
Reconstructing Dynamic Gene Regulatory Networks Using f-Divergence from Time-Series scRNA-Seq Data


Abstract
1. Introduction
2. Materials and Methods
2.1. f-Divergence-Based Temporal Variation Estimation
2.2. Granger Causality for Directed Network Inference
2.3. Regularization Methods for Slowly Changing Sparse Network Inference
2.3.1. Smoothly Clipped Absolute Deviation Penalty
2.3.2. Minimax Concave Penalty
2.4. Partial Correlation and Time-Varying Regulatory Network Inference Algorithm
| Algorithm 1 f-divergence-based dynamic gene regulatory network inference algorithm. |
| Input: Time-series scRNA-seq data matrix ; percentage of randomly sampled single cells ; number of samples ; f-divergence measures; regularization methods . |
| Output: Time-varying gene regulatory networks. |
Step 1: Random sampling and temporal variation calculation of genes using f-divergence
|
3. Results
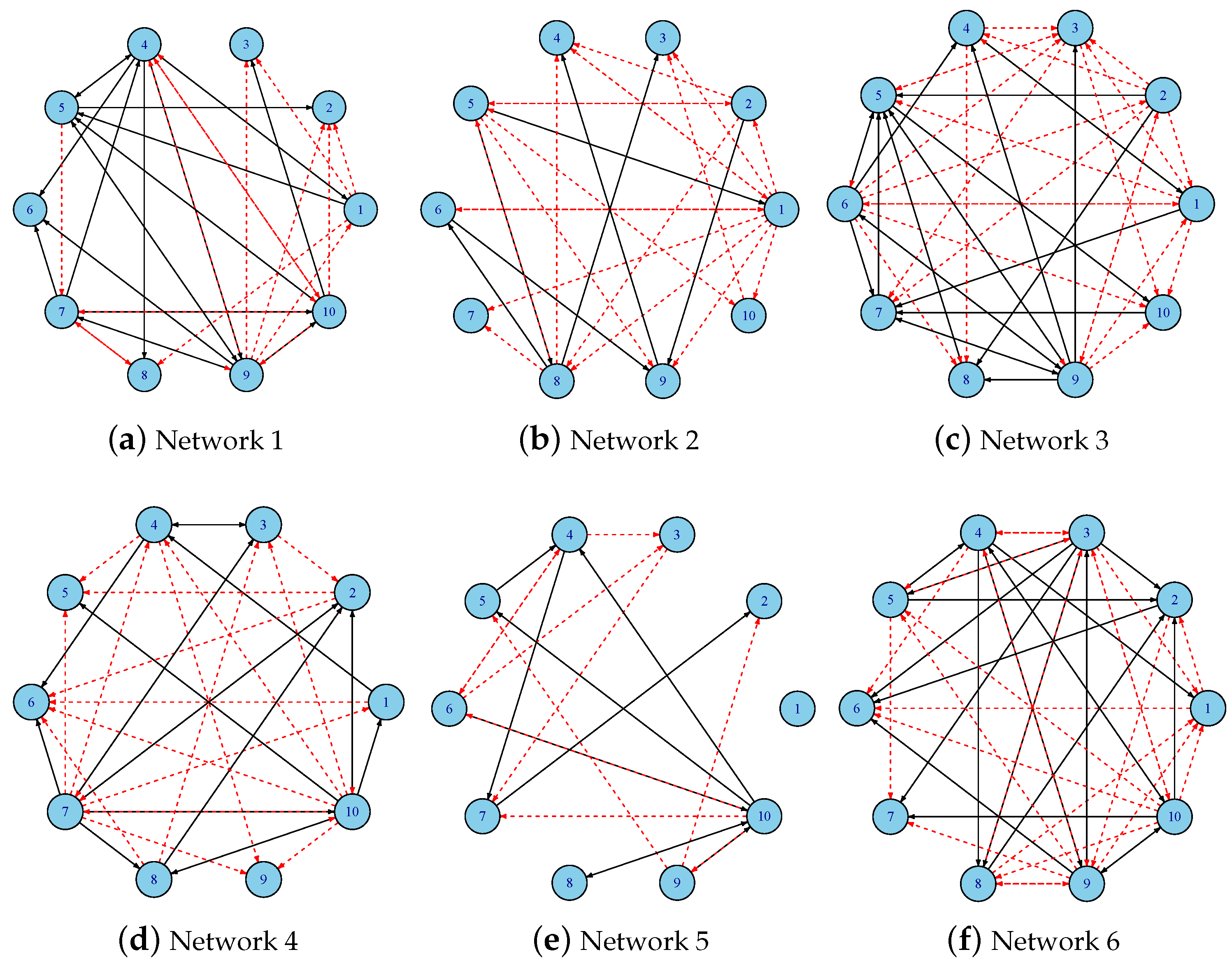
3.1. Datasets and Parameter Configuration
3.2. Evaluation Metrics
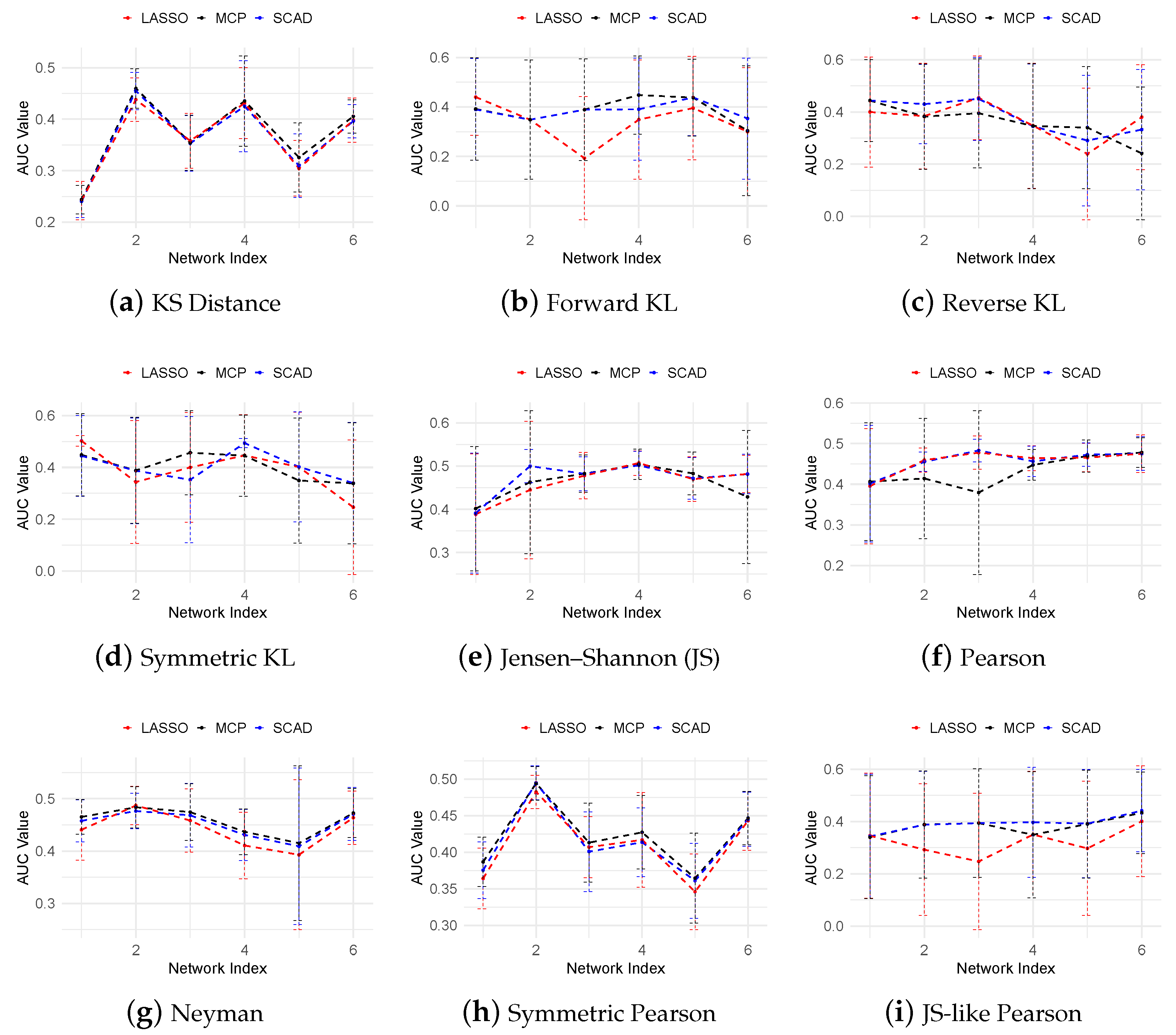
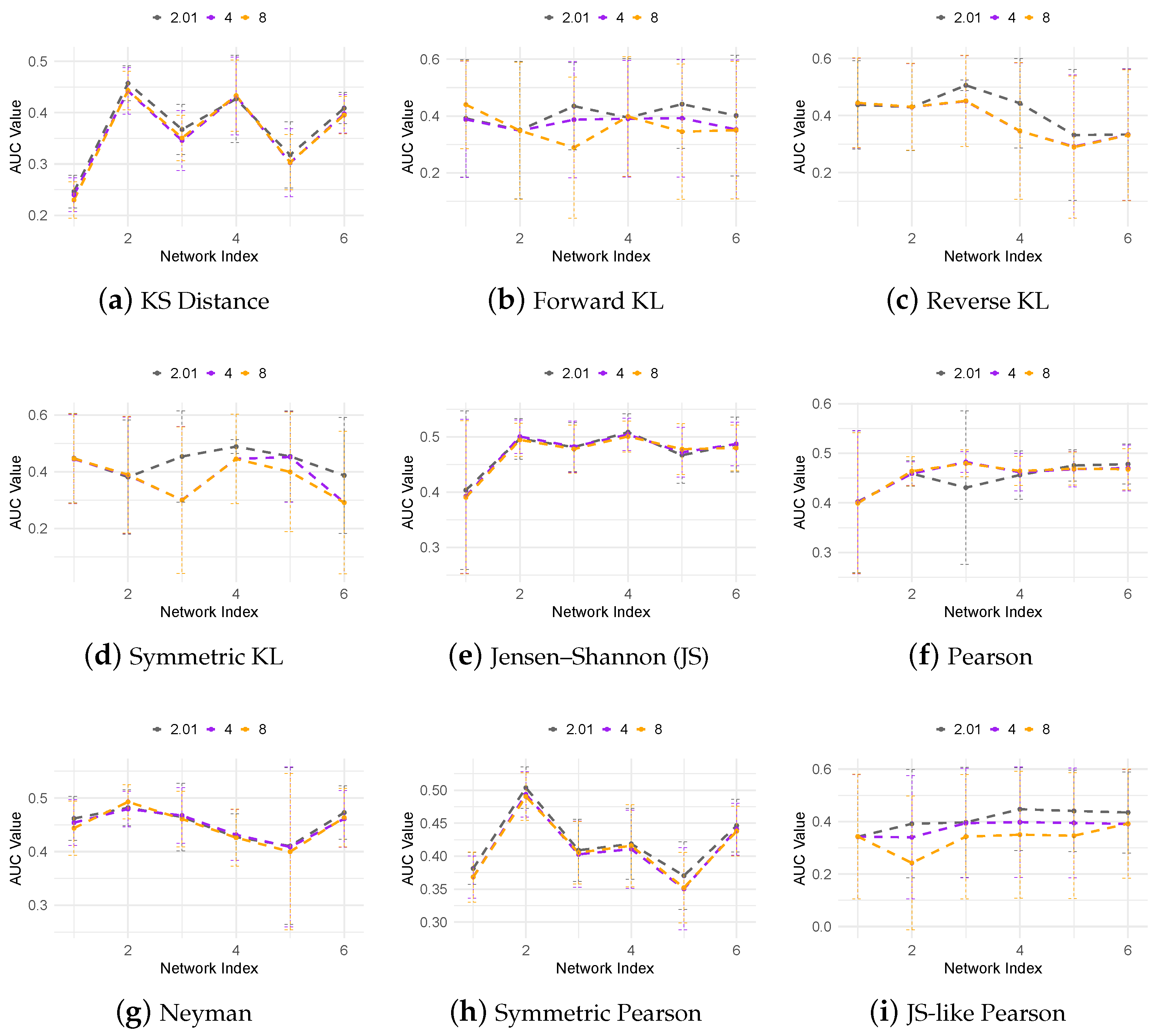
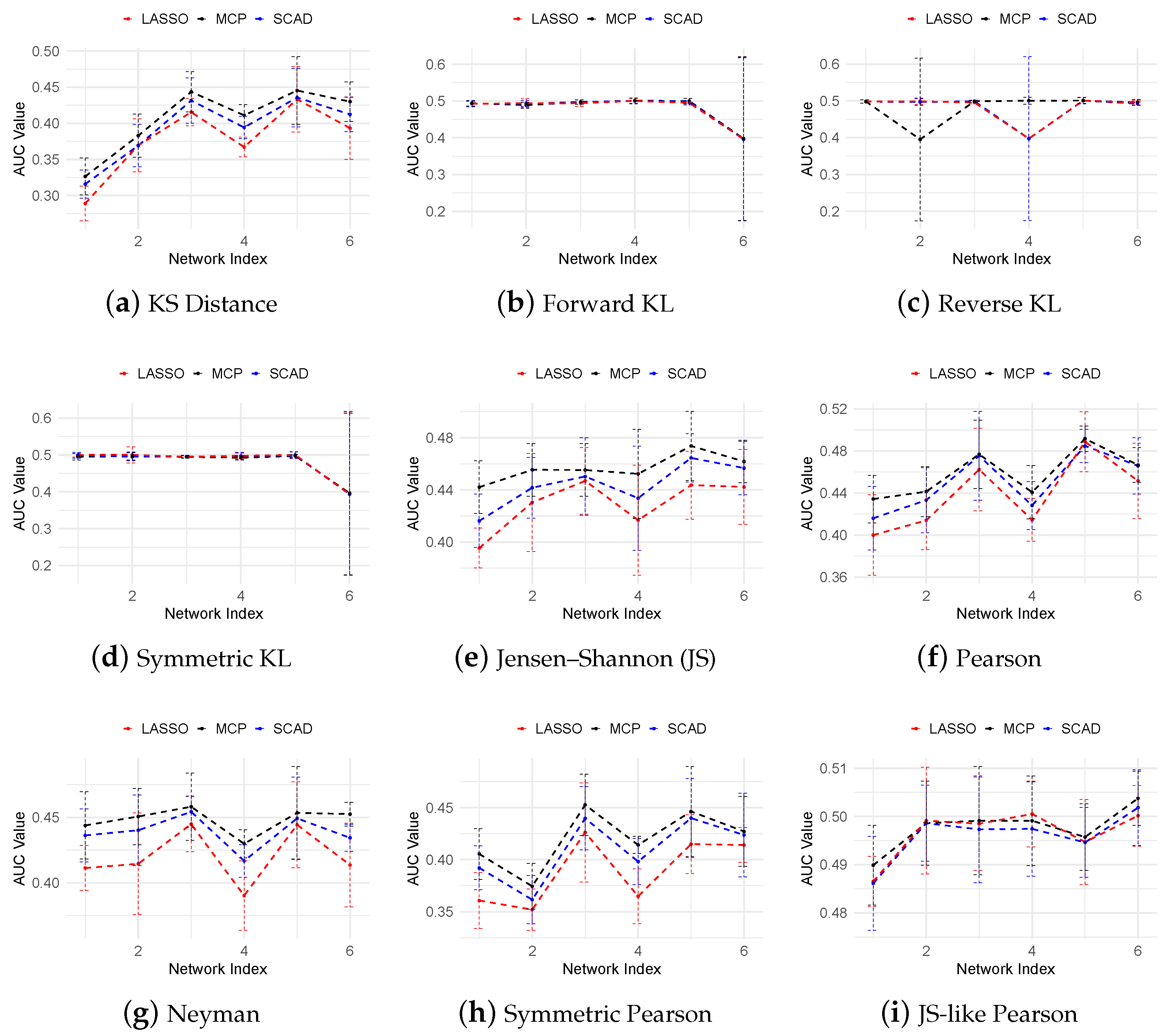
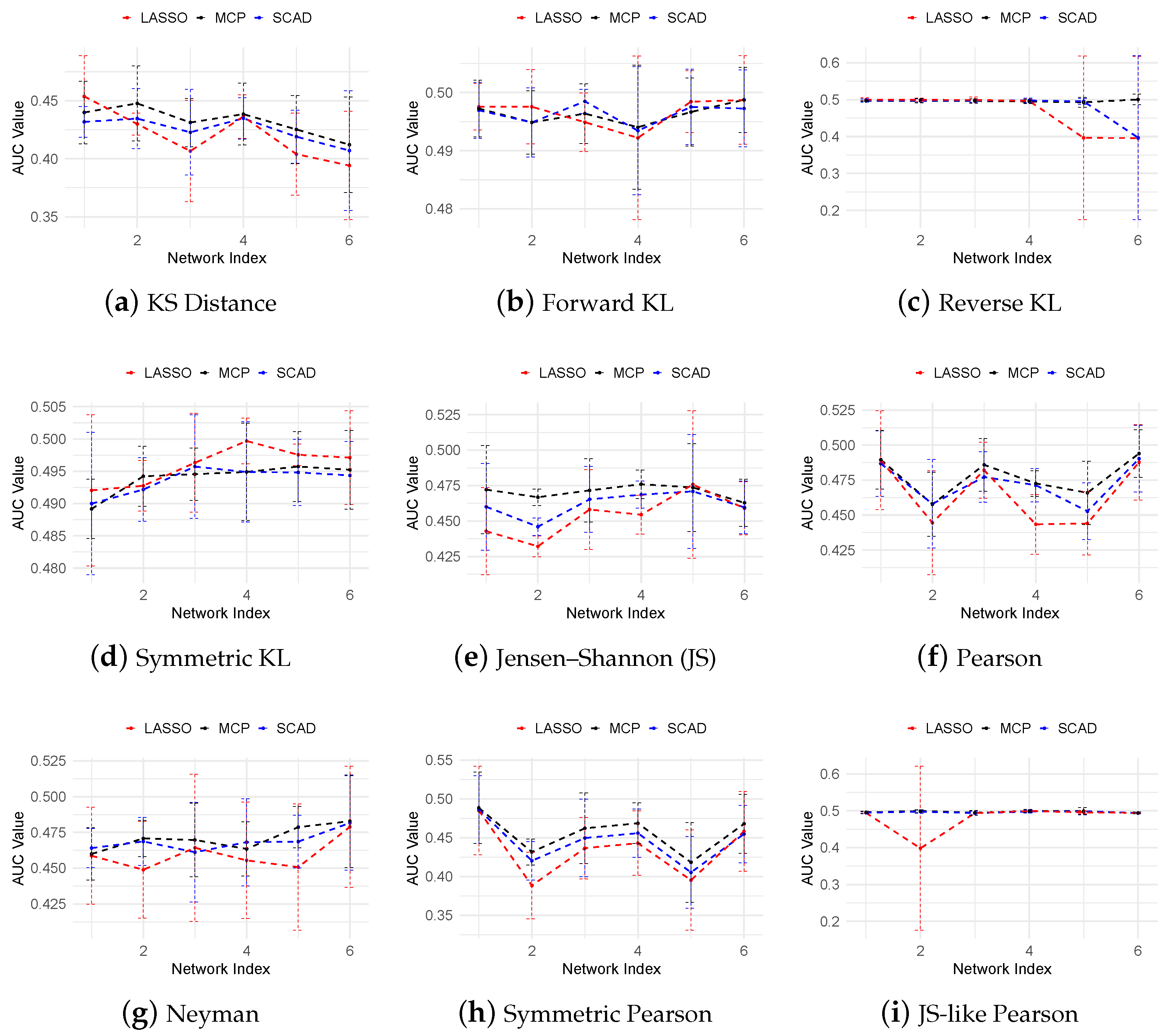
3.3. Regulatory Network Inference from In Silico Dataset
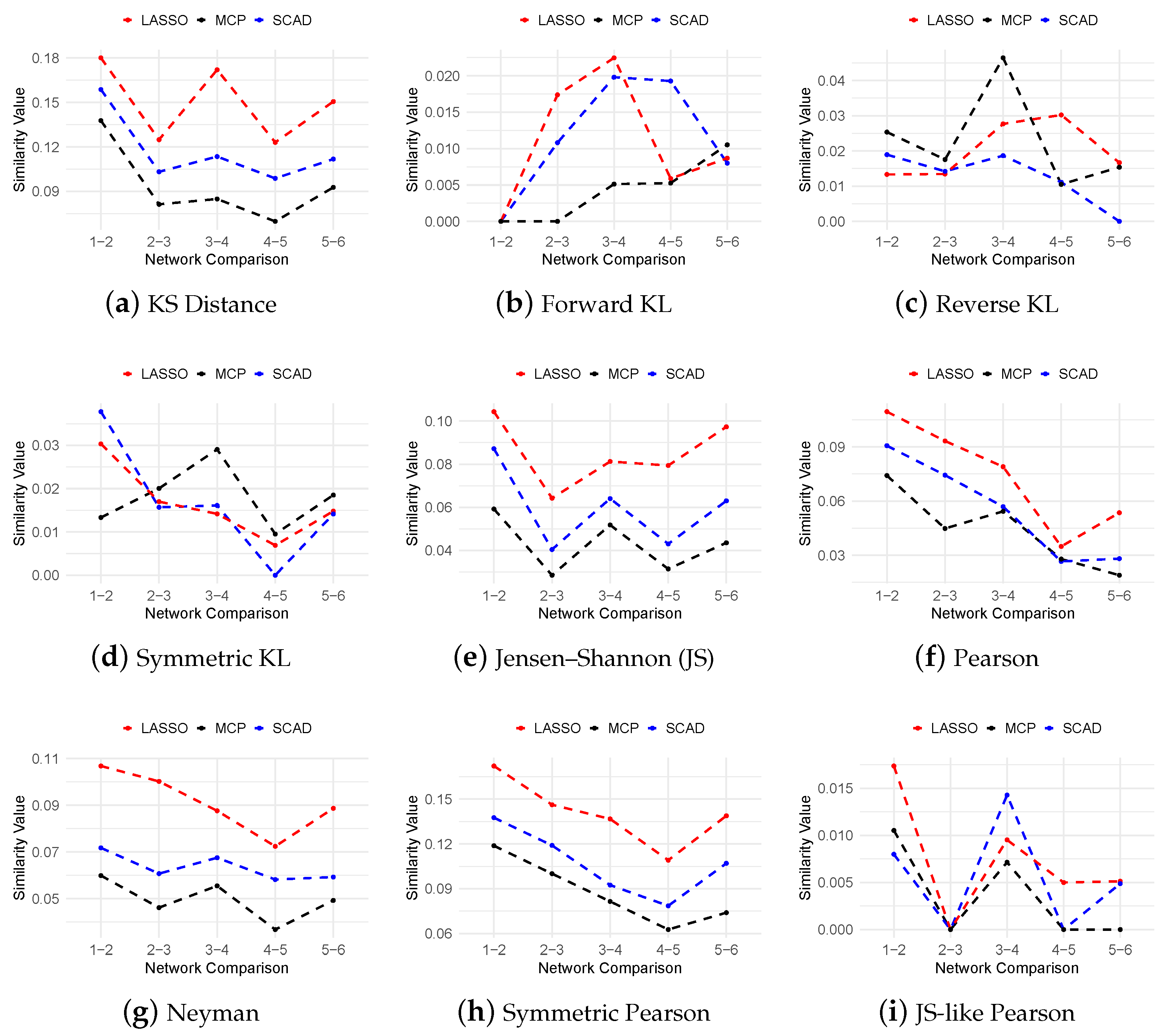
3.4. Inferring Dynamic GRNs Driving THP-1 Differentiation
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| scRNA-seq | single-cell RNA sequencing |
| GRN | gene regulatory network |
| AUROC | area under the receiver operating characteristic curve |
| LASSO | least absolute shrinkage and selection operator |
| MCP | minimax concave penalty |
| SCAD | smoothly clipped absolute deviation |
References
- Yosef, N.; Shalek, A.K.; Gaublomme, J.T.; Jin, H.; Lee, Y.; Awasthi, A.; Wu, C.; Karwacz, K.; Xiao, S.; Jorgolli, M.; et al. Dynamic Regulatory Network Controlling TH17 Cell Differentiation. Nature 2013, 496, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Karlebach, G.; Shamir, R. Modelling and analysis of gene regulatory networks. Nat. Rev. Mol. Cell Biol. 2008, 9, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Bar-Joseph, Z.; Gitter, A.; Simon, I. Studying and Modelling Dynamic Biological Processes Using Time-Series Gene Expression Data. Nat. Rev. Genet. 2012, 13, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Ajmal, H.B.; Madden, M.G. Dynamic Bayesian Network Learning to Infer Sparse Models From Time Series Gene Expression Data. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2794–2805. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Irrthum, A.; Wehenkel, L.; Geurts, P. Inferring regulatory networks from expression data using tree-based methods. PLoS ONE 2010, 5, e12776. [Google Scholar] [CrossRef]
