
The N-Linked Glycosylation Site N201 in eel Lutropin/Choriogonadotropin Receptor Is Uniquely Indispensable for cAMP Responsiveness and Receptor Surface Loss, but Not pERK1/2 Activity

 ,
, 
Abstract
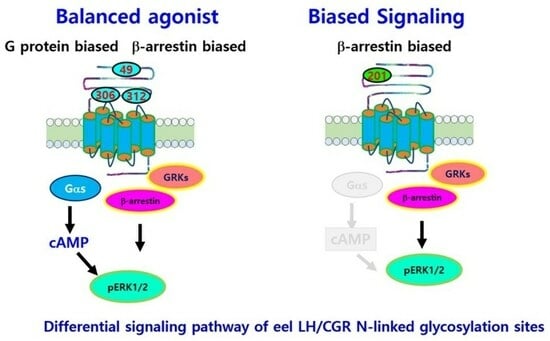
1. Introduction
2. Materials and Methods
2.1. Construction of Glycosylation Mutants of eel LH/CGR cDNA
2.2. Transient Transfection into CHO-K1 and HEK 293 Cells
2.3. cAMP Analysis Using Homogeneous Time-Revolved Fluorescence
2.4. Agonist-Induced Loss of Cell-Surface Expression of Receptors
2.5. Measurement of pERK1/2 Activity Using HTRF
2.6. Analysis of pERK1/2 Activation by Western Blotting
2.7. Data Analysis
3. Results
3.1. Expression at the Cell Surface After Mutation of Putative N-Linked Glycosylation Sites
3.2. Biological Activities of eel LH/CGR-Wt and N-Linked Glycosylation Mutants
3.3. Loss of Cell-Surface Receptors
3.4. pERK1/2 Activation in HEK293 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shiraishi, K.; Ascoli, M. Lutropin/choriogonadotropin stimulate the proliferation of primary cultures of rat Leydig cells through a pathway that involves activation of the extracellularly regulated kinase 1/2 cascade. Endocrinology 2007, 148, 3214–3225. [Google Scholar] [CrossRef] [PubMed]
- Ji, I.; Slaughter, R.G.; Ji, T.H. N-linked oligosaccharides are not required for hormone binding of the lutropin receptor in a Leydig tumor cell line and rat granulosa cells. Endocrinology 1990, 127, 494–496. [Google Scholar] [CrossRef]
- Ascoli, M. Potential Leydig cell mitogenic signals generated by the wild-type and constitutively active mutants of the lutropin/choriogonadotropin receptor (LHR). Mol. Cell. Endocrinol. 2007, 260–262, 244–248. [Google Scholar] [CrossRef]
- Kara, E.; Crepieux, P.; Gauthier, C.; Martinat, N.; Piketty, V.; Guillou, F.; Reiter, E. A phosphorylation cluster of five serine and threonine residues in the C-terminus of the follicle-stimulating hormone receptor is important for desensitization but not for β-arrestin-mediated ERK activation. Mol. Endocrinol. 2006, 20, 3014–3026. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Wang, J.; Plouffe, B.; Smith, J.S.; Yamani, L.; Kaur, S.; Jean-Charles, P.Y.; Gauthier, C.; Lee, M.H.; Pani, B.; et al. Manifold roles of β-arrestin in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal. 2018, 11, 549. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, A.; Cheng, N.; Trejo, J. Post-translational modifications of G protein-coupled receptors control cellular signaling dynamics in space and time. Pharmacol. Rev. 2020, 73, 120–151. [Google Scholar] [CrossRef] [PubMed]
- Goth, C.K.; Petaja-Repo, U.E.; Rosenkilde, M.M. G protein-coupled receptors in the sweet spot: Glycosylation and other post-translational modifications. ACS Pharmacol. Trans. Sci. 2020, 3, 237–245. [Google Scholar] [CrossRef]
- Liu, X.; Davis, D.; Segaloff, D.L. Distribution of potential sites for N-linked glycosylation does not impair hormone binding to the lutropin/choriogonadotropin receptor if Asn-173 is left intact. J. Biol. Chem. 1993, 268, 1513–1516. [Google Scholar] [CrossRef]
- Petaja-Repo, U.E.; Merz, W.E.; Rajaniemi, H.J. Significance of the carbohydrate moiety of the rat ovarian luteinizing hormone/chorionic gonadotropin receptor for ligand-binding specificity and signal transduction. Biochem. J. 1993, 292, 839–844. [Google Scholar] [CrossRef]
