
Progress in Transcriptomics and Metabolomics in Plant Responses to Abiotic Stresses
Abstract
1. Introduction
2. Transcriptomics
2.1. Comparative Analysis of Transcriptomic Research Technologies
2.2. Application of Transcriptomics in Plant Stress Resistance Research
3. Metabolomics
3.1. Characteristics of Metabolomic Research Techniques
3.2. Application of Metabolomics in Plant Stress Resistance
4. Studies on Transcriptomics and Metabolomics of Plant Responses to Different Abiotic Stresses
4.1. Studies on Transcriptomics and Metabolomics of Plant Responses to Temperature Stress
4.1.1. Heat Stress
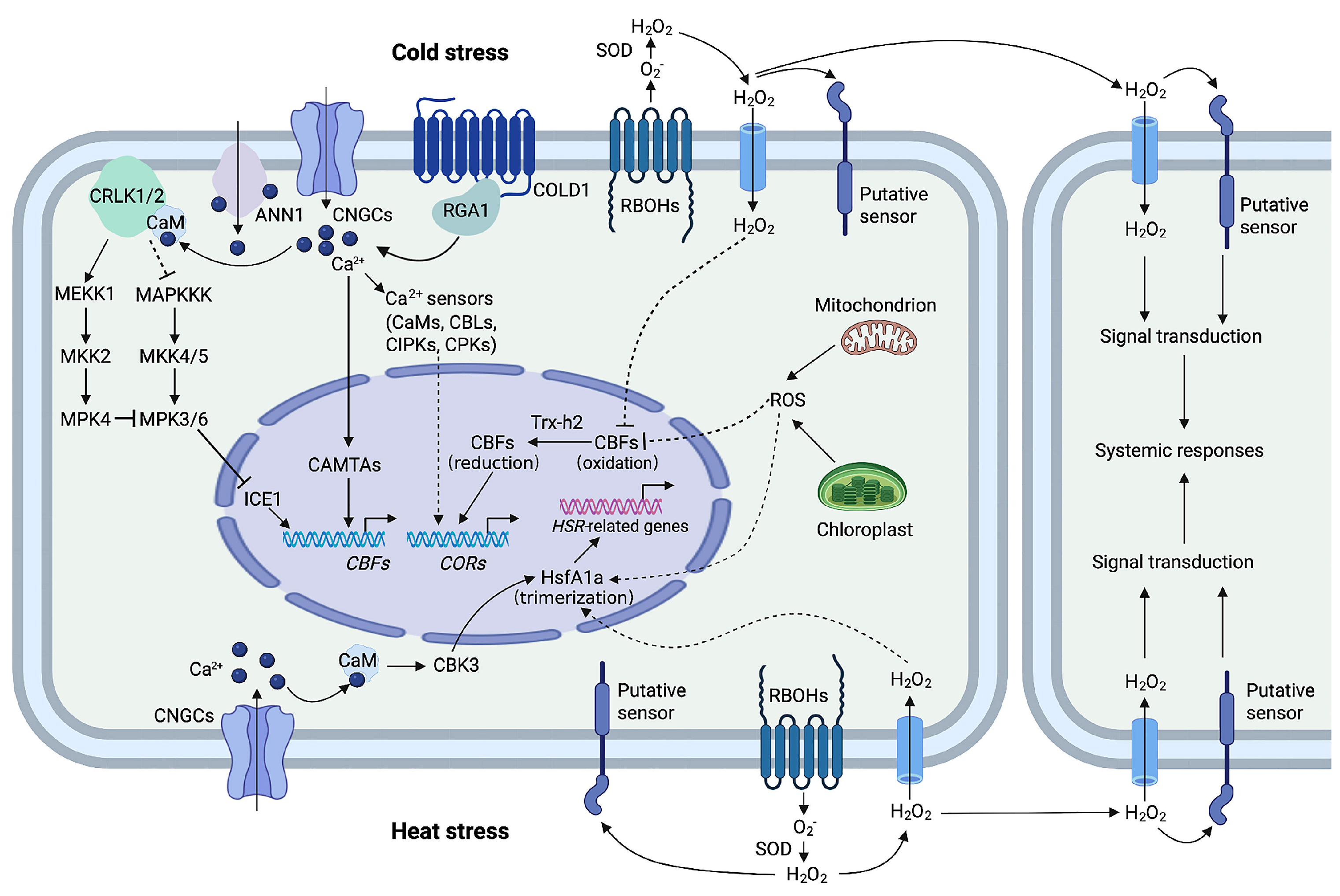
4.1.2. Cold Stress
4.2. Transcriptomic and Metabolomic Studies of Plant Responses to Water Stress
4.2.1. Drought Stress
4.2.2. Waterlogging Stress
4.3. Research on Transcriptomics and Metabolomics of Plant Responses to Heavy Metal Stress
4.4. Research on Transcriptomics and Metabolomics of Plant Response to Salt Stress
4.5. Emerging Insights into Other Abiotic Stressors
5. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manickam, S.; Rajagopalan, V.R.; Kambale, R.; Rajasekaran, R.; Kanagarajan, S.; Muthurajan, R. Current Initiatives and Future Prospects. Curr. Issues Mol. Biol. 2023, 5, 8894–8906. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ren, H.; Yao, X.; Wang, K.; Chang, J.; Shao, W. Metabolomics and Transcriptomics Analyses Reveal Regulatory Networks Associated with Fatty Acid Accumulation in Pecan Kernels. J. Agric. Food Chem. 2022, 70, 16010–16020. [Google Scholar] [CrossRef]
- Salam, U.; Ullah, S.; Tang, Z.H.; Elateeq, A.A.; Khan, Y.; Khan, J.; Khan, A.; Ali, S. Plant Metabolomics: An Overview of the Role of Primary and Secondary Metabolites against Different Environmental Stress Factors. Life 2023, 13, 706. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lv, J.; Liu, Z.; Liu, Y.; Song, J.; Ma, Y.; Ou, L.; Zhang, X.; Liang, C.; Wang, F.; et al. Integration of Transcriptomics and Metabolomics for Pepper (Capsicum annuum L.) in Response to Heat Stress. Int. J. Mol. Sci. 2019, 20, 5042. [Google Scholar] [CrossRef] [PubMed]
- Dubey, S.; Misra, P.; Dwivedi, S.; Chatterjee, S.; Bag, S.K.; Mantri, S.; Asif, M.H.; Rai, A.; Kumar, S.; Shri, M.; et al. Transcriptomic and metabolomic shifts in rice roots in response to Cr (VI) stress. BMC Genom. 2010, 11, 648. [Google Scholar] [CrossRef]
- Li, Y.; Shao, J.; Xie, Y.; Jia, L.; Fu, Y.; Xu, Z.; Zhang, N.; Feng, H.; Xun, W.; Liu, Y. Volatile compounds from beneficial rhizobacteria Bacillus spp. promote periodic lateral root development in Arabidopsis. Plant Cell Environ. 2021, 44, 1663–1678. [Google Scholar] [CrossRef]
- Gutierrez Reyes, C.D.; Alejo-Jacuinde, G.; Perez Sanchez, B.; Chavez Reyes, J.; Onigbinde, S.; Mogut, D.; Hernández-Jasso, I.; Calderón-Vallejo, D.; Quintanar, J.L.; Mechref, Y. Multi Omics Applications in Biological Systems. Curr. Issues Mol. Biol. 2024, 46, 5777–5793. [Google Scholar] [CrossRef]
- Zhang, E.; Zhu, X.; Wang, W.; Sun, Y.; Tian, X.; Chen, Z.; Mou, X.; Zhang, Y.; Wei, Y.; Fang, Z.; et al. Metabolomics reveals the response of hydroprimed maize to mitigate the impact of soil salinization. Front. Plant Sci. 2023, 14, 1109460. [Google Scholar] [CrossRef]
- Zhang, L.; Cui, D.; Ma, X.; Han, B.; Han, L. Comparative analysis of rice reveals insights into the mechanism of colored rice via widely targeted metabolomics. Food Chem. 2023, 15, 133926. [Google Scholar] [CrossRef]
- Yan, D.; Huang, L.; Mei, Z.; Bao, H.; Xie, Y.; Yang, C.; Gao, X. Untargeted metabolomics revealed the effect of soybean metabolites on poly(γ-glutamic acid) production in fermented natto and its metabolic pathway. J. Sci. Food Agric. 2024, 104, 1298–1307. [Google Scholar] [CrossRef]
- Sato, H.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Complex plant responses to drought and heat stress under climate change. Plant J. 2024, 117, 1873–1892. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.; Gong, Z.; Zhu, J.K. Abiotic stress responses in plants. Nat. Rev. Genet. 2022, 23, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Belaffif, M.B.; Brown, M.C.; Marcial, B.; Baysal, C.; Swaminathan, K. New strategies to advance plant transformation. Curr. Opin. Biotechnol. 2025, 91, 103241. [Google Scholar] [CrossRef]
- Sperdouli, I. Heavy Metal Toxicity Effects on Plants. Toxics 2022, 10, 715. [Google Scholar] [CrossRef]
- Ahmdikhah, A.; Safaeizadeh, M.; Tehranian, A.S. Responses of rice plant to multiple abiotic stresses revealed by transcriptome meta-analysis and identification of novel genetic factors. Sci. Rep. 2025, 15, 8248. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Z.; Wang, Z.; Fu, X.; Li, Z.; Li, J.; Xu, Z.; Cen, B. Large-scale bulk and single-cell RNA sequencing combined with machine learning reveals glioblastoma-associated neutrophil heterogeneity and establishes a VEGFA+ neutrophil prognostic model. Biol. Direct 2025, 20, 45. [Google Scholar] [CrossRef]
- Wang, J.; Ye, F.; Chai, H.; Jiang, Y.; Wang, T.; Ran, X.; Xia, Q.; Xu, Z.; Fu, Y.; Zhang, G.; et al. Advances and applications in single-cell and spatial genomics. Sci. China Life Sci. 2025, 68, 1226–1282. [Google Scholar]
- Zhu, M.; Hsu, C.W.; Peralta Ogorek, L.L.; Taylor, I.W.; La Cavera, S.; Oliveira, D.M.; Verma, L.; Mehra, P.; Mijar, M.; Sadanandom, A.; et al. Single-cell transcriptomics reveal how root tissues adapt to soil stress. Nature 2025. [Google Scholar] [CrossRef]