- Schulz, M.H.; Devanny, W.E.; Gitter, A.; Zhong, S.; Ernst, J.; Bar-Joseph, Z. DREM 2.0: Improved Reconstruction of Dynamic Regulatory Networks from Time-Series Expression Data. BMC Syst. Biol. 2012, 6, 104. [Google Scholar] [CrossRef]
- Shmulevich, I.; Dougherty, E.R.; Kim, S.; Zhang, W. Probabilistic Boolean networks: A rule-based uncertainty model for gene regulatory networks. Bioinformatics 2002, 18, 261–274. [Google Scholar] [CrossRef]
- Liang, J.; Han, J. Stochastic Boolean networks: An efficient approach to modeling gene regulatory networks. BMC Syst. Biol. 2012, 6, 113. [Google Scholar] [CrossRef]
- Gebert, J.; Radde, N.; Weber, G.W. Modeling gene regulatory networks with piecewise linear differential equations. Eur. J. Oper. Res. 2007, 181, 1148–1165. [Google Scholar] [CrossRef]
- Bansal, M.; Gatta, G.D.; Di Bernardo, D. Inference of gene regulatory networks and compound mode of action from time course gene expression profiles. Bioinformatics 2006, 22, 815–822. [Google Scholar] [CrossRef]
- Chen, K.C.; Wang, T.Y.; Tseng, H.H.; Huang, C.Y.F.; Kao, C.Y. A stochastic differential equation model for quantifying transcriptional regulatory network in Saccharomyces cerevisiae. Bioinformatics 2005, 21, 2883–2890. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Liu, Y.; Shen, L.C.; Yan, H.; Song, J.; Yu, D.J. GMFGRN: A matrix factorization and graph neural network approach for gene regulatory network inference. Briefings Bioinform. 2024, 25, bbad529. [Google Scholar] [CrossRef] [PubMed]
- Ochs, M.F.; Fertig, E.J. Matrix factorization for transcriptional regulatory network inference. In Proceedings of the 2012 IEEE Symposium on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), San Diego, CA, USA, 9–12 May 2012; pp. 387–396. [Google Scholar]
- Friedman, N.; Koller, D. Being Bayesian about network structure. A Bayesian approach to structure discovery in Bayesian networks. Mach. Learn. 2003, 50, 95–125. [Google Scholar] [CrossRef]
