 268–273. [Google Scholar] [CrossRef]
- Lopez-Tobon, A.; Shyti, R.; Villa, C.E.; Cheroni, C.; Fuentes-Bravo, P.; Trattaro, S.; Caporale, N.; Troglio, F.; Tenderini, E.; Mihailovich, M.; et al. GTF2I dosage regulates neuronal differentiation and social behavior in 7q11.23 neurodevelopmental disorders. Sci. Adv. 2023, 9, eadh2726. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kao, C.; Lim, K.K.; Chen, E.S. Emerging Epigenetic Therapeutics and Diagnostics for Autism Spectrum Disorder. Curr. Issues Mol. Biol. 2025, 47, 491. https://doi.org/10.3390/cimb47070491
Kao C, Lim KK, Chen ES. Emerging Epigenetic Therapeutics and Diagnostics for Autism Spectrum Disorder. Current Issues in Molecular Biology. 2025; 47(7):491. https://doi.org/10.3390/cimb47070491
Chicago/Turabian StyleKao, Cassie, Kim Kiat Lim, and Ee Sin Chen. 2025. "Emerging Epigenetic Therapeutics and Diagnostics for Autism Spectrum Disorder" Current Issues in Molecular Biology 47, no. 7: 491. https://doi.org/10.3390/cimb47070491
APA StyleKao, C., Lim, K. K., & Chen, E. S. (2025). Emerging Epigenetic Therapeutics and Diagnostics for Autism Spectrum Disorder. Current Issues in Molecular Biology, 47(7), 491. https://doi.org/10.3390/cimb47070491