- Balasubramanian, V.K.; Veličković, D.; Rubio Wilhelmi, M.D.M.; Anderton, C.R.; Stewart, C.N., Jr.; DiFazio, S.; Blumwald, E.; Ahkami, A.H. Spatiotemporal metabolic responses to water deficit stress in distinct leaf cell-types of poplar. Front. Plant Sci. 2024, 15, 1346853. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, W.; Qiu, J.; Zhou, K.; Yu, G.; Zhang, Y.; Wang, X.; Jiao, Y.; Wang, X.; Hu, S.; et al. Metabolic marker-assisted genomic prediction improves hybrid breeding. Plant Commun. 2025, 6, 101199. [Google Scholar] [CrossRef] [PubMed]
- Velculescu, V.E.; Zhang, L.; Zhou, W.; Vogelstein, J.; Basrai, M.A.; Bassett, D.E.; Hieter, P.; Vogelstein, B.; Kinzler, K.W. Characterization of the yeast transcriptome. Cell 1997, 88, 243–251. [Google Scholar] [CrossRef]
- Park, H.E.; Jo, S.H.; Lee, R.H.; Macks, C.P.; Ku, T.; Park, J.; Lee, C.W.; Hur, J.K.; Sohn, C.H. Technical Aspects of Recent Developments and Their Applications in Neuroscience and Cancer Research. Adv. Sci. 2023, 10, e2206939. [Google Scholar] [CrossRef]
- Mathur, S.; Singh, D.; Ranjan, R. Recent advances in plant translational genomics for crop improvement. Adv. Protein Chem. Struct. Biol. 2024, 139, 335–382. [Google Scholar]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Kharchenko, P.V. The triumphs and limitations of computational methods for scRNA-seq. Nat. Methods 2021, 18, 723–732. [Google Scholar] [CrossRef]
- Bowtell, D.D. Options available–from start to finish–for obtaining expression data by microarray. Nat. Genet. 1999, 21, 25–32. [Google Scholar] [CrossRef]
- Nigam, D.; Kumar, S.; Mishra, D.C.; Rai, A.; Smita, S.; Saha, A. Synergistic regulatory networks mediated by microRNAs and transcription factors under drought, heat and salt stresses in Oryza Sativa spp. Gene 2015, 555, 127–139. [Google Scholar] [CrossRef]
- Parkinson, J.; Blaxter, M. Expressed sequence tags: An overview. Methods Mol. Biol. 2009, 533, 1–12. [Google Scholar]
- Makkar, G.S.; Bhatia, D.; Suri, K.S.; Kaur, S. Insect resistance in Rice (Oryza sativa L.): Overview on current breeding interventions. Int. J. Trop. Insect Sci. 2019, 39, 259–272. [Google Scholar] [CrossRef]
- Tuteja, R.; Tuteja, N. Serial analysis of gene expression (SAGE): Unraveling the bioinformatics tools. Bioessays 2004, 26, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Yue, K.; Lingling, L.; Xie, J.; Coulter, J.A.; Luo, Z. Synthesis and regulation of auxin and abscisic acid in maize. Plant Signal. Behav. 2021, 16, 1891756. [Google Scholar] [CrossRef]
- Reinartz, J.; Bruyns, E.; Lin, J.Z.; Burcham, T.; Brenner, S.; Bowen, B.; Kramer, M.; Woychik, R. Massively parallel signature sequencing (MPSS) as a tool for in-depth quantitative gene expression profiling in all organisms. Brief. Funct. Genom. Proteomic 2002, 1, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Bishop, R.; Shah, T.; Pelle, R.; Hoyle, D.; Pearson, T.; Haines, L.; Brass, A.; Hulme, H.; Graham, S.P.; Taracha, E.L.; et al. Analysis of the transcriptome of the protozoan Theileria parva using MPSS reveals that the majority of genes are transcriptionally active in the schizont stage. Nucleic Acids Res. 2005, 33, 5503–5511. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Ma, Z.; Jia, Y.; Min, Y.; Fang, X.; Yan, H.; Ma, Q.; Cai, R. Maize ZmWRKY71 gene positively regulates drought tolerance through reactive oxygen species homeostasis. Plant Physiol. Biochem. 2025, 219, 109399. [Google Scholar] [CrossRef]
- Bawa, G.; Liu, Z.; Yu, X.; Qin, A.; Sun, X. Single-Cell RNA Sequencing for Plant Research: Insights and Possible Benefits. Int. J. Mol. Sci. 2022, 23, 4497. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Ma, W.; Chen, R.; Li, S.T.; Wang, Q.; Wei, C.; Hong, Y.; Sun, H.X.; Cheng, Q.; Zhao, J.; et al. Multiome in the Same Cell Reveals the Impact of Osmotic Stress on Arabidopsis Root Tip Development at Single-Cell Level. Adv. Sci. 2024, 11, e2308384. [Google Scholar] [CrossRef]
- Aspholm, E.E.; Lidman, J.; Burmann, B.M. Structural basis of substrate recognition and allosteric activation of the proapoptotic mitochondrial HtrA2 protease. Nat. Commun. 2024, 15, 4592. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, Y.; Xie, Z.; Yu, B.; Sun, Y.; Huang, J. OsNAC016 regulates plant architecture and drought tolerance by interacting with the kinases GSK2 and SAPK8. Plant Physiol. 2022, 189, 1296–1313. [Google Scholar] [CrossRef]
- Feng, H.; Fan, W.; Liu, M.; Huang, J.; Li, B.; Sang, Q.; Song, B. Cross-species single-nucleus analysis reveals the potential role of whole-genome duplication in the evolution of maize flower development. BMC Genom. 2025, 26, 3. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, H.; Gao, S.; He, K.; Yu, T.; Gao, S.; Wang, J.; Li, H. Unveiling Salt Tolerance Mechanisms in Plants: Integrating the KANMB Machine Learning Model With Metabolomic and Transcriptomic Analysis. Adv. Sci. 2025, e2417560. [Google Scholar] [CrossRef]
- Yan, L.; Baoxiang, W.; Jingfang, L.; Zhiguang, S.; Ming, C.; Yungao, X.; Bo, X.; Bo, Y.; Jian, L.; Jinbo, L.; et al. A novel SAPK10-WRKY87-ABF1 biological pathway synergistically enhance abiotic stress tolerance in transgenic rice (Oryza sativa). Plant Physiol. Biochem. 2021, 168, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Zhuang, J.; Zhang, Z.; Chen, W.; Xu, H.; Zhao, M.; Ma, D. Multi-omics approach reveals the contribution of OsSEH1 to rice cold tolerance. Front. Plant Sci. 2023, 13, 1110724. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Z.; Wang, Y.; Wang, J.; Xiao, M.; Liu, H.; Quan, R.; Zhang, H.; Huang, R.; Zhu, L.; et al. Cellulose synthase-like protein OsCSLD4 plays an important role in the response of rice to salt stress by mediating abscisic acid biosynthesis to regulate osmotic stress tolerance. Plant Biotechnol. J. 2022, 20, 468–484. [Google Scholar] [CrossRef]
