
Revolution of Circulating Tumor DNA: From Bench Innovations to Bedside Implementations

Abstract
1. Introduction
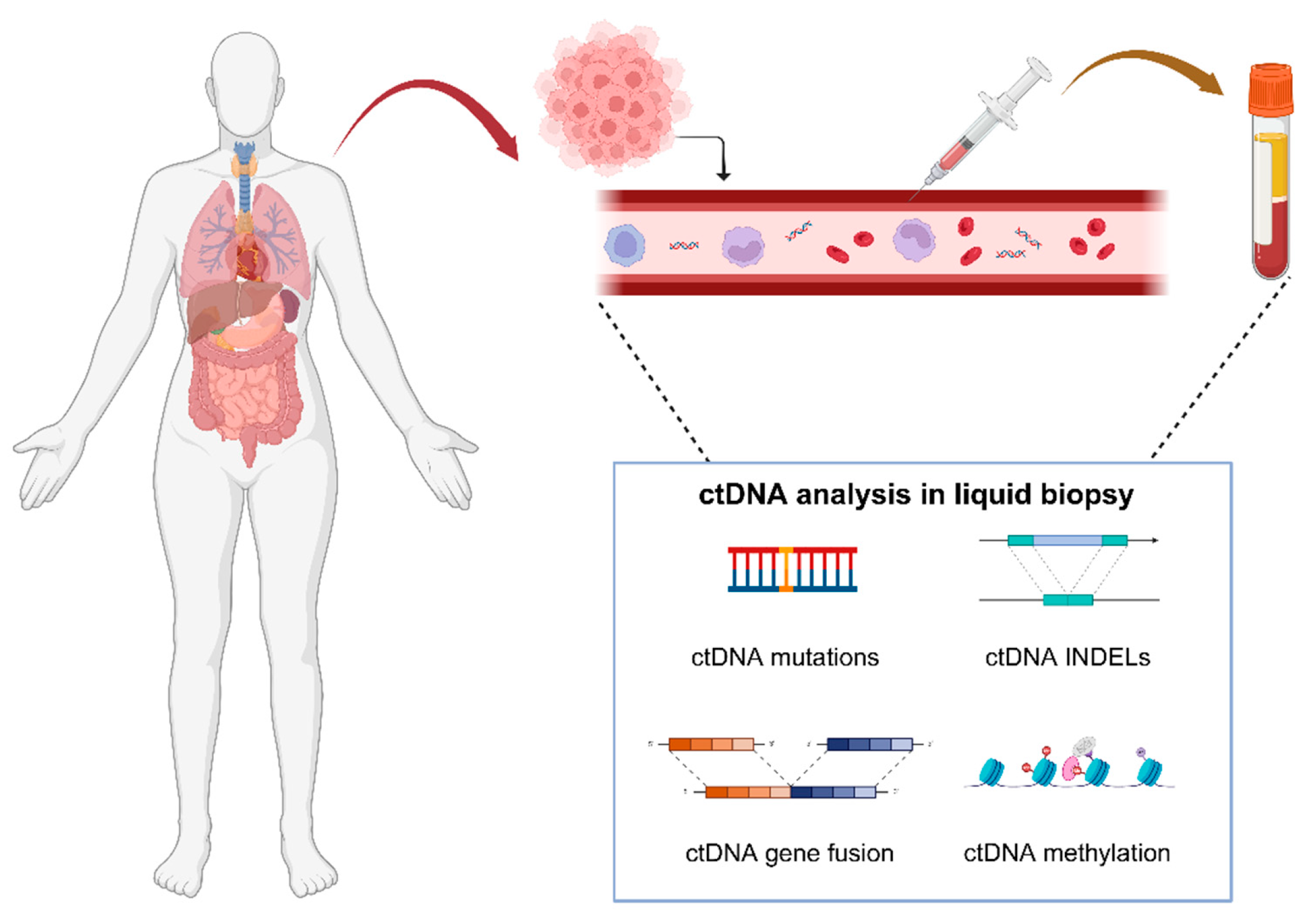
2. General Description of ctDNA
2.1. Origins
2.2. Features
2.3. Clinical Applications
3. Detecting Techniques for ctDNA
3.1. dPCRS
3.2. NGS
3.3. Comparisons
4. Applications of ctDNA in Clinical Practice
4.1. Early Screening

{kind=link}
| Methodology | Purpose | Study | Cancer Type | Total Sample | Conclusion | References/ClinicalTrial.gov Identifier |
|---|---|---|---|---|---|---|
| Mutation detection | Early detection | CancerSEEK | Ovarian, liver, gastric, pancreatic, esophageal, colorectal, lung, and breast cancers | 1817 | Medium sensitivity: 70% Medium specificity: 99% | [74] |
| Multi-cancer early detection | DETECT-A | Multiple cancers | 10,006 | Medium sensitivity: 27.1% Medium specificity: 98.9% | [75] | |
| Multi-cancer early detection | ASCEND | Multiple cancers | 4620 | / | NCT04213326 | |
| Methylation detection | Multi-cancer early detection | CCGA | Multiple cancers | 4077 | Medium sensitivity: 51.5% Medium specificity: 99.5% | [78] |
| Multi-cancer early detection | STRIVE | Breast cancer and other invasive cancers, including hematologic malignancies | 100,000 | / | NCT03085888 | |
| Multi-cancer early detection | SUMMIT | Multiple cancers | 13,000 | / | NCT03934866 | |
| Multi-cancer early detection | PATHFINDER | Multiple cancers | 6621 | Medium sensitivity: 38.0% Medium specificity: 99.1% | [79] | |
| Multi-cancer early detection | PATHFINDER2 | Multiple cancers | 35,885 | / | NCT05155605 | |
| Multi-cancer early detection | SYMPLIFY | Multiple cancers | 5461 | Medium sensitivity: 66.3% Medium specificity: 98.4% | [80] | |
| Multi-cancer early detection | NHS-Galleri | Multiple cancers | 140,000 | / | NCT05611632 | |
| Early detection | K-DETEK | Stomach, esophageal, colorectal, lung, or liver cancer | 100,501 | Medium sensitivity: 88.0% Medium specificity: 96.0% | [82] | |
| Multi-cancer early detection | / | Multiple cancers | 50,000 | / | NCT05673018 | |
| Early detection | / | Lung cancer | 600 | / | NCT05432128 | |
| Multi-cancer early detection | CADENCE | Multiple cancers | 15,000 | / | NCT05633342 | |
| Multi-cancer early detection | CHARM2 | Hereditary cancer syndromes | 1000 | / | NCT06726642 | |
| Multi-cancer early detection | ProSight | Lung cancer, colorectal cancer, liver cancer, gastric cancer, and esophageal cancer | 2527 | / | NCT06790355 | |
| Multi-cancer early detection | / | Gastric cancer | 540 | / | NCT04511559 | |
| Multi-cancer early detection | / | Lung cancer | 300 | / | NCT03685669 | |
| Multi-cancer early detection | / | Esophageal squamous cell carcinoma | 300 | / | NCT03922230 | |
| Mutation and methylation detection | Multi-cancer early detection | ASCEND-PANCREATIC | Multiple cancers | 7062 | / | NCT05556603 |
| Early detection | / | Lung cancer | 900 | / | NCT04814407 | |
| Multi-cancer early detection | / | Non-small cell lung cancer | 400 | / | NCT03301961 | |
| Fragment detection | Early detection | DELFI | Breast, colorectal, lung, ovarian, pancreatic, gastric, or bile duct cancer | 481 | Medium sensitivity: 73.0% Medium specificity: 98.0% | [73] |
| Early detection | DELFI-L101 | Lung cancer | 342 | Medium sensitivity: 84.0% Medium specificity: 53.0% | [81] | |
| Early detection | DELFI-L201 | Lung cancer | 15,000 | / | NCT05306288 |
4.2. Postoperative MRD Detection
4.3. Therapeutic Assessment
5. Conclusions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. [Nuclear Acids In Human Blood Plasma]. Comptes Rendus Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Corcoran, R.B.; Chabner, B.A. Application of Cell-free DNA Analysis to Cancer Treatment. N. Engl. J. Med. 2018, 379, 1754–1765. [Google Scholar] [CrossRef] [PubMed]