- Zhang, R.; Cai, H.; Fatima, N.; Buczko, E.; Dufau, M.L. Functional glycosylation sites of the rat luteinizing hormone receptor required for ligand binding. J. Biol. Chem. 1995, 270, 21722–21728. [Google Scholar] [CrossRef]
- Davis, D.P.; Liu, X.; Segaloff, D.L. Identification of the sites of N-linked glycosylation on the follicle-stimulating hormone (FSH) receptor and assessment of their role in the FSH receptor function. Mol. Endocrinol. 1995, 9, 159–170. [Google Scholar] [PubMed]
- Davis, D.P.; Rozell, T.G.; Liu, X.; Segaloff, D.L. The six-N-linked carbohydrates of the lutropin/choriogonadotropin receptor are not absolutely required for correct folding, cell surface expression, hormone binding, or signal transduction. Mol. Endocrinol. 1997, 11, 550–562. [Google Scholar] [CrossRef]
- Davis, D.P.; Segaloff, D.L. N-linked carbohydrates on G protein-coupled receptors: Mapping sites of attachment and determining functional roles. Methods Enzymol. 2002, 343, 200–212. [Google Scholar] [PubMed]
- Russo, D.; Chazenbalk, G.D.; Nagayama, Y.; Wadsworth, H.L.; Rapoport, B. Site-directed mutagenesis of the human thyrotropin receptor: Role of asparagine-linked oligosaccharides in the expression of a functional receptor. Mol. Endocrinol. 1991, 5, 29–33. [Google Scholar] [CrossRef]
- Byambaragchaa, M.; Seong, H.K.; Choi, S.H.; Kim, D.J.; Kang, M.H.; Min, K.S. Constitutively activating mutants of equine LH/CGR constitutively induce signal transduction and inactivating mutations impair biological activity and cell-surface receptor loss in vitro. Int. J. Mol. Sci. 2021, 22, 10723. [Google Scholar] [CrossRef]
- Byambaragchaa, M.; Park, H.K.; Kim, D.J.; Lee, J.H.; Kang, M.H.; Min, K.S. The N-linked glycosylation site N191 is necessary for PKA signal transduction in eel follicle-stimulating hormone receptor. Int. J. Mol. Sci. 2022, 23, 12792. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Byambaragchaa, M.; Kim, D.J.; Lee, J.H.; Kang, M.H.; Min, K.S. Specific signal transduction of constitutively activating (D576G) and inactivating (R476H) mutants of agonist-stimulated luteinizing hormone receptor in eel. Int. J. Mol. Sci. 2023, 24, 9133. [Google Scholar] [CrossRef] [PubMed]
- Kahsai, A.W.; Shah, K.S.; Shim, P.J.; Lee, M.A.; Shreiber, B.N.; Schwalb, A.M.; Zhang, X.; Kwon, H.Y.; Huang, L.Y.; Soderblom, E.J.; et al. Signal transduction at GPCRs: Allosteric activation of the ERK MAPK by β-arrestin. Proc. Natl. Acad. Sci. USA 2023, 120, e2303794120. [Google Scholar] [CrossRef]
- Ascoli, M.; Fanelli, F.; Segaloff, D.L. The lutropin/choriogonadotropin, a 2002 perspective. Endocr. Rev. 2002, 23, 141–174. [Google Scholar] [CrossRef]
- Zhang, R.; Tsai-Morris, C.H.; Kitamura, M.; Buchzko, E.; Dufau, M.L. Changes in binding activity of luteinizing hormone receptors by site directed mutagenesis of potential glycosylation sites. Biochem. Biophys. Res. Commun. 1991, 181, 804–808. [Google Scholar] [CrossRef]
- Arnhold, I.J.; Lofrano-Porto, A.; Latronico, A.C. Inactivating mutations of luteinizing hormone β-subunit or luteinizing hormone receptor cause oligo-amenorrhea and infertility in women. Horm. Res. 2009, 71, 75–82. [Google Scholar] [CrossRef]