- Beal, M.J.; Falciani, F.; Ghahramani, Z.; Rangel, C.; Wild, D.L. A Bayesian approach to reconstructing genetic regulatory networks with hidden factors. Bioinformatics 2005, 21, 349–356. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, S.W.; Guo, W.F.; Wei, Z.G.; Chen, L. Inference of gene regulatory network based on local Bayesian networks. PLoS Comput. Biol. 2016, 12, e1005024. [Google Scholar] [CrossRef]
- Zou, M.; Conzen, S.D. A new dynamic Bayesian network (DBN) approach for identifying gene regulatory networks from time course microarray data. Bioinformatics 2005, 21, 71–79. [Google Scholar] [CrossRef]
- Shermin, A.; Orgun, M.A. Using dynamic Bayesian networks to infer gene regulatory networks from expression profiles. In Proceedings of the 2009 ACM symposium on Applied Computing, Honolulu, HI, USA, 9–12 March 2009; pp. 799–803. [Google Scholar]
- Gong, H.; Klinger, J.; Damazyn, K.; Li, X.; Huang, S. A novel procedure for statistical inference and verification of gene regulatory subnetwork. BMC Bioinform. 2015, 16, S7. [Google Scholar] [CrossRef]
- Abegaz, F.; Wit, E. Sparse time series chain graphical models for reconstructing genetic networks. Biostatistics 2013, 14, 586–599. [Google Scholar] [CrossRef]
- Menéndez, P.; Kourmpetis, Y.A.; ter Braak, C.J.; van Eeuwijk, F.A. Gene regulatory networks from multifactorial perturbations using Graphical Lasso: Application to the DREAM4 challenge. PLoS ONE 2010, 5, e14147. [Google Scholar] [CrossRef]
- Furqan, M.S.; Siyal, M.Y. Elastic-net copula granger causality for inference of biological networks. PLoS ONE 2016, 11, e0165612. [Google Scholar] [CrossRef]
- Huynh-Thu, V.A.; Sanguinetti, G. Combining tree-based and dynamical systems for the inference of gene regulatory networks. Bioinformatics 2015, 31, 1614–1622. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Patel, Y.; Wang, Y. TRENDY: Gene Regulatory Network Inference Enhanced by Transformer. Bioinformatics 2025, 41, btaf314. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.S. Single-cell RNA sequencing for the study of development, physiology and disease. Nat. Rev. Nephrol. 2018, 14, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559–1582. [Google Scholar] [CrossRef]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Goren, A.; Weitz, D.A.; Bernstein, B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015, 33, 1165–1172. [Google Scholar] [CrossRef]