- Reams, A.B.; Roth, J.R. Mechanisms of gene duplication and amplification. Cold Spring Harb. Perspect. Biol. 2015, 7, a016592. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Koch, A.; Joosten, S.C.; Feng, Z.; de Ruijter, T.C.; Draht, M.X.; Melotte, V.; Smits, K.M.; Veeck, J.; Herman, J.G.; Van Neste, L.; et al. Analysis of DNA methylation in cancer: Location revisited. Nat. Rev. Clin. Oncol. 2018, 15, 459–466. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Ignatiadis, M.; Sledge, G.W.; Jeffrey, S.S. Liquid biopsy enters the clinic–implementation issues and future challenges. Nat. Rev. Clin. Oncol. 2021, 18, 297–312. [Google Scholar] [CrossRef]
- Nikanjam, M.; Kato, S.; Kurzrock, R. Liquid biopsy: Current technology and clinical applications. J. Hematol. Oncol. 2022, 15, 131. [Google Scholar] [CrossRef]
- Zhang, L.; Parvin, R.; Fan, Q.; Ye, F. Emerging digital PCR technology in precision medicine. Biosens. Bioelectron. 2022, 211, 114344. [Google Scholar] [CrossRef] [PubMed]
- Sreejith, K.R.; Ooi, C.H.; Jin, J.; Dao, D.V.; Nguyen, N.T. Digital polymerase chain reaction technology—Recent advances and future perspectives. Lab. Chip 2018, 18, 3717–3732. [Google Scholar] [CrossRef] [PubMed]
- Xuan, J.; Yu, Y.; Qing, T.; Guo, L.; Shi, L. Next-generation sequencing in the clinic: Promises and challenges. Cancer Lett. 2013, 340, 284–295. [Google Scholar] [CrossRef]
- Agathangelidis, A.; Vlachonikola, E.; Davi, F.; Langerak, A.W.; Chatzidimitriou, A. High-Throughput immunogenetics for precision medicine in cancer. Semin. Cancer Biol. 2022, 84, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Campos-Carrillo, A.; Weitzel, J.N.; Sahoo, P.; Rockne, R.; Mokhnatkin, J.V.; Murtaza, M.; Gray, S.W.; Goetz, L.; Goel, A.; Schork, N.; et al. Circulating tumor DNA as an early cancer detection tool. Pharmacol. Ther. 2020, 207, 107458. [Google Scholar] [CrossRef]
- Hackshaw, A.; Clarke, C.A.; Hartman, A.R. New genomic technologies for multi-cancer early detection: Rethinking the scope of cancer screening. Cancer Cell 2022, 40, 109–113. [Google Scholar] [CrossRef]
- Rubinstein, W.S.; Patriotis, C.; Dickherber, A.; Han, P.K.J.; Katki, H.A.; LeeVan, E.; Pinsky, P.F.; Prorok, P.C.; Skarlupka, A.L.; Temkin, S.M.; et al. Cancer screening with multicancer detection tests: A translational science review. CA Cancer J. Clin. 2024, 74, 368–382. [Google Scholar] [CrossRef]
- Moding, E.J.; Nabet, B.Y.; Alizadeh, A.A.; Diehn, M. Detecting Liquid Remnants of Solid Tumors: Circulating Tumor DNA Minimal Residual Disease. Cancer Discov. 2021, 11, 2968–2986. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Tao, X.Y.; Li, Q.Q.; Zeng, Y. Clinical application of liquid biopsy in colorectal cancer: Detection, prediction, and treatment monitoring. Mol. Cancer 2024, 23, 145. [Google Scholar] [CrossRef] [PubMed]
- Aucamp, J.; Bronkhorst, A.J.; Badenhorst, C.P.S.; Pretorius, P.J. The diverse origins of circulating cell-free DNA in the human body: A critical re-evaluation of the literature. Biol. Rev. Camb. Philos. Soc. 2018, 93, 1649–1683. [Google Scholar] [CrossRef]
- Thierry, A.R.; El Messaoudi, S.; Gahan, P.B.; Anker, P.; Stroun, M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016, 35, 347–376. [Google Scholar] [CrossRef]
- Gahan, P.B.; Stroun, M. The virtosome-a novel cytosolic informative entity and intercellular messenger. Cell Biochem. Funct. 2010, 28, 529–538. [Google Scholar] [CrossRef]
- Stadler, J.C.; Belloum, Y.; Deitert, B.; Sementsov, M.; Heidrich, I.; Gebhardt, C.; Keller, L.; Pantel, K. Current and Future Clinical Applications of ctDNA in Immuno-Oncology. Cancer Res. 2022, 82, 349–358. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Hurley, J.; Roberts, D.; Chakrabortty, S.K.; Enderle, D.; Noerholm, M.; Breakefield, X.O.; Skog, J.K. Exosome-based liquid biopsies in cancer: Opportunities and challenges. Ann. Oncol. 2021, 32, 466–477. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, Z.; Lei, J. CTC, ctDNA, and Exosome in Thyroid Cancers: A Review. Int. J. Mol. Sci. 2023, 24, 13767. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Rahadiani, N.; Stephanie, M.; Manatar, A.F.; Krisnuhoni, E. The Diagnostic Utility of cfDNA and ctDNA in Liquid Biopsies for Gastrointestinal Cancers over the Last Decade. Oncol. Res. Treat. 2025, 48, 125–141. [Google Scholar] [CrossRef]
