_Kim.png)
Balancing Microglial Density and Activation in Central Nervous System Development and Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Microglial Function in Neurodevelopment
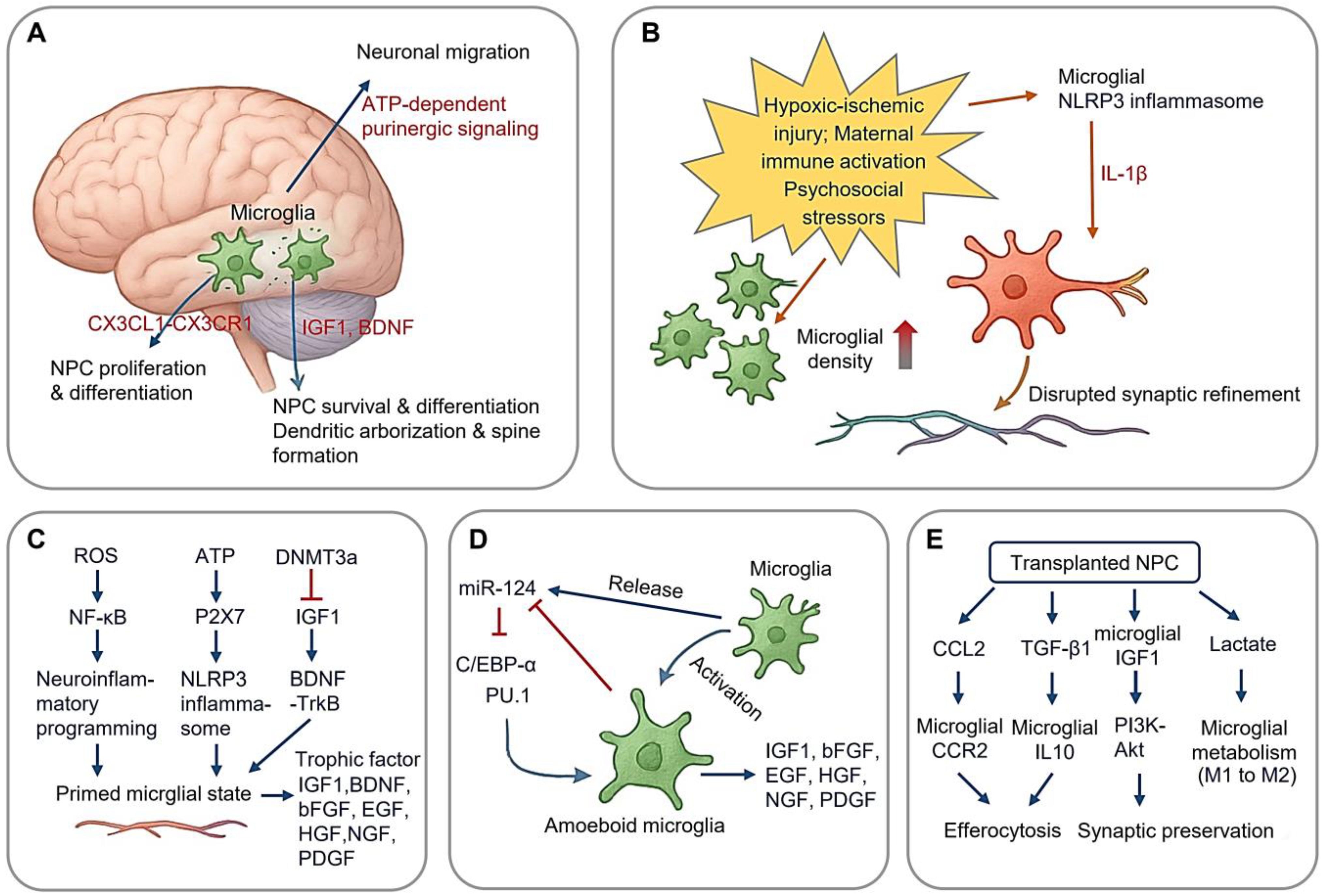
2.1. Regulation of Neurogenesis
2.1.1. Perinatal Stress and Neuroinflammatory Programming
2.1.2. Microglial miR-124 in Neurogenesis