- Ding, J.; Sharon, N.; Bar-Joseph, Z. Temporal modelling using single-cell transcriptomics. Nat. Rev. Genet. 2022, 23, 355–368. [Google Scholar] [CrossRef]
- Wagner, A.; Regev, A.; Yosef, N. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 2016, 34, 1145–1160. [Google Scholar] [CrossRef]
- Kharchenko, P.V.; Silberstein, L.; Scadden, D.T. Bayesian approach to single-cell differential expression analysis. Nat. Methods 2014, 11, 740–742. [Google Scholar] [CrossRef]
- Woodhouse, S.; Piterman, N.; Wintersteiger, C.M.; Göttgens, B.; Fisher, J. SCNS: A graphical tool for reconstructing executable regulatory networks from single-cell genomic data. BMC Syst. Biol. 2018, 12, 59. [Google Scholar] [CrossRef]
- Lim, C.Y.; Wang, H.; Woodhouse, S.; Piterman, N.; Wernisch, L.; Fisher, J.; Göttgens, B. BTR: Training asynchronous Boolean models using single-cell expression data. BMC Bioinform. 2016, 17, 355. [Google Scholar] [CrossRef]
- Matsumoto, H.; Kiryu, H.; Furusawa, C.; Ko, M.S.; Ko, S.B.; Gouda, N.; Hayashi, T.; Nikaido, I. SCODE: An efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics 2017, 33, 2314–2321. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Kiryu, H. SCOUP: A probabilistic model based on the Ornstein–Uhlenbeck process to analyze single-cell expression data during differentiation. BMC Bioinform. 2016, 17, 232. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.E.; Stumpf, M.P.; Babtie, A.C. Gene regulatory network inference from single-cell data using multivariate information measures. Cell Syst. 2017, 5, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Moerman, T.; Aibar Santos, S.; Bravo González-Blas, C.; Simm, J.; Moreau, Y.; Aerts, J.; Aerts, S. GRNBoost2 and Arboreto: Efficient and scalable inference of gene regulatory networks. Bioinformatics 2019, 35, 2159–2161. [Google Scholar] [CrossRef]
- Shu, H.; Zhou, J.; Lian, Q.; Li, H.; Zhao, D.; Zeng, J.; Ma, J. Modeling gene regulatory networks using neural network architectures. Nat. Comput. Sci. 2021, 1, 491–501. [Google Scholar] [CrossRef]
- Nguyen, H.; Tran, D.; Tran, B.; Pehlivan, B.; Nguyen, T. A comprehensive survey of regulatory network inference methods using single cell RNA sequencing data. Briefings Bioinform. 2021, 22, bbaa190. [Google Scholar] [CrossRef]