- Li, Y.; Fan, Z.; Meng, Y.; Liu, S.; Zhan, H. Blood-based DNA methylation signatures in cancer: A systematic review. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166583. [Google Scholar] [CrossRef] [PubMed]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Mouliere, F.; Robert, B.; Arnau Peyrotte, E.; Del Rio, M.; Ychou, M.; Molina, F.; Gongora, C.; Thierry, A.R. High fragmentation characterizes tumour-derived circulating DNA. PLoS ONE 2011, 6, e23418. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, I.; Elst, H.J.; Van Laere, S.J.; Maes, H.; Huget, P.; van Dam, P.; Van Marck, E.A.; Vermeulen, P.B.; Dirix, L.Y. The presence of circulating total DNA and methylated genes is associated with circulating tumour cells in blood from breast cancer patients. Br. J. Cancer 2009, 100, 1277–1286. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Pantel, K.; Kemper, B.; Beeger, C.; Otterbach, F.; Kimmig, R.; Kasimir-Bauer, S. Comparative evaluation of cell-free tumor DNA in blood and disseminated tumor cells in bone marrow of patients with primary breast cancer. Breast Cancer Res. 2009, 11, R71. [Google Scholar] [CrossRef]
- Agostini, M.; Pucciarelli, S.; Enzo, M.V.; Del Bianco, P.; Briarava, M.; Bedin, C.; Maretto, I.; Friso, M.L.; Lonardi, S.; Mescoli, C.; et al. Circulating cell-free DNA: A promising marker of pathologic tumor response in rectal cancer patients receiving preoperative chemoradiotherapy. Ann. Surg. Oncol. 2011, 18, 2461–2468. [Google Scholar] [CrossRef]
- Czeiger, D.; Shaked, G.; Eini, H.; Vered, I.; Belochitski, O.; Avriel, A.; Ariad, S.; Douvdevani, A. Measurement of circulating cell-free DNA levels by a new simple fluorescent test in patients with primary colorectal cancer. Am. J. Clin. Pathol. 2011, 135, 264–270. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Alix-Panabières, C.; Müller, I.; Letang, N.; Vendrell, J.P.; Rebillard, X.; Pantel, K. Cell-free tumor DNA in blood plasma as a marker for circulating tumor cells in prostate cancer. Clin. Cancer Res. 2009, 15, 1032–1038. [Google Scholar] [CrossRef]
- Ellinger, J.; Bastian, P.J.; Haan, K.I.; Heukamp, L.C.; Buettner, R.; Fimmers, R.; Mueller, S.C.; von Ruecker, A. Noncancerous PTGS2 DNA fragments of apoptotic origin in sera of prostate cancer patients qualify as diagnostic and prognostic indicators. Int. J. Cancer 2008, 122, 138–143. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef]
- Yang, Y.C.; Wang, D.; Jin, L.; Yao, H.W.; Zhang, J.H.; Wang, J.; Zhao, X.M.; Shen, C.Y.; Chen, W.; Wang, X.L.; et al. Circulating tumor DNA detectable in early- and late-stage colorectal cancer patients. Biosci. Rep. 2018, 38, BSR20180322. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Pantel, K. Liquid Biopsy: From Discovery to Clinical Application. Cancer Discov. 2021, 11, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Lone, S.N.; Nisar, S.; Masoodi, T.; Singh, M.; Rizwan, A.; Hashem, S.; El-Rifai, W.; Bedognetti, D.; Batra, S.K.; Haris, M.; et al. Liquid biopsy: A step closer to transform diagnosis, prognosis and future of cancer treatments. Mol. Cancer 2022, 21, 79. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Ye, L.; Wang, D.; Han, L.; Zhou, S.; Wang, H.; Dai, W.; Wang, Y.; Luo, W.; Wang, R.; et al. Early Detection of Molecular Residual Disease and Risk Stratification for Stage I to III Colorectal Cancer via Circulating Tumor DNA Methylation. JAMA Oncol. 2023, 9, 770–778. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Wang, Y.; Christie, M.; Simons, K.; Lee, M.; Wong, R.; Kosmider, S.; Ananda, S.; McKendrick, J.; et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019, 5, 1710–1717. [Google Scholar] [CrossRef]
- Cohen, S.A.; Liu, M.C.; Aleshin, A. Practical recommendations for using ctDNA in clinical decision making. Nature 2023, 619, 259–268. [Google Scholar] [CrossRef]
- Bianchi, D.W.; Chiu, R.W.K. Sequencing of Circulating Cell-free DNA during Pregnancy. N. Engl. J. Med. 2018, 379, 464–473. [Google Scholar] [CrossRef]
- Levitsky, J.; Kandpal, M.; Guo, K.; Kleiboeker, S.; Sinha, R.; Abecassis, M. Donor-derived cell-free DNA levels predict graft injury in liver transplant recipients. Am. J. Transplant. 2022, 22, 532–540. [Google Scholar] [CrossRef]