- Takasaki, H.; Maruyama, K.; Kidokoro, S.; Ito, Y.; Fujita, Y.; Shinozaki, K.; Yamaguchi-Shinozaki, K.; Nakashima, K. The abiotic stress-responsive NAC-type transcription factor OsNAC5 regulates stress-inducible genes and stress tolerance in rice. Mol. Genet. Genom. 2010, 284, 173–183. [Google Scholar] [CrossRef]
- Zhang, H.; Li, G.; Hu, D.; Zhang, Y.; Zhang, Y.; Shao, H.; Zhao, L.; Yang, R.; Guo, X. Functional characterization of maize heat shock transcription factor gene ZmHsf01 in thermotolerance. PeerJ 2020, 8, e8926. [Google Scholar] [CrossRef]
- Feng, S.; Yue, R.; Tao, S.; Yang, Y.; Zhang, L.; Xu, M.; Wang, H.; Shen, C. Genome-wide identification, expression analysis of auxin-responsive GH3 family genes in maize (Zea mays L.) under abiotic stresses. J. Integr. Plant Biol. 2015, 57, 783–795. [Google Scholar] [CrossRef]
- Jiang, H.; Shi, Y.; Liu, J.; Li, Z.; Fu, D.; Wu, S.; Li, M.; Yang, Z.; Shi, Y.; Lai, J.; et al. Natural polymorphism of ZmICE1 contributes to amino acid metabolism that impacts cold tolerance in maize. Nat. Plants 2022, 8, 1176–1190. [Google Scholar] [CrossRef] [PubMed]
- Djemal, R.; Khoudi, H. The ethylene-responsive transcription factor of durum wheat, TdSHN1, confers cadmium, copper, and zinc tolerance to yeast and transgenic tobacco plants. Protoplasma 2022, 259, 19–31. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, S.; Liu, W.; Zhai, Z.; Pan, Y.; Liu, C.; Chern, M.; Wang, H.; Huang, M.; Zhang, Z.; et al. A chlorophyll a oxygenase 1 gene ZmCAO1 contributes to grain yield and waterlogging tolerance in maize. J. Exp. Bot. 2021, 72, 3155–3167. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; King, R.D.; Altmann, T.; Fiehn, O. Application of metabolomics to plant genotype discrimination using statistics and machine learning. Bioinformatics 2002, 18, S241–S248. [Google Scholar] [CrossRef]
- Peters, K.; Worrich, A.; Weinhold, A.; Alka, O.; Balcke, G.; Birkemeyer, C.; Bruelheide, H.; Calf, O.W.; Dietz, S.; Dührkop, K.; et al. Current Challenges in Plant Eco-Metabolomics. Int. J. Mol. Sci. 2018, 19, 1385. [Google Scholar] [CrossRef]
- Ribbenstedt, A.; Ziarrusta, H.; Benskin, J.P. Development, characterization and comparisons of targeted and non-targeted metabolomics methods. PLoS ONE 2018, 13, e0207082. [Google Scholar] [CrossRef]
- Song, E.H.; Kim, H.J.; Jeong, J.; Chung, H.J.; Kim, H.Y.; Bang, E.; Hong, Y.S. A (1)H HR-MAS NMR-Based Metabolomic Study for Metabolic Characterization of Rice Grain from Various Oryza sativa L. Cultivars. J. Agric. Food Chem. 2016, 64, 3009–3016. [Google Scholar] [CrossRef]
- Choudhury, F.K.; Pandey, P.; Meitei, R.; Cardona, D.; Gujar, A.C.; Shulaev, V. GC-MS/MS Profiling of Plant Metabolites. Methods Mol. Biol. 2022, 2396, 101–115. [Google Scholar]
- Zhang, X.; Chen, T.; Li, Z.; Wang, X.; Bao, H.; Zhao, C.; Zhao, X.; Lu, X.; Xu, G. Fine-Scale Characterization of Plant Diterpene Glycosides Using Energy-Resolved Untargeted LC-MS/MS Metabolomics Analysis. J. Am. Soc. Mass. Spectrom. 2024, 35, 603–612. [Google Scholar] [CrossRef]
- Chi, Z.; Yang, L. Advances in chiral separation and analysis by capillary electrophoresis-mass spectrometry. Chin. J. Chromatogr. 2022, 40, 509–519. [Google Scholar] [CrossRef]
- Ratcliffe, R.G.; Shachar-Hill, Y. Measuring multiple fluxes through plant metabolic networks. Plant J. 2006, 45, 490–511. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Bamba, T.; Harada, K.; Fukusaki, E.; Kobayashi, A. Time-course metabolic profiling in Arabidopsis thaliana cell cultures after salt stress treatment. J. Exp. Bot. 2007, 58, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Dudareva, N.; Klempien, A.; Muhlemann, J.K.; Kaplan, I. Biosynthesis, function and metabolic engineering of plant volatile organic compounds. New Phytol. 2013, 198, 16–32. [Google Scholar] [CrossRef]
- Zhou, S.; Feng, D.; Zhou, Y.; Duan, H.; He, Y.; Jiang, Y.; Yan, W. Characteristic Volatile Organic Compound Analysis of Different Cistanches Based on HS-GC-IMS. Molecules 2022, 27, 6789. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H.; et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef]
- Harrieder, E.M.; Kretschmer, F.; Böcker, S.; Witting, M. Current state-of-the-art of separation methods used in LC-MS based metabolomics and lipidomics. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2022, 1188, 123069. [Google Scholar] [CrossRef]
- Ramautar, R.; Somsen, G.W.; De Jong, G.J. CE-MS for metabolomics: Developments and applications in the period 2016–2018. Electrophoresis 2019, 40, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Bagwe, K.; Gould, N.; Johnson, K.R.; Ivanov, A.R. Single-cell omic molecular profiling using capillary electrophoresis-mass spectrometry. Trends Anal. Chem. 2023, 165, 117117. [Google Scholar] [CrossRef]
- Joshi, J.; Hasnain, G.; Logue, T.; Lynch, M.; Wu, S.; Guan, J.C.; Alseekh, S.; Fernie, A.R.; Hanson, A.D.; McCarty, D.R. A Core Metabolome Response of Maize Leaves Subjected to Long-Duration Abiotic Stresses. Metabolites 2021, 11, 797. [Google Scholar] [CrossRef]
- Bheemanahalli, R.; Impa, S.M.; Krassovskaya, I.; Vennapusa, A.R.; Gill, K.S.; Obata, T.; Jagadish, S.V.K. Enhanced N-metabolites, ABA and IAA-conjugate in anthers instigate heat sensitivity in spring wheat. Physiol. Plant 2020, 169, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Zhang, J.; Cao, J.; Li, X.; Li, S.; Liu, C.; Wang, L. Metabolic Insight into Cold Stress Response in Two Contrasting Maize Lines. Life 2022, 12, 282. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, M.; Blein-Nicolas, M.; Prigent, S.; Bernillon, S.; Deborde, C.; Balliau, T.; Maucourt, M.; Jacob, D.; Ballias, P.; Bénard, C.; et al. Maize metabolome and proteome responses to controlled cold stress partly mimic early-sowing effects in the field and differ from those of Arabidopsis. Plant Cell Environ. 2021, 44, 1504–1521. [Google Scholar] [CrossRef] [PubMed]