- Tsai, M.J.; Wang, J.R.; Ho, S.J.; Shu, L.S.; Huang, W.L.; Ho, S.Y. GREMA: Modelling of emulated gene regulatory networks with confidence levels based on evolutionary intelligence to cope with the underdetermined problem. Bioinformatics 2020, 36, 3833–3840. [Google Scholar] [CrossRef]
- Papili Gao, N.; Ud-Dean, S.M.; Gandrillon, O.; Gunawan, R. SINCERITIES: Inferring gene regulatory networks from time-stamped single cell transcriptional expression profiles. Bioinformatics 2018, 34, 258–266. [Google Scholar] [CrossRef]
- Song, Q.; Ruffalo, M.; Bar-Joseph, Z. Using single cell atlas data to reconstruct regulatory networks. Nucleic Acids Res. 2023, 51, e38. [Google Scholar] [CrossRef]
- Chen, J.; Cheong, C.; Lan, L.; Zhou, X.; Liu, J.; Lyu, A.; Cheung, W.K.; Zhang, L. DeepDRIM: A deep neural network to reconstruct cell-type-specific gene regulatory network using single-cell RNA-seq data. Briefings Bioinform. 2021, 22, bbab325. [Google Scholar] [CrossRef]
- Fan, Y.; Ma, X. Gene regulatory network inference using 3D convolutional neural network. Proc. AAAI Conf. Artif. Intell. 2021, 35, 99–106. [Google Scholar] [CrossRef]
- Dautle, M.; Zhang, S.; Chen, Y. scTIGER: A Deep-Learning Method for Inferring Gene Regulatory Networks from Case versus Control scRNA-seq Datasets. Int. J. Mol. Sci. 2023, 24, 13339. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Wang, H.; Zhang, Y.; Wilkins, M.R.; Canete, P.F.; Yu, D.; Yang, Y.; Zhang, W. Inferring gene regulatory networks by hypergraph generative model. Cell Rep. Methods 2025, 5, 101026. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Dautle, M.; Gao, R.; Zhang, S.; Chen, Y. Inferring gene regulatory networks from time-series scRNA-seq data via GRANGER causal recurrent autoencoders. Briefings Bioinform. 2025, 26, bbaf089. [Google Scholar] [CrossRef]
- Roman-Vicharra, C.; Cai, J.J. Quantum gene regulatory networks. npj Quantum Inf. 2023, 9, 67. [Google Scholar] [CrossRef]
- Fujita, A.; Sato, J.R.; Garay-Malpartida, H.M.; Morettin, P.A.; Sogayar, M.C.; Ferreira, C.E. Time-varying modeling of gene expression regulatory networks using the wavelet dynamic vector autoregressive method. Bioinformatics 2007, 23, 1623–1630. [Google Scholar] [CrossRef]
- Fujita, A.; Sato, J.R.; Garay-Malpartida, H.M.; Yamaguchi, R.; Miyano, S.; Sogayar, M.C.; Ferreira, C.E. Modeling gene expression regulatory networks with the sparse vector autoregressive model. BMC Syst. Biol. 2007, 1, 39. [Google Scholar] [CrossRef]