- Oellerich, M.; Sherwood, K.; Keown, P.; Schütz, E.; Beck, J.; Stegbauer, J.; Rump, L.C.; Walson, P.D. Liquid biopsies: Donor-derived cell-free DNA for the detection of kidney allograft injury. Nat. Rev. Nephrol. 2021, 17, 591–603. [Google Scholar] [CrossRef]
- Ren, J.; Jiang, L.; Liu, X.; Liao, Y.; Zhao, X.; Tang, F.; Yu, H.; Shao, Y.; Wang, J.; Wen, L.; et al. Heart-specific DNA methylation analysis in plasma for the investigation of myocardial damage. J. Transl. Med. 2022, 20, 36. [Google Scholar] [CrossRef]
- Gevensleben, H.; Garcia-Murillas, I.; Graeser, M.K.; Schiavon, G.; Osin, P.; Parton, M.; Smith, I.E.; Ashworth, A.; Turner, N.C. Noninvasive detection of HER2 amplification with plasma DNA digital PCR. Clin. Cancer Res. 2013, 19, 3276–3284. [Google Scholar] [CrossRef]
- Wu, J.; Tang, B.; Qiu, Y.; Tan, R.; Liu, J.; Xia, J.; Zhang, J.; Huang, J.; Qu, J.; Sun, J.; et al. Clinical validation of a multiplex droplet digital PCR for diagnosing suspected bloodstream infections in ICU practice: A promising diagnostic tool. Crit. Care 2022, 26, 243. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.J.Z.; Morovic, W.; DeMeules, M.; Stahl, B.; Sindelar, C.W. Absolute Enumeration of Probiotic Strains Lactobacillus acidophilus NCFM(®) and Bifidobacterium animalis subsp. lactis Bl-04 (®) via Chip-Based Digital PCR. Front. Microbiol. 2018, 9, 704. [Google Scholar] [CrossRef]
- Chen, L.; Ding, J.; Yuan, H.; Chen, C.; Li, Z. Deep-dLAMP: Deep Learning-Enabled Polydisperse Emulsion-Based Digital Loop-Mediated Isothermal Amplification. Adv. Sci. 2022, 9, e2105450. [Google Scholar] [CrossRef]
- Wang, Y.H.; Song, Z.; Hu, X.Y.; Wang, H.S. Circulating tumor DNA analysis for tumor diagnosis. Talanta 2021, 228, 122220. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Diamandis, E.P.; Crown, J. Circulating tumor DNA (ctDNA) as a pan-cancer screening test: Is it finally on the horizon? Clin. Chem. Lab. Med. 2021, 59, 1353–1361. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Hess, J.F.; Kohl, T.A.; Kotrová, M.; Rönsch, K.; Paprotka, T.; Mohr, V.; Hutzenlaub, T.; Brüggemann, M.; Zengerle, R.; Niemann, S.; et al. Library preparation for next generation sequencing: A review of automation strategies. Biotechnol. Adv. 2020, 41, 107537. [Google Scholar] [CrossRef]
- Rodriguez, R.; Krishnan, Y. The chemistry of next-generation sequencing. Nat. Biotechnol. 2023, 41, 1709–1715. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gerlach, D.; Misale, S.; Petronczki, M.; Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov. 2022, 12, 924–937. [Google Scholar] [CrossRef]
- Hanrahan, A.J.; Chen, Z.; Rosen, N.; Solit, D.B. BRAF—A tumour-agnostic drug target with lineage-specific dependencies. Nat. Rev. Clin. Oncol. 2024, 21, 224–247. [Google Scholar] [CrossRef] [PubMed]
- Pomari, E.; Piubelli, C.; Perandin, F.; Bisoffi, Z. Digital PCR: A new technology for diagnosis of parasitic infections. Clin. Microbiol. Infect. 2019, 25, 1510–1516. [Google Scholar] [CrossRef]
- Tan, L.L.; Loganathan, N.; Agarwalla, S.; Yang, C.; Yuan, W.; Zeng, J.; Wu, R.; Wang, W.; Duraiswamy, S. Current commercial dPCR platforms: Technology and market review. Crit. Rev. Biotechnol. 2023, 43, 433–464. [Google Scholar] [CrossRef]
- Lim, J.S.J.; Janku, F.; Yap, T.A. Circulating tumor DNA-From bench to bedside. Curr. Probl. Cancer 2017, 41, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Lancaster, H.L.; Heuvelmans, M.A.; Oudkerk, M. Low-dose computed tomography lung cancer screening: Clinical evidence and implementation research. J. Intern. Med. 2022, 292, 68–80. [Google Scholar] [CrossRef]
- Silvestri, G.A.; Young, R.P.; Tanner, N.T.; Mazzone, P. Screening Low-Risk Individuals for Lung Cancer: The Need May Be Present, but the Evidence of Benefit Is Not. J. Thorac. Oncol. 2024, 19, 1155–1163. [Google Scholar] [CrossRef]
- Milner, D.A., Jr.; Lennerz, J.K. Technology and Future of Multi-Cancer Early Detection. Life 2024, 14, 833. [Google Scholar] [CrossRef] [PubMed]
- Young, M.R.; Wagner, P.D.; Ghosh, S.; Rinaudo, J.A.; Baker, S.G.; Zaret, K.S.; Goggins, M.; Srivastava, S. Validation of Biomarkers for Early Detection of Pancreatic Cancer: Summary of The Alliance of Pancreatic Cancer Consortia for Biomarkers for Early Detection Workshop. Pancreas 2018, 47, 135–141. [Google Scholar] [CrossRef]