- Soto, A.G.; Trejo, J. N-linked glycosylation of protease-activated receptor-1 second extracellular loop: A critical determinant for ligand-induced receptor activation and internalization. J. Biol. Chem. 2010, 285, 18781–18793. [Google Scholar] [CrossRef] [PubMed]
- Soto, A.G.; Smith, T.H.; Chen, B.; Bhattacharya, S.; Cordova, I.C.; Kenakin, T.; Vaidehi, N.; Trejo, J. N-linked glycosylation of protease-activated receptor-1 at extracellular loop2 regulates G-protein signaling bias. Proc. Natl. Acad. Sci. USA 2015, 112, E3600–E3608. [Google Scholar] [CrossRef] [PubMed]
- Compton, S.J.; Sandhu, S.; Wijesuriya, S.J.; Hollenberg, M.D. Glycosylation of human proteinase-activated receptor-2 (hPAR2): Role in cell surface expression and signaling. Biochem. J. 2002, 368, 495–505. [Google Scholar] [CrossRef]
- Michineau, S.; Muller, L.; Pizard, A.; Alhenc-Gélas, F.; Rajerison, R.M. N-linked glycosylation of the human bradykinin B2 receptor is required for optimal cell surface expression and coupling. Biol. Chem. 2004, 385, 49–57. [Google Scholar] [CrossRef]
- Marada, S.; Navarro, G.; Truong, A.; Stewart, D.P.; Arensdorf, A.M.; Nachtergaele, S.; Angelats, E.; Opferman, J.T.; Rohatgi, R.; McCormick, P.J.; et al. Functional divergence in the role of N-linked glycosylation in smoothened signaling. PLoS Genet. 2015, 11, e1005473. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Nakagawa, S.; Miyake, T.; Setsu, G.; Kunisue, S.; Goto, K.; Hirasawa, A.; Okamura, H.; Yamaguchi, Y.; Doi, M. Identification and functional characterization of N-linked glycosylation of the orphan G protein-coupled receptor Gpr176. Sci. Rep. 2020, 10, 4429. [Google Scholar]
- Van Koppen, C.J.; Nathanson, N.M. Site-directed mutagenesis of the m2 muscarinic acetylcholine receptor. Analysis of the role of N-glycosylation in receptor expression and function. J. Biol. Chem. 1990, 265, 20887–20892. [Google Scholar] [CrossRef]
- Fukushima, Y.; Oka, Y.; Saitoh, T.; Katagiri, H.; Asano, T.; Matsuhashi, N.; Takata, K.; van Breda, E.; Yazaki, Y.; Sugano, K. Structural and functional analysis of the canine histamine H2 receptor by site-directed mutagenesis: N-glycosylation is not vital for its action. Biochem. J. 1995, 310, 553–558. [Google Scholar] [CrossRef]
- Kozielewicz, P.; Alomar, H.; Yusof, S.; Grafton, G.; Cooper, A.J.; Curnow, S.J.; Ironside, J.W.; Pall, H.; Barnes, N.M. N-glycosylation and expression in human tissues of the orphan GPR61 receptor. FEBS Open Bio 2017, 7, 1982–1993. [Google Scholar] [CrossRef]
- Nakagawa, T.; Takahashi, C.; Matsuzaki, H.; Takeyama, S.; Sato, S.; Sato, A.; Kuroda, Y.; Higashi, H. N-glycan-dependent cell-surface expression of the P2Y2 receptor and N-glycan-independent distribution to lipid rafts. Biochem. Biophys. Res. Commun. 2017, 485, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Norskov-Lauritsen, L.; Jorgensen, S.; Brauner-Osborne, H. N-glycosylation and disulfide bonding affects GPRC6A receptor expression, function, and dimerization. FEBS Lett. 2015, 589, 588–597. [Google Scholar] [CrossRef]
- Janezic, E.M.; Lauer, S.M.; Williams, R.G.; Chungyoun, M.; Lee, K.S.; Navaluna, E.; Lau, H.T.; Ong, S.E.; Hague, C. N-glycosylation of α1D-adrenergic receptor N-terminal domain is required for correct trafficking, function, and biogenesis. Sci. Rep. 2020, 10, 7209. [Google Scholar] [CrossRef]