- Duran Garzon, C.; Lequart, M.; Rautengarten, C.; Bassard, S.; Sellier-Richard, H.; Baldet, P.; Heazlewood, J.L.; Gibon, Y.; Domon, J.M.; Giauffret, C.; et al. Regulation of carbon metabolism in two maize sister lines contrasted for chilling tolerance. J. Exp. Bot. 2020, 71, 356–369. [Google Scholar] [CrossRef]
- Jian, H.; Xie, L.; Wang, Y.; Cao, Y.; Wan, M.; Lv, D.; Li, J.; Lu, K.; Xu, X.; Liu, L. Characterization of cold stress responses in different rapeseed ecotypes based on metabolomics and transcriptomics analyses. PeerJ 2020, 8, e8704. [Google Scholar] [CrossRef]
- Itam, M.; Mega, R.; Tadano, S.; Abdelrahman, M.; Matsunaga, S.; Yamasaki, Y.; Akashi, K.; Tsujimoto, H. Metabolic and physiological responses to progressive drought stress in bread wheat. Sci. Rep. 2020, 10, 17189. [Google Scholar] [CrossRef]
- Ackah, M.; Shi, Y.; Wu, M.; Wang, L.; Guo, P.; Guo, L.; Jin, X.; Li, S.; Zhang, Q.; Qiu, C.; et al. Metabolomics Response to Drought Stress in Morus alba L. Variety Yu-711. Plants 2021, 10, 1636. [Google Scholar] [CrossRef]
- Hong, Y.; Ni, S.J.; Zhang, G.P. Transcriptome and metabolome analysis reveals regulatory networks and key genes controlling barley malting quality in responses to drought stress. Plant Physiol. Biochem. 2020, 152, 1–11. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Yao, L.; Li, B.; Ma, X.; Si, E.; Yang, K.; Li, C.; Shang, X.; Meng, Y.; et al. Combined proteomic and metabolomic analysis of the molecular mechanism underlying the response to salt stress during seed germination in barley. Int. J. Mol. Sci. 2022, 23, 10515. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Chen, X.; Li, H.; Jiang, Q. Metabolomic analysis of wheat response to salt stress. J. Agric. Sci. Technol. 2023, 25, 43–56. (In Chinese) [Google Scholar]
- Gao, L.; Jia, B.; Zhang, W.; Li, A.; Li, T.; Jin, D.; Liu, Y.; Liu, H.; Wu, Y. Physiological characteristics and metabonomics analysis of blueberry leaves under salt stress. Plant Physiol. J. 2022, 58, 155–164. (In Chinese) [Google Scholar]
- Tan, B.; Tan, X.; Liu, C.; Li, Y. Effects of lead stress on rice (Oryza sativa L.) growth and metabolism in the rhizosphere microenvironment: The role of eicosanoid compounds. Plant Growth Regul. 2022, 96, 483–495. [Google Scholar] [CrossRef]
- Lai, J.L.; Deng, Z.X.; Ji, X.H.; Luo, X.G. Absorption and interaction mechanisms of uranium & cadmium in purple sweet potato (Ipomoea batatas L.). J. Hazard. Mater. 2020, 400, 123264. [Google Scholar]
- Zhang, J.; Yang, Q.; Liu, Q.; Liu, S.; Zhu, Y.; Yao, J.; Wang, H.; Guan, W. The responses of harmful dinoflagellate Karenia mikimotoi to simulated ocean acidification at the transcriptional level. Harmful Algae 2022, 111, 102167. [Google Scholar] [CrossRef]
- Prieto, J.; García-Cañaveras, J.C.; León, M.; Sendra, R.; Ponsoda, X.; Izpisúa Belmonte, J.C.; Lahoz, A.; Torres, J. c-MYC Triggers Lipid Remodelling During Early Somatic Cell Reprogramming to Pluripotency. Stem Cell Rev. Rep. 2021, 17, 2245–2261. [Google Scholar] [CrossRef] [PubMed]
- Khosravi-Nejad, F.; Khavari-Nejad, R.A.; Moradi, F.; Najafi, F. Cytokinin and abscisic acid alleviate drought stress through changing organic acids profile, ion immolation, and fatty acid profile to improve yield of wheat (Triticum aestivum L.) cultivars. Physiol. Mol. Biol. Plants 2022, 28, 1119–1129. [Google Scholar] [CrossRef]
- Sairam, R.; Srivastava, G. Changes in antioxidant activity in sub-cellular fractions of tolerant and susceptible wheat genotypes in response to long term salt stress. Plant Sci. 2002, 162, 897–904. [Google Scholar] [CrossRef]
- Ohashi, M.; Ishiyama, K.; Kusano, M.; Fukushima, A.; Kojima, S.; Hayakawa, T.; Yamaya, T. Reduction in sucrose contents by downregulation of fructose-1,6-bisphosphatase 2 causes tiller outgrowth cessation in rice mutants lacking glutamine synthetase1;2. Rice 2018, 11, 65. [Google Scholar] [CrossRef]
- Kopecká, R.; Kameniarová, M.; Černý, M.; Brzobohatý, B.; Novák, J. Abiotic Stress in Crop Production. Int. J. Mol. Sci. 2023, 24, 6603. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, J.; Li, R.; Ge, Y.; Li, Y.; Li, R. Plants’ Response to Abiotic Stress: Mechanisms and Strategies. Int. J. Mol. Sci. 2023, 24, 10915. [Google Scholar] [CrossRef]
- Ding, Y.; Yang, S. Surviving and thriving: How plants perceive and respond to temperature stress. Dev. Cell 2022, 57, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Kan, Y.; Mu, X.R.; Gao, J.; Lin, H.X.; Lin, Y. The molecular basis of heat stress responses in plants. Mol. Plant 2023, 16, 1612–1634. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xiao, Y.; Chang, H.; Sun, S.; Wang, J.; Liang, Q.; Wu, Q.; Wu, J.; Qin, Y.; Chen, J.; et al. The Regulatory Network of Sweet Corn (Zea mays L.) Seedlings under Heat Stress Revealed by Transcriptome and Metabolome Analysis. Int. J. Mol. Sci. 2023, 24, 10845. [Google Scholar] [CrossRef]
- Hu, T.; Sun, X.Y.; Zhao, Z.J.; Amombo, E.; Fu, J.M. High temperature damage to fatty acids and carbohydrate metabolism in tall fescue by coupling deep transcriptome and metabolome analysis. Ecotoxicol. Environ. Saf. 2020, 203, 110943. [Google Scholar] [CrossRef]
- Guo, J.; Gu, X.; Lu, W.; Lu, D. Multiomics analysis of kernel development in response to short-term heat stress at the grain formation stage in waxy maize. J. Exp. Bot. 2021, 72, 6291–6304. [Google Scholar] [CrossRef]