- Nossov, V.; Amneus, M.; Su, F.; Lang, J.; Janco, J.M.; Reddy, S.T.; Farias-Eisner, R. The early detection of ovarian cancer: From traditional methods to proteomics. Can we really do better than serum CA-125? Am. J. Obstet. Gynecol. 2008, 199, 215–223. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef]
- Killock, D. Diagnosis: CancerSEEK and destroy—A blood test for early cancer detection. Nat. Rev. Clin. Oncol. 2018, 15, 133. [Google Scholar] [CrossRef] [PubMed]
- Cristiano, S.; Leal, A.; Phallen, J.; Fiksel, J.; Adleff, V.; Bruhm, D.C.; Jensen, S.; Medina, J.E.; Hruban, C.; White, J.R.; et al. Genome-wide cell-free DNA fragmentation in patients with cancer. Nature 2019, 570, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Lennon, A.M.; Buchanan, A.H.; Kinde, I.; Warren, A.; Honushefsky, A.; Cohain, A.T.; Ledbetter, D.H.; Sanfilippo, F.; Sheridan, K.; Rosica, D.; et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020, 369, eabb9601. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, A.; Liu, M.C.; Klein, E.A.; Venn, O.; Hubbell, E.; Beausang, J.F.; Gross, S.; Melton, C.; Fields, A.P.; Liu, Q.; et al. Evaluation of cell-free DNA approaches for multi-cancer early detection. Cancer Cell 2022, 40, 1537–1549.e1512. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef]
- Schrag, D.; Beer, T.M.; McDonnell, C.H., 3rd; Nadauld, L.; Dilaveri, C.A.; Reid, R.; Marinac, C.R.; Chung, K.C.; Lopatin, M.; Fung, E.T.; et al. Blood-based tests for multicancer early detection (PATHFINDER): A prospective cohort study. Lancet 2023, 402, 1251–1260. [Google Scholar] [CrossRef]
- Nicholson, B.D.; Oke, J.; Virdee, P.S.; Harris, D.A.; O’Doherty, C.; Park, J.E.; Hamady, Z.; Sehgal, V.; Millar, A.; Medley, L.; et al. Multi-cancer early detection test in symptomatic patients referred for cancer investigation in England and Wales (SYMPLIFY): A large-scale, observational cohort study. Lancet Oncol. 2023, 24, 733–743. [Google Scholar] [CrossRef]
- Mazzone, P.J.; Bach, P.B.; Carey, J.; Schonewolf, C.A.; Bognar, K.; Ahluwalia, M.S.; Cruz-Correa, M.; Gierada, D.; Kotagiri, S.; Lloyd, K.; et al. Clinical Validation of a Cell-Free DNA Fragmentome Assay for Augmentation of Lung Cancer Early Detection. Cancer Discov. 2024, 14, 2224–2242. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef] [PubMed]
- Pieters, R.; de Groot-Kruseman, H.; Van der Velden, V.; Fiocco, M.; van den Berg, H.; de Bont, E.; Egeler, R.M.; Hoogerbrugge, P.; Kaspers, G.; Van der Schoot, E.; et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J. Clin. Oncol. 2016, 34, 2591–2601. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Bundred, J.R.; Michael, S.; Stuart, B.; Cutress, R.I.; Beckmann, K.; Holleczek, B.; Dahlstrom, J.E.; Gath, J.; Dodwell, D.; Bundred, N.J. Margin status and survival outcomes after breast cancer conservation surgery: Prospectively registered systematic review and meta-analysis. BMJ (Clin. Res. Ed.) 2022, 378, e070346. [Google Scholar] [CrossRef]
- Glasspool, R.M.; Evans, T.R. Clinical imaging of cancer metastasis. Eur. J. Cancer 2000, 36, 1661–1670. [Google Scholar] [CrossRef]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant tumour cells, their niches and the influence of immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Swanton, C. Early stage NSCLC—Challenges to implementing ctDNA-based screening and MRD detection. Nat. Rev. Clin. Oncol. 2018, 15, 577–586. [Google Scholar] [CrossRef]
- Nakamura, Y.; Kaneva, K.; Lo, C.; Neems, D.; Freaney, J.E.; Boulos, H.; Hyun, S.W.; Islam, F.; Yamada-Hanff, J.; Driessen, T.M.; et al. A Tumor-Naïve ctDNA Assay Detects Minimal Residual Disease in Resected Stage II or III Colorectal Cancer and Predicts Recurrence: Subset Analysis from the GALAXY Study in CIRCULATE-Japan. Clin. Cancer Res. 2025, 31, 328–338. [Google Scholar] [CrossRef]
- Zhong, R.; Gao, R.; Fu, W.; Li, C.; Huo, Z.; Gao, Y.; Lu, Y.; Li, F.; Ge, F.; Tu, H.; et al. Accuracy of minimal residual disease detection by circulating tumor DNA profiling in lung cancer: A meta-analysis. BMC Med. 2023, 21, 180. [Google Scholar] [CrossRef]