- Piketty, V.; Kara, E.; Guillou, F.; Reiter, E.; Crepiux, P. Follicle-stimulating hormone (FSH) activates extracellular signal-regulated kinase phosphorylation independently of beta-arrestin- and dynamin-mediated FSH receptor internalization. RBE 2006, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signaling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.J.; Baillie, G.S.; Kohout, T.A.; McPhee, I.; Magiera, M.M.; Ang, K.L.; Miller, W.E.; Mclean, A.J.; Conti, M.; Houslay, M.D.; et al. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science 2002, 298, 834–836. [Google Scholar] [CrossRef]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of receptor signals by beta-arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Jean-Charles, P.V.; Kaur, S.; Shenoy, S.K. GPCR signaling via β-arrestin-dependent mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 142–158. [Google Scholar] [CrossRef]
- Pakharukova, N.; Masoudi, A.; Pani, B.; Staus, D.P.; Lefkowitz, R.J. Allosteric activation of proto-oncogene kinase Src by GPCR-beta-arrestin complexes. J. Biol. Chem. 2020, 295, 16773–16784. [Google Scholar] [CrossRef]
- Zang, Y.; Kahsai, A.W.; Pakharukova, N.; Huang, L.Y.; Lefkowitz, R.J. The GPCR-beta-arrestin complex allosterically activates C-Raf by binding its amino terminus. J. Biol. Chem. 2021, 297, 101369. [Google Scholar] [CrossRef]
- Hirakawa, T.; Galet, C.; Ascoli, M. MA-10 cells transfected with the human lutropin/choriogonadotropin receptor (hLHR): A novel experimental paradigm to study the functional properties of the hLHR. Endocrinology 2002, 143, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Tai, P.; Shiraishi, K.; Ascoli, M. Activation of the lutropin/choriogonadotropin receptor inhibits apoptosis of immature Leydig cells in primary culture. Endocrinology 2009, 150, 3766–3773. [Google Scholar] [CrossRef] [PubMed]
- Tranchant, T.; Durand, G.; Gauthier, C.; Crepieux, P.; Ulloa-Aguirre, A.; Royere, D.; Reiter, E. Preferential β-arrestin signaling at low receptor density revealed by functional characterization of the human FSH receptor A189V mutation. Mol. Cell. Endocrinol. 2011, 331, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Uchida, H.; Maruyama, T.; Kajitani, T.; Oda, H.; Miyazaki, K.; Kagami, M.; Yoshimura, Y. Molecular analysis of a mutated FSH receptor detected in a patient with spontaneous ovarian hyperstimulation syndrome. PLoS ONE 2013, 8, e75478. [Google Scholar] [CrossRef]
- Ayoub, M.A.; Yvinec, R.; Jegot, G.; Dias, J.A.; Poli, S.M.; Poupon, A.; Crepieux, P.; Reiter, E. Profiling of FSHR negative allosteric modulators on LH/CGR reveals biased antagonism with implication in steroidogenesis. Mol. Cell. Endocrinol. 2016, 436, 10–22. [Google Scholar] [CrossRef]
- De Pascali, F.; Reiter, E. Beta-arrestins and biased signaling in gonadotropin receptors. Minerva Ginecol. 2018, 70, 525–538. [Google Scholar] [CrossRef]
- Landomiel, F.; De Pascali, F.; Raynaud, P.; Jean-Alphonse, F.; Yvinec, R.; Pellissier, L.P.; Boszon, V.; Bruneau, G.; Crepieux, P.; Poupon, A.; et al. Biased signaling and allosteric modulation at the FSHR. Front. Endocrinol. 2019, 10, 148. [Google Scholar] [CrossRef]