- Grzegorczyk, M. A non-homogeneous dynamic Bayesian network with a hidden Markov model dependency structure among the temporal data points. Mach. Learn. 2016, 102, 155–207. [Google Scholar] [CrossRef]
- Grzegorczyk, M.; Husmeier, D.; Edwards, K.D.; Ghazal, P.; Millar, A.J. Modelling non-stationary gene regulatory processes with a non-homogeneous Bayesian network and the allocation sampler. Bioinformatics 2008, 24, 2071–2078. [Google Scholar] [CrossRef]
- Richards, H.; Wang, Y.; Si, T.; Zhang, H.; Gong, H. Intelligent Learning and Verification of Biological Networks. In Advances in Artificial Intelligence, Computation, and Data Science: For Medicine and Life Science; Springer: Berlin/Heidelberg, Germany, 2021; pp. 3–28. [Google Scholar]
- Dondelinger, F.; Lèbre, S.; Husmeier, D. Non-Homogeneous Dynamic Bayesian Networks with Bayesian Regularization for Inferring Gene Regulatory Networks with Gradually Time-Varying Structure. Mach. Learn. 2013, 90, 191–230. [Google Scholar] [CrossRef]
- Ahmed, A.; Xing, E.P. Recovering time-varying networks of dependencies in social and biological studies. Proc. Natl. Acad. Sci. USA 2009, 106, 11878–11883. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wang, Y. Hidden Markov Induced Dynamic Bayesian Network for Recovering Time Evolving Gene Regulatory Networks. Sci. Rep. 2015, 5, 17841. [Google Scholar] [CrossRef]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Favera, R.D.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006, 7, S7. [Google Scholar] [CrossRef] [PubMed]
- Wit, E.C.; Abbruzzo, A. Inferring slowly-changing dynamic gene-regulatory networks. BMC Bioinform. 2015, 16, S5. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Braun, R. Time-lagged Ordered Lasso for network inference. BMC Bioinform. 2018, 19, 545. [Google Scholar] [CrossRef]
- Zhang, Y.; Chang, X.; Liu, X. Inference of gene regulatory networks using pseudo-time series data. Bioinformatics 2021, 37, 2423–2431. [Google Scholar] [CrossRef]
- Hallac, D.; Park, Y.; Boyd, S.; Leskovec, J. Network inference via the time-varying graphical lasso. In Proceedings of the 23rd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Halifax, NS, Canada, 13–17 August 2017; Volume 2017, pp. 205–213. [Google Scholar]