- Parikh, A.R.; Van Seventer, E.E.; Siravegna, G.; Hartwig, A.V.; Jaimovich, A.; He, Y.; Kanter, K.; Fish, M.G.; Fosbenner, K.D.; Miao, B.; et al. Minimal Residual Disease Detection using a Plasma-only Circulating Tumor DNA Assay in Patients with Colorectal Cancer. Clin. Cancer Res. 2021, 27, 5586–5594. [Google Scholar] [CrossRef]
- Henriksen, T.V.; Demuth, C.; Frydendahl, A.; Nors, J.; Nesic, M.; Rasmussen, M.H.; Reinert, T.; Larsen, O.H.; Jaensch, C.; Løve, U.S.; et al. Unraveling the potential clinical utility of circulating tumor DNA detection in colorectal cancer-evaluation in a nationwide Danish cohort. Ann. Oncol. 2024, 35, 229–239. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Schøler, L.V.; Reinert, T.; Ørntoft, M.W.; Kassentoft, C.G.; Árnadóttir, S.S.; Vang, S.; Nordentoft, I.; Knudsen, M.; Lamy, P.; Andreasen, D.; et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clin. Cancer Res. 2017, 23, 5437–5445. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef]
- Peng, M.; Huang, Q.; Yin, W.; Tan, S.; Chen, C.; Liu, W.; Tang, J.; Wang, X.; Zhang, B.; Zou, M.; et al. Circulating Tumor DNA as a Prognostic Biomarker in Localized Non-small Cell Lung Cancer. Front. Oncol. 2020, 10, 561598. [Google Scholar] [CrossRef]
- Provencio, M.; Serna-Blasco, R.; Nadal, E.; Insa, A.; García-Campelo, M.R.; Casal Rubio, J.; Dómine, M.; Majem, M.; Rodríguez-Abreu, D.; Martínez-Martí, A.; et al. Overall Survival and Biomarker Analysis of Neoadjuvant Nivolumab Plus Chemotherapy in Operable Stage IIIA Non-Small-Cell Lung Cancer (NADIM phase II trial). J. Clin. Oncol. 2022, 40, 2924–2933. [Google Scholar] [CrossRef]
- Xu, J.; Wan, R.; Cai, Y.; Cai, S.; Wu, L.; Li, B.; Duan, J.; Cheng, Y.; Li, X.; Wang, X.; et al. Circulating tumor DNA-based stratification strategy for chemotherapy plus PD-1 inhibitor in advanced non-small-cell lung cancer. Cancer Cell 2024, 42, 1598–1613.e1594. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Bao, H.; Fan, X.; Xia, F.; Wan, J.; Shen, L.; Guan, Y.; Bao, H.; Wu, X.; et al. Utility of ctDNA in predicting response to neoadjuvant chemoradiotherapy and prognosis assessment in locally advanced rectal cancer: A prospective cohort study. PLoS Med. 2021, 18, e1003741. [Google Scholar] [CrossRef]
- Tie, J.; Cohen, J.D.; Lahouel, K.; Lo, S.N.; Wang, Y.; Kosmider, S.; Wong, R.; Shapiro, J.; Lee, M.; Harris, S.; et al. Circulating Tumor DNA Analysis Guiding Adjuvant Therapy in Stage II Colon Cancer. N. Engl. J. Med. 2022, 386, 2261–2272. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Q.; Nie, R.C.; Huang, Y.S.; Chen, Y.B.; Wang, S.Y.; Sun, X.W.; Li, Y.F.; Liu, Z.K.; Chen, Y.X.; Yao, Y.C.; et al. Residual circulating tumor DNA after adjuvant chemotherapy effectively predicts recurrence of stage II-III gastric cancer. Cancer Commun. 2023, 43, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Magbanua, M.J.M.; Swigart, L.B.; Wu, H.T.; Hirst, G.L.; Yau, C.; Wolf, D.M.; Tin, A.; Salari, R.; Shchegrova, S.; Pawar, H.; et al. Circulating tumor DNA in neoadjuvant-treated breast cancer reflects response and survival. Ann. Oncol. 2021, 32, 229–239. [Google Scholar] [CrossRef]
- Magbanua, M.J.M.; Brown Swigart, L.; Ahmed, Z.; Sayaman, R.W.; Renner, D.; Kalashnikova, E.; Hirst, G.L.; Yau, C.; Wolf, D.M.; Li, W.; et al. Clinical significance and biology of circulating tumor DNA in high-risk early-stage HER2-negative breast cancer receiving neoadjuvant chemotherapy. Cancer Cell 2023, 41, 1091–1102.e1094. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Swift, C.; Jenkins, B.; Kilburn, L.; Coakley, M.; Beaney, M.; Fox, L.; Goddard, K.; Garcia-Murillas, I.; Proszek, P.; et al. Results of the c-TRAK TN trial: A clinical trial utilising ctDNA mutation tracking to detect molecular residual disease and trigger intervention in patients with moderate- and high-risk early-stage triple-negative breast cancer. Ann. Oncol. 2023, 34, 200–211. [Google Scholar] [CrossRef]