- Xiang, N.; Hu, J.G.; Yan, S.; Guo, X. Plant Hormones and Volatiles Response to Temperature Stress in Sweet Corn (Zea mays L.) Seedlings. J. Agric. Food Chem. 2021, 69, 6779–6790. [Google Scholar] [CrossRef]
- Kidokoro, S.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Transcriptional regulatory network of plant cold-stress responses. Trends Plant Sci. 2022, 27, 922–935. [Google Scholar] [CrossRef]
- Guo, Q.; Li, X.; Niu, L.; Jameson, P.E.; Zhou, W. Transcription-associated metabolomic adjustments in maize occur during combined drought and cold stress. Plant Physiol. 2021, 186, 677–695. [Google Scholar] [CrossRef]
- Xu, J.; Chen, Z.; Wang, F.; Jia, W.; Xu, Z. Combined transcriptomic and metabolomic analyses uncover rearranged gene expression and metabolite metabolism in tobacco during cold acclimation. Sci. Rep. 2020, 10, 5242. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lu, X.; Duan, P.; Liang, Y.; Cui, J. Integrating transcriptome and metabolome analyses of the response to cold stress in pumpkin (Cucurbita maxima). PLoS ONE 2021, 16, e0249108. [Google Scholar] [CrossRef]
- Barickman, T.C.; Simpson, C.R.; Sams, C.E. Waterlogging Causes Early Modification in the Physiological Performance, Carotenoids, Chlorophylls, Proline, and Soluble Sugars of Cucumber Plants. Plants 2019, 8, 160. [Google Scholar] [CrossRef]
- Bárzana, G.; Carvajal, M. Genetic regulation of water and nutrient transport in water stress tolerance in roots. J. Biotechnol. 2020, 324, 134–142. [Google Scholar] [CrossRef]
- Ozturk, M.; Turkyilmaz Unal, B.; García-Caparrós, P.; Khursheed, A.; Gul, A.; Hasanuzzaman, M. Osmoregulation and its actions during the drought stress in plants. Physiol. Plant 2021, 172, 1321–1335. [Google Scholar] [CrossRef]
- Mathan, J.; Singh, A.; Ranjan, A. Sucrose transport in response to drought and salt stress involves ABA-mediated induction of OsSWEET13 and OsSWEET15 in rice. Physiol. Plant 2021, 171, 620–637. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tiwari, M.; Tang, Y.; Wang, L.; Yang, S.; Long, H.; Guo, J.; Wang, Y.; Wang, H.; Yang, Q.; et al. Metabolomic and transcriptomic analyses reveal that sucrose synthase regulates maize pollen viability under heat and drought stress. Ecotoxicol. Environ. Saf. 2022, 246, 114191. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Xia, R.; Chen, C.; Shang, X.; Ge, F.; Wei, H.; Chen, H.; Wu, Y.; Xie, Q. ZmbHLH124 identified in maize recombinant inbred lines contributes to drought tolerance in crops. Plant Biotechnol. J. 2021, 19, 2069–2081. [Google Scholar] [CrossRef]
- Li, Y.; Su, Z.; Lin, Y.; Xu, Z.; Bao, H.; Wang, F.; Liu, J.; Hu, S.; Wang, Z.; Yu, X.; et al. Utilizing transcriptomics and metabolomics to unravel key genes and metabolites of maize seedlings in response to drought stress. BMC Plant Biol. 2024, 24, 34. [Google Scholar] [CrossRef]
- Waadt, R.; Seller, C.A.; Hsu, P.K.; Takahashi, Y.; Munemasa, S.; Schroeder, J.I. Plant hormone regulation of abiotic stress responses. Nat. Rev. Mol. Cell Biol. 2022, 23, 516. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Fan, X.H.; Wang, Q.; Yin, Z.G.; Sheng, X.W.; Chen, J.; Zhou, Y.B.; Chenm, M.; Ma, Y.Z.; Ma, J.; et al. Genomic Analysis of Soybean PP2A-B Family and Its Effects on Drought and Salt Tolerance. Front. Plant Sci. 2022, 12, 784038. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Hou, K.; Zhang, H.; Wang, X.; Wu, W. Integrating transcriptomics and metabolomics to studies key metabolism, pathways and candidate genes associated with drought-tolerance in Carthamus tinctorius L. Under drought stress. Ind. Crops Prod. 2020, 151, 112465. [Google Scholar] [CrossRef]
- Huang, C.; Qin, A.; Gao, Y.; Ma, S.; Liu, Z.; Zhao, B.; Ning, D.; Zhang, K.; Gong, W.; Sun, M.; et al. Effects of water deficit at different stages on growth and ear quality of waxy maize. Front. Plant Sci. 2023, 14, 1069551. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Jiang, F.; Yin, J.; Wang, Y.; Li, Y.; Yu, X.; Song, X.; Ottosen, C.O.; Rosenqvist, E.; Mittler, R.; et al. ROS-mediated waterlogging memory, induced by priming, mitigates photosynthesis inhibition in tomato under waterlogging stress. Front. Plant Sci. 2023, 14, 1238108. [Google Scholar] [CrossRef]
- Mansoor, S.; Ali Wani, O.; Lone, J.K.; Manhas, S.; Kour, N.; Alam, P.; Ahmad, A.; Ahmad, P. Reactive Oxygen Species in Plants: From Source to Sink. Antioxidants 2022, 11, 225. [Google Scholar] [CrossRef]
- Luan, H.; Li, H.; Li, Y.; Chen, C.; Li, S.; Wang, Y.; Yang, J.; Xu, M.; Shen, H.; Qiao, H.; et al. Transcriptome analysis of barley (Hordeum vulgare L.) under waterlogging stress, and overexpression of the HvADH4 gene confers waterlogging tolerance in transgenic Arabidopsis. BMC Plant Biol. 2023, 23, 62. [Google Scholar] [CrossRef]
- Feng, F.; Wang, Q.; Jiang, K.; Lei, D.; Huang, S.; Wu, H.; Yue, G.; Wang, B. Transcriptome analysis reveals ZmERF055 contributes to waterlogging tolerance in sweetcorn. Plant Physiol. Biochem. 2023, 204, 108087. [Google Scholar] [CrossRef]
- Hong, B.; Zhou, B.; Peng, Z.; Yao, M.; Wu, J.; Wu, X.; Guan, C.; Guan, M. Tissue-Specific Transcriptome and Metabolome Analysis Reveals the Response Mechanism of Brassica napus to Waterlogging Stress. Int. J. Mol. Sci. 2023, 24, 6015. [Google Scholar] [CrossRef]
- Zheng, Q.; Li, G.; Wang, H.; Zhou, Z. The relationship between ethylene-induced autophagy and reactive oxygen species in Arabidopsis root cells during the early stages of waterlogging stress. PeerJ 2023, 11, e15404. [Google Scholar] [CrossRef]