- Dallakyan, A.; Kim, R.; Pourahmadi, M. Time series graphical lasso and sparse VAR estimation. Comput. Stat. Data Anal. 2022, 176, 107557. [Google Scholar] [CrossRef]
- Wang, L.; Trasanidis, N.; Wu, T.; Dong, G.; Hu, M.; Bauer, D.E.; Pinello, L. Dictys: Dynamic gene regulatory network dissects developmental continuum with single-cell multiomics. Nat. Methods 2023, 20, 1368–1378. [Google Scholar] [CrossRef]
- Kamimoto, K.; Hoffmann, C.M.; Morris, S.A. CellOracle: Dissecting cell identity via network inference and in silico gene perturbation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Si, T.; Koch, L.; Roberts, S.; Gong, H. Time-Varying Gene Regulatory Networks Inference Using KL Divergence from Single Cell Data. In Proceedings of the 17th International Conference on Bioinformatics and Biomedical Technology (Accepted), Hangzhou, China, 23–26 May 2025. [Google Scholar]
- Si, T.; Hopkins, Z.; Yanev, J.; Hou, J.; Gong, H. A novel f-divergence based generative adversarial imputation method for scRNA-seq data analysis. PLoS ONE 2023, 18, e0292792. [Google Scholar] [CrossRef]
- Liu, W.S.; Si, T.; Kriauciunas, A.; Snell, M.; Gong, H. Bidirectional f-Divergence-Based Deep Generative Method for Imputing Missing Values in Time-Series Data. Stats 2025, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Granger, C.W. Investigating causal relations by econometric models and cross-spectral methods. Econom. J. Econom. Soc. 1969, 37, 424–438. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. B Methodol. 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Fan, J.; Li, R. Variable selection via nonconcave penalized likelihood and its oracle properties. J. Am. Stat. Assoc. 2001, 96, 1348–1360. [Google Scholar] [CrossRef]
- Zhang, C.H. Nearly unbiased variable selection under minimax concave penalty. Ann. Stat. 2010, 38, 894–942. [Google Scholar] [CrossRef]
- Pinna, A.; Soranzo, N.; De La Fuente, A. From knockouts to networks: Establishing direct cause-effect relationships through graph analysis. PLoS ONE 2010, 5, e12912. [Google Scholar] [CrossRef]
- Higham, D.J. An algorithmic introduction to numerical simulation of stochastic differential equations. SIAM Rev. 2001, 43, 525–546. [Google Scholar] [CrossRef]
- Kouno, T.; de Hoon, M.; Mar, J.C.; Tomaru, Y.; Kawano, M.; Carninci, P.; Suzuki, H.; Hayashizaki, Y.; Shin, J.W. Temporal dynamics and transcriptional control using single-cell gene expression analysis. Genome Biol. 2013, 14, R118. [Google Scholar] [CrossRef]