- Annala, M.; Taavitsainen, S.; Khalaf, D.J.; Vandekerkhove, G.; Beja, K.; Sipola, J.; Warner, E.W.; Herberts, C.; Wong, A.; Fu, S.; et al. Evolution of Castration-Resistant Prostate Cancer in ctDNA during Sequential Androgen Receptor Pathway Inhibition. Clin. Cancer Res. 2021, 27, 4610–4623. [Google Scholar] [CrossRef]
| cfDNA (Cell-Free DNA) | ctDNA (Circulating Tumor DNA) | References | |
|---|---|---|---|
| General Description | All DNA fragments | DNA fragments from cancer cells | [22,23,24,25] |
| Source | Originates from a wide range of cells, including normal, inflammatory, necrotic, and tumor cells | Mainly originates from tumor cells | [22,23,24,25,26,27,28] |
| Positive Population | Both healthy individuals and patients | Just cancer patients | [29,30,31] |
| Specificity | Non-specific; does not carry mutations and can derive from various physiological processes | Highly specific; usually carries tumor-related mutations and methylation | / |
| Length | 100 bp–21 kbp | Less than 100 bp | [32,33] |
| Plasma Concentration | |||
| Healthy Individuals | 1–10 ng/mL | Undetectable | [23,34,35,36,37,38,39,40,41] |
| Cancer Patients | 10–1000 ng/mL | 0.01–100 ng/mL | |
| Proportion of Total cfDNA | 100% (includes both ctDNA and DNA from normal cells) | <1–10% (can reach up to 40% in some advanced cancers) | [20] |
| Applications | Prenatal diagnosis, organ transplant monitoring, and detection of inflammatory diseases | Early screening of cancer, tumor profiling, monitoring of treatment resistance, recurrence detection | [19,20,42,43,44,45,46,47,48,49,50] |
| Clinical Significance | Reflects the overall cellular state in the body and can be used for various disease studies | Reflects tumor burden, mutation status, and treatment response | / |
| dPCR | NGS | References | |
|---|---|---|---|
| Basic Principle | Determines the absolute copy number of target DNA by analyzing endpoint fluorescence signals in micro-reaction units | Reads DNA sequence information using high-throughput sequencing technology | [51,54,57] |
| Sensitivity | Extremely high, capable of detecting mutation frequencies as low as 0.1% or even lower | Relatively high, capable of detecting low-frequency mutations, but limited by sequencing depth | [56] |
| Quantification Accuracy | Absolute quantification, independent of standard curves | Relative quantification, dependent on sequencing depth and data normalization | [55,57,59] |
| Sample Requirement | Low, small amounts of DNA are sufficient for detection | Requires high-quality and relatively large amounts of DNA | [57,58] |
| Detection Range | Suitable for detecting single or a small number of gene variations | Suitable for large-scale genomic analysis, covering SNPs, Indels, CNVs, and other genetic variations | [51,56,60,61] |
| Data Analysis | Simple and fast | Requires complex bioinformatics analysis | [63] |
| Experimental Cost | Low | High | [63] |
| Clinical Situations Applied | Low-frequency mutations, high-sensitivity quantification scenarios, and limited-sample contexts | Pan-cancer screening with multi-gene panels, tumor heterogeneity profiling through subclonal variant detection, immunotherapy biomarker evaluation, and exploration of unknown resistance mechanisms involving emerging mutations or fusion genes | [51,54,56,57,58,60,61] |
| Cancer Type | Purpose | Stage | Methodology | Sampling Time Points | Total Sample | Conclusion | References |
|---|---|---|---|---|---|---|---|
| Lung cancer | Determine the efficacy of neoadjuvant chemotherapy plus nivolumab | Stage IIIA | Oncomine tumor mutation load assay | Before and after neoadjuvant treatment (before surgery) | 46 | ctDNA levels were significantly associated with OS and outperformed radiologic assessments in the prediction of survival and proved the efficacy of neoadjuvant chemotherapy plus nivolumab in resectable NSCLC | [100] |
| Establish a ctDNA-based stratification strategy for immunochemotherapy in patients with NSCLC and evaluate its reproducibility and reliability | / | High-throughput panel-based deep-next-generation sequencing and low-pass whole genome sequencing | / | 460 | Proposed a potential therapeutic algorithm based on the ctDNA-based stratification strategy and shed light on the individualized management of immune–chemotherapies for patients with advanced NSCLC | [101] | |