- Geng, S.; Lin, Z.; Xie, S.; Xiao, J.; Wang, H.; Zhao, X.; Zhou, Y.; Duan, L. Ethylene enhanced waterlogging tolerance by changing root architecture and inducing aerenchyma formation in maize seedlings. J. Plant Physiol. 2023, 287, 154042. [Google Scholar] [CrossRef]
- Sreeratree, J.; Butsayawarapat, P.; Chaisan, T.; Somta, P.; Juntawong, P. RNA-Seq Reveals Waterlogging-Triggered Root Plasticity in Mungbean Associated with Ethylene and Jasmonic Acid Signal Integrators for Root Regeneration. Plants 2022, 11, 930. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, H.; Zhang, X.; Wang, Y.; Zhu, H.; Zhao, Y.; Ma, Q. Identification and characterization of waterlogging-responsive genes in the parental line of maize hybrid An’nong 876. Genet. Mol. Biol. 2024, 46, e20230026. [Google Scholar] [CrossRef] [PubMed]
- Owusu, A.G.; Lv, Y.P.; Liu, M.; Wu, Y.; Li, C.L.; Guo, N.; Li, D.H.; Gao, J.S. Transcriptomic and metabolomic analyses reveal the potential mechanism of waterlogging resistance in cotton (Gossypium hirsutum L.). Front. Plant Sci. 2023, 14, 1088537. [Google Scholar] [CrossRef] [PubMed]
- Majhi, S.; Sikdar Née Bhakta, M. How heavy metal stress affects the growth and development of pulse crops: Insights into germination and physiological processes. 3 Biotech 2023, 13, 155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zheng, S.; Liu, R.; Lu, L.; Zhang, C.; Zhang, L.; Yant, L.; Wu, Y. The genome-wide impact of cadmium on microRNA and mRNA expression in contrasting Cd responsive wheat genotypes. BMC Genom. 2019, 20, 615. [Google Scholar] [CrossRef]
- Mwamba, T.M.; Islam, F.; Ali, B.; Lwalaba, J.L.W.; Gill, R.A.; Zhang, F.; Farooq, M.A.; Ali, S.; Ulhassan, Z.; Huang, Q.; et al. Comparative metabolomic responses of low- and high-cadmium accumulating genotypes reveal the cadmium adaptive mechanism in Brassica napus. Chemosphere 2020, 250, 126308. [Google Scholar] [CrossRef]
- Zhao, W.; Chen, Z.; Yang, X.; Sheng, L.; Mao, H.; Zhu, S. Integrated transcriptomics and metabolomics reveal key metabolic pathway responses in Pistia stratiotes under Cd stress. J. Hazard. Mater. 2023, 452, 131214. [Google Scholar] [CrossRef]
- Sharma, A.; Kapoor, D.; Gautam, S.; Landi, M.; Kandhol, N.; Araniti, F.; Ramakrishnan, M.; Satish, L.; Singh, V.P.; Sharma, P.; et al. Heavy metal induced regulation of plant biology: Recent insights. Physiol. Plant 2022, 174, e13688. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Li, J.; Yang, Y.; Jiang, C.; Guo, Y. Designing salt stress-resilient crops: Current progress and future challenges. J. Integr. Plant Biol. 2024, 66, 303–329. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Q.; Liu, M.; Zhou, H.; Ma, C.; Wang, P. Regulation of Plant Responses to Salt Stress. Int. J. Mol. Sci. 2021, 22, 4609. [Google Scholar] [CrossRef]
- Fu, H.; Yang, Y. How Plants Tolerate Salt Stress. Curr. Issues Mol. Biol. 2023, 45, 5914–5934. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, X.; Lu, X.; Zhao, B.; Yang, Y.; Liu, J. Integrative analysis of transcriptome and metabolome reveal mechanism of tolerance to salt stress in oat (Avena sativa L.). Plant Physiol. Biochem. 2021, 160, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Chen, G.; Ma, L.; Zhang, C.; Yan, H.; Bao, J.; Nai, G.; Wang, W.; Chen, B.; Ma, S.; et al. Integrated transcriptome and metabolome analysis reveals antioxidant machinery in grapevine exposed to salt and alkali stress. Physiol. Plant 2023, 175, e13950. [Google Scholar] [CrossRef]
- Han, M.; Cui, R.; Wang, D.; Huang, H.; Rui, C.; Malik, W.A.; Wang, J.; Zhang, H.; Xu, N.; Liu, X.; et al. Combined transcriptomic and metabolomic analyses elucidate key salt-responsive biomarkers to regulate salt tolerance in cotton. BMC Plant Biol. 2023, 23, 245. [Google Scholar] [CrossRef]
- Jin, J.; Wang, J.; Li, K.; Wang, S.; Qin, J.; Zhang, G.; Na, X.; Wang, X.; Bi, Y. Integrated Physiological, Transcriptomic, and Metabolomic Analyses Revealed Molecular Mechanism for Salt Resistance in Soybean Roots. Int. J. Mol. Sci. 2021, 22, 12848. [Google Scholar] [CrossRef]
- Shu, J.; Ma, X.; Ma, H.; Huang, Q.; Zhang, Y.; Guan, M.; Guan, C. Transcriptomic, proteomic, metabolomic, and functional genomic approaches of Brassica napus L. during salt stress. PLoS ONE 2022, 17, e0262587. [Google Scholar] [CrossRef]
- Ma, L.; Han, R.; Yang, Y.; Liu, X.; Li, H.; Zhao, X.; Li, J.; Fu, H.; Huo, Y.; Sun, L.; et al. Phytochromes enhance SOS2-mediated PIF1 and PIF3 phosphorylation and degradation to promote Arabidopsis salt tolerance. Plant Cell 2023, 35, 2997–3020. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, J.; Zhang, J.; Zhang, W.; Zheng, L.; Borjigin, T.; Wang, Y. Nitric oxide alleviates salt-induced stress damage by regulating the ascorbate-glutathione cycle and Na+/K+ homeostasis in Nitraria tangutorum Bobr. Plant Physiol. Biochem. 2022, 173, 46–58. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Yang, F.; Zhou, W.; Mao, S.; Lin, J.; Yan, X. Untargeted LC-MS-based metabolomics revealed specific metabolic changes in cotyledons and roots of Ricinus communis during early seedling establishment under salt stress. Plant Physiol. Biochem. 2021, 163, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ke, X.; Yang, X.; Liu, Y.; Hou, X. Plants response to light stress. J. Genet. Genom. 2022, 49, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Zhang, H.; Yu, C.; Luo, N.; Yan, J.; Zheng, S.; Hu, Q.; Zhang, D.; Kou, L.; Meng, X.; et al. Low phosphorus promotes NSP1-NSP2 heterodimerization to enhance strigolactone biosynthesis and regulate shoot and root architecture in rice. Mol. Plant 2023, 16, 1811–1831. [Google Scholar] [CrossRef] [PubMed]