- Breheny, P.; Huang, J. Coordinate descent algorithms for nonconvex penalized regression, with applications to biological feature selection. Ann. Appl. Stat. 2011, 5, 232. [Google Scholar] [CrossRef]
- Tomaru, Y.; Simon, C.; Forrest, A.R.; Miura, H.; Kubosaki, A.; Hayashizaki, Y.; Suzuki, M. Regulatory interdependence of myeloid transcription factors revealed by Matrix RNAi analysis. Genome Biol. 2009, 10, R121. [Google Scholar] [CrossRef]
- Qin, R.; Wang, Y. ImputeGAN: Generative adversarial network for multivariate time series imputation. Entropy 2023, 25, 137. [Google Scholar] [CrossRef] [PubMed]
- Si, T.; Wang, Y.; Zhang, L.; Richmond, E.; Ahn, T.H.; Gong, H. Multivariate Time Series Change-Point Detection with a Novel Pearson-like Scaled Bregman Divergence. Stats 2024, 7, 462–480. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Duan, Z. Finder: A novel approach of change point detection for multivariate time series. Appl. Intell. 2022, 52, 2496–2509. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Divergence Function |
|---|---|
| Forward KL | |
| Reverse KL | |
| Symmetric KL | |
| Jensen–Shannon (JS) | |
| Pearson | |
| Neyman | |
| Symmetric Pearson | |
| JS-like Pearson |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhang, L.; Si, T.; Roberts, S.; Wang, Y.; Gong, H. Reconstructing Dynamic Gene Regulatory Networks Using f-Divergence from Time-Series scRNA-Seq Data. Curr. Issues Mol. Biol. 2025, 47, 408. https://doi.org/10.3390/cimb47060408
Wang Y, Zhang L, Si T, Roberts S, Wang Y, Gong H. Reconstructing Dynamic Gene Regulatory Networks Using f-Divergence from Time-Series scRNA-Seq Data. Current Issues in Molecular Biology. 2025; 47(6):408. https://doi.org/10.3390/cimb47060408
Chicago/Turabian StyleWang, Yunge, Lingling Zhang, Tong Si, Sarah Roberts, Yuqi Wang, and Haijun Gong. 2025. "Reconstructing Dynamic Gene Regulatory Networks Using f-Divergence from Time-Series scRNA-Seq Data" Current Issues in Molecular Biology 47, no. 6: 408. https://doi.org/10.3390/cimb47060408
APA StyleWang, Y., Zhang, L., Si, T., Roberts, S., Wang, Y., & Gong, H. (2025). Reconstructing Dynamic Gene Regulatory Networks Using f-Divergence from Time-Series scRNA-Seq Data. Current Issues in Molecular Biology, 47(6), 408. https://doi.org/10.3390/cimb47060408