| Breast Cancer | Predict pCR and risk of metastatic recurrence | Early Stage | WES | At pretreatment (T0); 3 weeks after initiation of paclitaxel (T1); between paclitaxel and anthracycline regimens (T2); or prior to surgery (T3) | 84 | Personalized monitoring of ctDNA during new adjuvant chemotherapy (NAC) may aid in the real-time assessment of treatment response and help fine-tune a pathologic complete response (pCR) as a surrogate endpoint of survival | [105] |
| Examine the predictive and prognostic value of ctDNA | Early Stage | Multiplex PCR | At pretreatment (T0); 3 weeks after the initiation of treatment (T1); at 12 weeks, between paclitaxel-based and anthracycline (AC) regimens (T2); and after NAC before surgery (T3) | 283 | Maximized and fine-tuned the use of ctDNA as a biomarker of response and survival in patients with high-risk early-stage breast cancer receiving NAC | [106] | |
| Assess the utility of prospective ctDNA surveillance in TNBC and the activity of pembrolizumab in patients with ctDNA detected [ctDNA positive (ctDNA+)] | Early Stage | dPCR | Three-monthly blood sampling to 12 months (18 months if the samples were missed due to coronavirus disease) after initial therapy | 208 | Emphasized the importance of commencing ctDNA testing early, with more sensitive and/or frequent ctDNA testing regimes, as well as the activity of pembrolizumab | [107] | |
| Colorectal Cancer | Explore the value of circulating tumor DNA (ctDNA) in combination with MRI in the prediction of pCR before surgery and investigate the utility of ctDNA in risk stratification and prognostic prediction for patients undergoing nCRT and total mesorectal excision (TME) | Advanced Stage | Deep-targeted panel sequencing | At baseline, during nCRT, and after surgery | 119 | Combining ctDNA and MRI can improve the predictive performance, and combining ctDNA with high-risk features can stratify patients with a high risk of recurrence | [102] |
| Assess whether a ctDNA-guided approach could reduce the use of adjuvant chemotherapy without compromising recurrence risk | Stage II | Safe-sequencing system | At week 4 and week 7, after surgery | 455 | A ctDNA-guided strategy could reduce adjuvant chemotherapy use without increasing the recurrence risk in stage II colon cancer | [103] | |
| Prostate Cancer | Determine the acquired genomic contributors to cross-resistance | Metastatic castration-resistant prostate cancer | Deep-targeted and whole-exome sequencing | At baseline and progression time points | 458 | The dominant AR genotype continues to evolve during sequential lines of AR inhibition and drives acquired resistance in patients with mCRPC | [108] |
| Gastric Cancer | Evaluate the predictive value of ctDNA in disease recurrence after adjuvant chemotherapy | Stage II/III | Targeted sequencing panel | Perioperatively and within 3 months after adjuvant chemotherapy | 100 | Residual ctDNA after ACT effectively predicts high recurrence risk in stage II/III GC, and the combination of tissue-based and circulating tumor features could achieve better risk prediction | [104] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, X.; Su, J.; Wang, Z. Revolution of Circulating Tumor DNA: From Bench Innovations to Bedside Implementations. Curr. Issues Mol. Biol. 2025, 47, 428. https://doi.org/10.3390/cimb47060428
Yan X, Su J, Wang Z. Revolution of Circulating Tumor DNA: From Bench Innovations to Bedside Implementations. Current Issues in Molecular Biology. 2025; 47(6):428. https://doi.org/10.3390/cimb47060428
Chicago/Turabian StyleYan, Xuehan, Juncheng Su, and Zheng Wang. 2025. "Revolution of Circulating Tumor DNA: From Bench Innovations to Bedside Implementations" Current Issues in Molecular Biology 47, no. 6: 428. https://doi.org/10.3390/cimb47060428
APA StyleYan, X., Su, J., & Wang, Z. (2025). Revolution of Circulating Tumor DNA: From Bench Innovations to Bedside Implementations. Current Issues in Molecular Biology, 47(6), 428. https://doi.org/10.3390/cimb47060428