- Kamble, U.; Mishra, C.N.; Govindan, V.; Sharma, A.K.; Pawar, S.; Kumar, S.; Krishnappa, G.; Gupta, O.P.; Singh, G.P.; Singh, G. Ensuring Nutritional Security in India through Wheat Biofortification: A Review. Genes 2022, 13, 2298. [Google Scholar] [CrossRef]
- Rosário-Filho, N.A.; Urrutia-Pereira, M.; D’Amato, G.; Cecchi, L.; Ansotegui, I.J.; Galán, C.; Pomés, A.; Murrieta-Aguttes, M.; Caraballo, L.; Rouadi, P.; et al. Air pollution and indoor settings. World Allergy Organ. J. 2021, 14, 100499. [Google Scholar] [CrossRef]
- Sytar, O.; Ghosh, S.; Malinska, H.; Zivcak, M.; Brestic, M. Physiological and molecular mechanisms of metal accumulation in hyperaccumulator plants. Physiol. Plant 2021, 173, 148–166. [Google Scholar] [CrossRef]
- Chen, X.; Wang, J.; Wang, R.; Zhang, D.; Chu, S.; Yang, X.; Hayat, K.; Fan, Z.; Cao, X.; Ok, Y.S.; et al. Insights into growth-promoting effect of nanomaterials: Using transcriptomics and metabolomics to reveal the molecular mechanisms of MWCNTs in enhancing hyperaccumulator under heavy metal(loid)s stress. J. Hazard. Mater. 2022, 439, 129640. [Google Scholar] [CrossRef]
- Pang, Z.; Lu, Y.; Zhou, G.; Hui, F.; Xu, L.; Viau, C.; Spigelman, A.F.; MacDonald, P.E.; Wishart, D.S.; Li, S.; et al. MetaboAnalyst 6.0: Towards a unified platform for metabolomics data processing, analysis and interpretation. Nucleic Acids Res. 2024, 52, W398–W406. [Google Scholar] [CrossRef]
- Cambiaghi, A.; Ferrario, M.; Masseroli, M. Analysis of metabolomic data: Tools, current strategies and future challenges for omics data integration. Brief. Bioinform. 2017, 18, 498–510. [Google Scholar] [CrossRef] [PubMed]
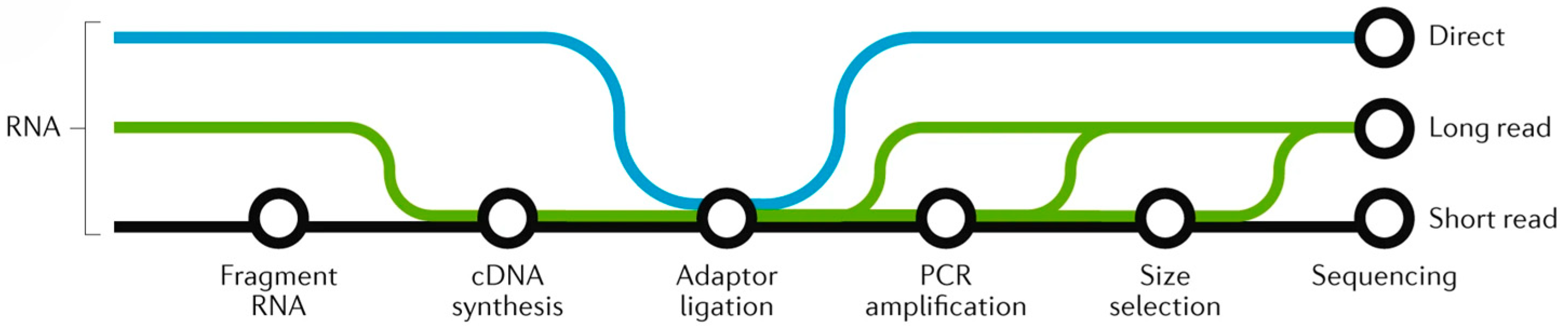

{kind=link}
{kind=link}
| Technology | Theory | Advantage | Limitation | Examples |
|---|---|---|---|---|
| Microassay [26] | Hybrid | 1. Fast speed 2. Low cost 3. Simple sample preparation 4. Flexible analysis range | 1. The sensitivity for detecting low-expression genes is insufficient 2. The sensitivity of hybridization technology is limited 3. The prerequisite work requires a high level of foundation 4. It is difficult to detect abnormal transcription products | Microarray technology was used for the expression analysis of genes in Arabidopsis thaliana responding to salt stress, and salt-tolerant-related genes were screened by fluorescence signal differences [27]. |
| EST [28] | Sanger | 1. Detection range is wide 2. Accuracy is high 3. Improves the efficiency of gene isolation | 1. The sequencing read length is short 2. The error rate is high 3. The sequencing throughput is low | Multiple gene fragments encoding protease inhibitors were identified through EST technology, providing a molecular basis for insect-resistant breeding [29]. |
| SAGE [30] | Sanger | 1. High-throughput detection 2. Can quantitatively evaluate gene expression levels 3. Gain a comprehensive understanding of gene expression regulation mechanisms | 1. High cost 2. Complex data processing 3. Relying on known gene databases, there are certain limitations in identifying unknown genes | The significant upregulation of ABA synthesis-related genes was discovered using SAGE technology, revealing the critical role of ABA in drought responses [31]. |
| MPSS [32] | Sanger | 1. High throughput 2. Quantitatively display the expression of genes within cells | 1. High cost 2. Complex operation 3. Difficulties in bioinformatics processing | In yeast transcriptome research, MPSS (Massively Parallel Signature Sequencing) technology is used for genome-wide quantitative analysis of gene expression [33]. |
| RNA sequencing [34] | High-throughput sequencing | 1. High throughput 2. High accuracy 3. Wide detection range 4. Low cost | 1. The sample preparation is cumbersome 2. It cannot reveal the heterogeneity of expression among single cells 3. The bioinformatics analysis tools are limited | RNA-sequencing analysis revealed that the expression patterns of genes associated with translation, membrane, and oxidoreductase activity pathways were altered under drought stress [35]. |
| scRNA-seq [36] | High-throughput sequencing | 1. High accuracy and specificity 2. Clarify cell function and localization | 1. High requirements for sample quality 2. High cost 3. Difficulties in data analysis/interpretation | Single-cell analysis of Arabidopsis root tips reveals specific transcriptional responses of epidermal and cortical cells under salt stress [37]. |
| Gene | Crops | Abiotic Stress | Gene Function | References |
|---|---|---|---|---|
| OsWRKY87 | Rice | Drought, salt stress | OsWRKY87 functions as a transcriptional activator | [42] |
| OsSEH1 | Rice | Cold stress | OsSEH1 regulates the expression and metabolite accumulation of genes related to phenylpropanoid and flavonoid biosynthesis, mediating ABA expression levels in response to cold stress | [43] |
| OsCSLD4 | Rice | Salt stress | OsCSLD4, a cell wall polysaccharide synthase, responds to salt stress through ABA-induced osmotic stress | [44] |
| OsNAC5 | Rice | Drought stress, cold stress | It enhances stress tolerance by upregulating the expression of the OsLEA3 gene | [45] |
| ZmHsf01 | Maize | Heat stress | Plays an important role in heat shock signal transduction and downstream gene expression | [46] |
| ZmNAC3 | Maize | Cold stress, salt stress | ZmNAC3 encodes a nuclear-targeted protein with a highly conserved NAC domain at its N-terminus | [47] |
| ZmICE1 | Maize | Cold stress | ZmICE1 regulates the expression of the DREB1 gene, inhibits the expression of ZmAS, and reduces Glu/Asn biosynthesis, thus alleviating the production of reactive oxygen species | [48] |