- De Pascali, F.; Ayoub, M.A.; Benevelli, R.; Sposini, S.; Lehoux, J.; Gallay, N.; Raynaud, P.; Landomiel, F.; Jean-Alphonse, F.; Gauthier, C.; et al. Pharmacological characterization of low molecular weight biased agonist at the follicle-stimulating hormone receptor. Int. J. Mol. Sci. 2021, 22, 9850. [Google Scholar] [CrossRef]
- Duan, J.; Xu, P.; Cheng, X.; Mao, C.; Crol, T.; He, X.; Shi, J.; Luan, X.; Yin, W.; You, E.; et al. Structures of full-length glycoprotein hormone receptor signalling complexes. Nature 2021, 598, 688–692. [Google Scholar] [CrossRef]
- Skapinker, E.; Aldbai, R.; Aucoin, E.; Clake, E.; Clark, M.; Ghokasian, D.; Kombargi, H.; Abraham, M.J.; Li, Y.; Bunsick, D.A.; et al. Artificial and natural sweeteners biased T1R2/T1R3 taste receptors transactivate glycosylated receptors on cancer cells to induce epithelial-mesenchymal transition of metastatic phenotype. Nutrients 2024, 16, 1840. [Google Scholar] [CrossRef]
- Bunsick, D.A.; Matsukobo, J.; Aldbai, R.; Baghaie, L.; Szewczuk, M.R. Functional selectivity of cannabinoid type 1 G protein-coupled receptor agonists in transactivating glycosylated receptors on cancer cells to induce epithelial-mesenchymal transition metastatic phenotype. Cells 2024, 13, 480. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| eel LH/CG Receptors | cAMP Responses | ||
|---|---|---|---|
| Basal a (nM/104 Cells) | EC50 (ng/mL) | Rmax b (nM/104 Cells) | |
| LH/CGR-wt | 0.1 ± 0.1 | 189.7 (1.0-fold) (159.1 to 235.0) c | 58.8 ± 1.8 (100%) |
| LH/CGR-N49Q | 1.6 ± 0.9 | 215.1 (0.88-fold) (173.2 to 283.7) | 70.1 ± 2.7 (119%) |
| LH/CGR-N201Q | - d | - | - |
| LH/CGR-N306Q | 0.1 ± 0.1 | 233.3 (0.81-fold) (182.2 to 324.1) | 37.8 ± 1.8 (64%) |
| LH/CGR-N312Q | 0.1 ± 0.2 | 235.0 (0.80-fold) (181.9 to 331.9) | 35.5 ± 1.8 (60%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byambaragchaa, M.; Kim, D.-W.; Park, S.H.; Kang, M.-H.; Min, K.-S. The N-Linked Glycosylation Site N201 in eel Lutropin/Choriogonadotropin Receptor Is Uniquely Indispensable for cAMP Responsiveness and Receptor Surface Loss, but Not pERK1/2 Activity. Curr. Issues Mol. Biol. 2025, 47, 345. https://doi.org/10.3390/cimb47050345
Byambaragchaa M, Kim D-W, Park SH, Kang M-H, Min K-S. The N-Linked Glycosylation Site N201 in eel Lutropin/Choriogonadotropin Receptor Is Uniquely Indispensable for cAMP Responsiveness and Receptor Surface Loss, but Not pERK1/2 Activity. Current Issues in Molecular Biology. 2025; 47(5):345. https://doi.org/10.3390/cimb47050345
Chicago/Turabian StyleByambaragchaa, Munkhzaya, Dong-Wan Kim, Sei Hyen Park, Myung-Hwa Kang, and Kwan-Sik Min. 2025. "The N-Linked Glycosylation Site N201 in eel Lutropin/Choriogonadotropin Receptor Is Uniquely Indispensable for cAMP Responsiveness and Receptor Surface Loss, but Not pERK1/2 Activity" Current Issues in Molecular Biology 47, no. 5: 345. https://doi.org/10.3390/cimb47050345
APA StyleByambaragchaa, M., Kim, D.-W., Park, S. H., Kang, M.-H., & Min, K.-S. (2025). The N-Linked Glycosylation Site N201 in eel Lutropin/Choriogonadotropin Receptor Is Uniquely Indispensable for cAMP Responsiveness and Receptor Surface Loss, but Not pERK1/2 Activity. Current Issues in Molecular Biology, 47(5), 345. https://doi.org/10.3390/cimb47050345