| TdSHN1 | Wheat | Heavy metal stress | Enhances cadmium tolerance by increasing the activity of superoxide dismutase and catalase | [49] |
| ZmCAO1 | Maize | Waterlogging stress | Mutation of ZmCAO1 leads to the downregulation of key photosynthetic genes, increased reactive oxygen species, and sensitivity to waterlogging | [50] |
| Technology | Targets | Advantage | Limitation | Examples |
|---|---|---|---|---|
| liNMR | Most compounds in metabolites | 1. Small sample size required 2. No sample preprocessing required 3. Accurate provision of metabolite structure information | 1. Low detection sensitivity and resolution 2. Difficult to detect low-abundance metabolites 3. High requirements for sample preparation | Plant holistic metabolic profiling, metabolic flux analysis, in situ/non-destructive metabolic studies [59,60]. |
| 1GC-MS | Volatile, gasifiable, or small molecules | 1. High-resolution and sensitivity 2. Ability to identify metabolite structures 3. Easy qualitative analysis of metabolites | 1. Unable to separate macromolecules 2. Cannot analyze thermally unstable and non-gasifiable substances 3. Complex and time-consuming derivatization preprocessing procedures | Plant primary metabolite analysis, volatile organic compound analysis, fatty acid profiling [61,62]. |
| LC-MS | High boiling point, non-volatile, non-derivatizable macromolecules | 1. High detection sensitivity 2. Fast analysis speed 3. Ability to separate metabolites with similar structures | 1. Limited database size 2. Limited types of metabolites analyzed 3. Not all metabolites can be accommodated by the same column material | Plant secondary metabolite analysis, lipidomics—phospholipids, glycolipids, phytohormone quantification [63,64]. |
| CE-MS | Trace, complex samples | 1. High detection sensitivity 2. Fast analysis speed 3. Small sample size required 4. Wide coverage of metabolites | 1. High requirements for equipment and devices 2. Small sample size, poor reproducibility of separation 3. Narrow linear range for quantitative analysis 4. Limited quantitative analysis due to narrow linear range | Highly polar/ionic compound analysis, analysis of minute samples (single-cell metabolomics) [65,66]. |
| Abiotic Stress | Crops | Metabolite | Change | References |
|---|---|---|---|---|
| Heat stress | Maize | Tryptophan, Threonine, Histidine, Raffinose, Galactitol, Lactitol | Upregulated | [67] |
| Heat stress | Wheat | N-based Amino Acids, ABA, IAA-conjugates | Upregulated | [68] |
| Cold stress | Maize | Guanosine 30, 50-Cyclic Monophosphate, Sophoroside-7-O-Glucoside, L-Lysine, L-Phenylalanine, L-Glutamine, Shanenol, Feruloyl Tartaric Acid | Upregulated | [69] |
| Cold stress | Maize | Trans-aconitate, Coumaroyl Hydroxycitrate, Geranyl Glucosyl Rhamnoside Rhamnoside, Caffeoylquinate, Ferroylquinate, (Iso)Vitexin, DIBOA-Glucoside | Upregulated | [70] |
| Cold stress | Maize | Chlorophyll, Glucose-6-Phosphate Dehydrogenase, Sucrose-to-Starch Ratio | Upregulated | [71] |
| Cold stress | Canola seed | Amino Acids, Organic Acids, Sugars | Upregulated | [72] |
| Drought stress | Wheat | 1-Aminocyclopropane-1-Carboxylic Acid, Asn, 5-HT, GABA, Cystine, Deoxyuridine, Tryptamine, Putrescine | Upregulated | [73] |
| Drought stress | Mulberry tree | Galactolipids, Phospholipids, Flavonoids, Cinnamic Acid, Amino Acids, Carbohydrates, Benzenoids, Organic Heterocyclic Compounds | Upregulated | [74] |
| Drought stress | Barley | Amino Acids, Sugars, Abscisic Acid, Jasmonic Acid, Ferulic Acid | Upregulated | [75] |
| Salt stress | Barley | Aminoacyl-tRNA Biosynthesis, Glycine, Serine, and Threonine Metabolism, Glyoxylate and Dicarboxylate Metabolism, Porphyrin and Chlorophyll Metabolism | Upregulated | [76] |
| Salt stress | Wheat | Amino Acids and Derivatives, Flavonoid Compounds, Organic Acids and Derivatives, Nucleotides and Derivatives, Lipids | Upregulated | [77] |
| Salt stress | Blueberry | Glycine, Malic Acid, Octadecanoic Acid, L-Threonic Acid | Upregulated | [78] |
| Heavy metal stress | Rice | Lipids, Eicosanoids | Upregulated | [79] |
| Heavy metal stress | Purple sweet potato | Glutathione, Tryptophan | Upregulated | [80] |
| Heat stress | Maize | Chlorophyll a, Glutathione (GSH) | Downregulated | [81] |
| Cold stress | Maize | Fructose-6-phosphate, Phosphatidylcholine | Downregulated | [82] |
| Drought stress | Wheat | Citric Acid, Malic Acid | egulaDownrted | [83] |
| Salt stress | Wheat | Riboflavin, Ascorbic Acid | Downregulated | [84] |
| Heavy metal stress | Rice | Glutamine, Succinic Acid | Downregulated | [85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, T.; Ma, X.; Zhang, J.; Cao, S.; Li, W.; Yang, G.; He, C. Progress in Transcriptomics and Metabolomics in Plant Responses to Abiotic Stresses. Curr. Issues Mol. Biol. 2025, 47, 421. https://doi.org/10.3390/cimb47060421
Yu T, Ma X, Zhang J, Cao S, Li W, Yang G, He C. Progress in Transcriptomics and Metabolomics in Plant Responses to Abiotic Stresses. Current Issues in Molecular Biology. 2025; 47(6):421. https://doi.org/10.3390/cimb47060421
Chicago/Turabian StyleYu, Tao, Xuena Ma, Jianguo Zhang, Shiliang Cao, Wenyue Li, Gengbin Yang, and Changan He. 2025. "Progress in Transcriptomics and Metabolomics in Plant Responses to Abiotic Stresses" Current Issues in Molecular Biology 47, no. 6: 421. https://doi.org/10.3390/cimb47060421
APA StyleYu, T., Ma, X., Zhang, J., Cao, S., Li, W., Yang, G., & He, C. (2025). Progress in Transcriptomics and Metabolomics in Plant Responses to Abiotic Stresses. Current Issues in Molecular Biology, 47(6), 421. https://doi.org/10.3390/cimb47060421