2.1.3. Apoptosis Regulation of Microglia in Development and AFB1-Induced Stress
2.1.4. Crosstalk Between NPC and Microglia Function During Neurogenesis
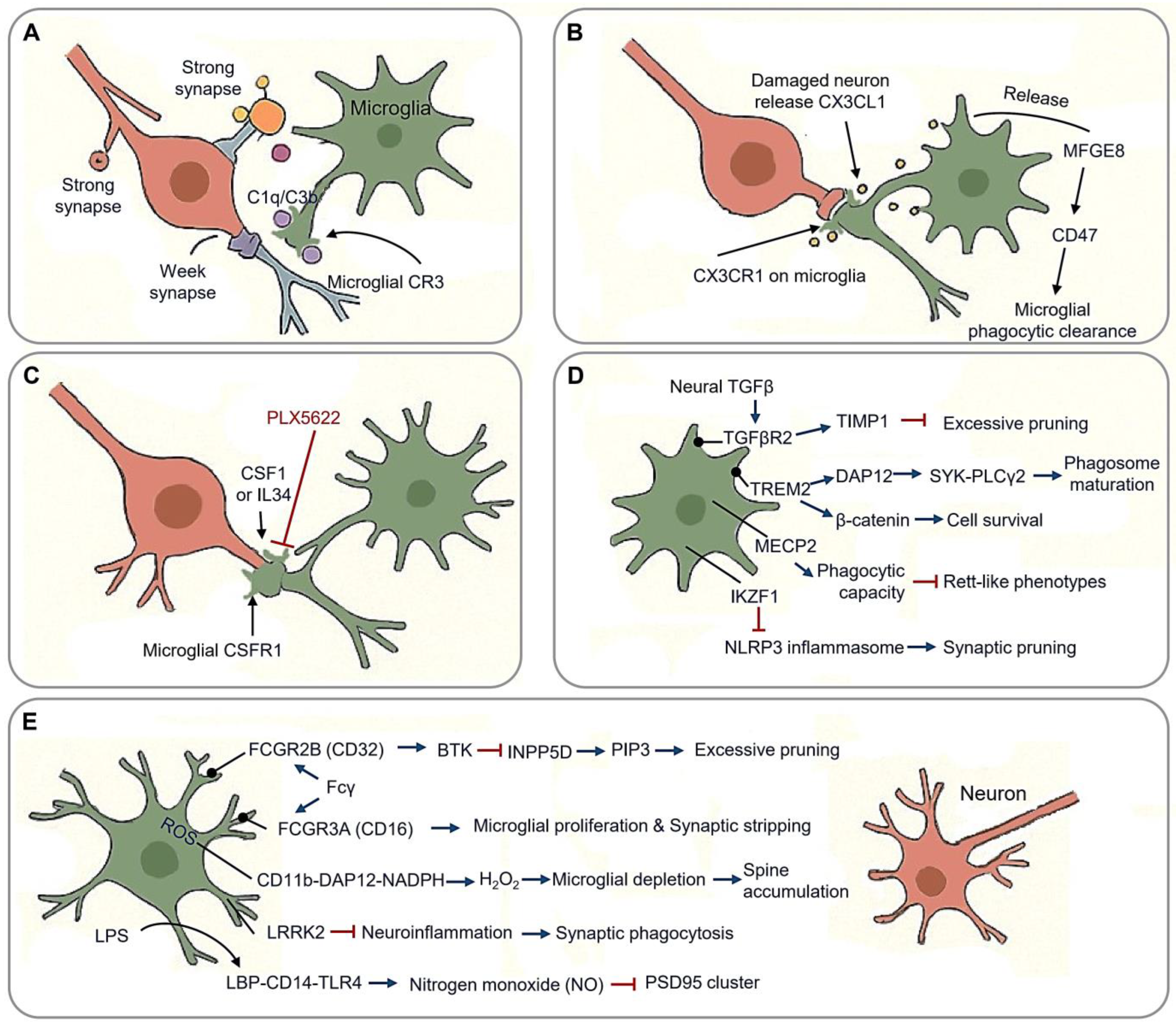
2.2. Synaptic Pruning in Neural Circuit Optimization
2.2.1. Molecular Machinery of Synaptic Pruning
2.2.2. Synaptic Stabilization vs. Neurodegenerative Transformation
3. Microglia in Neurological Diseases
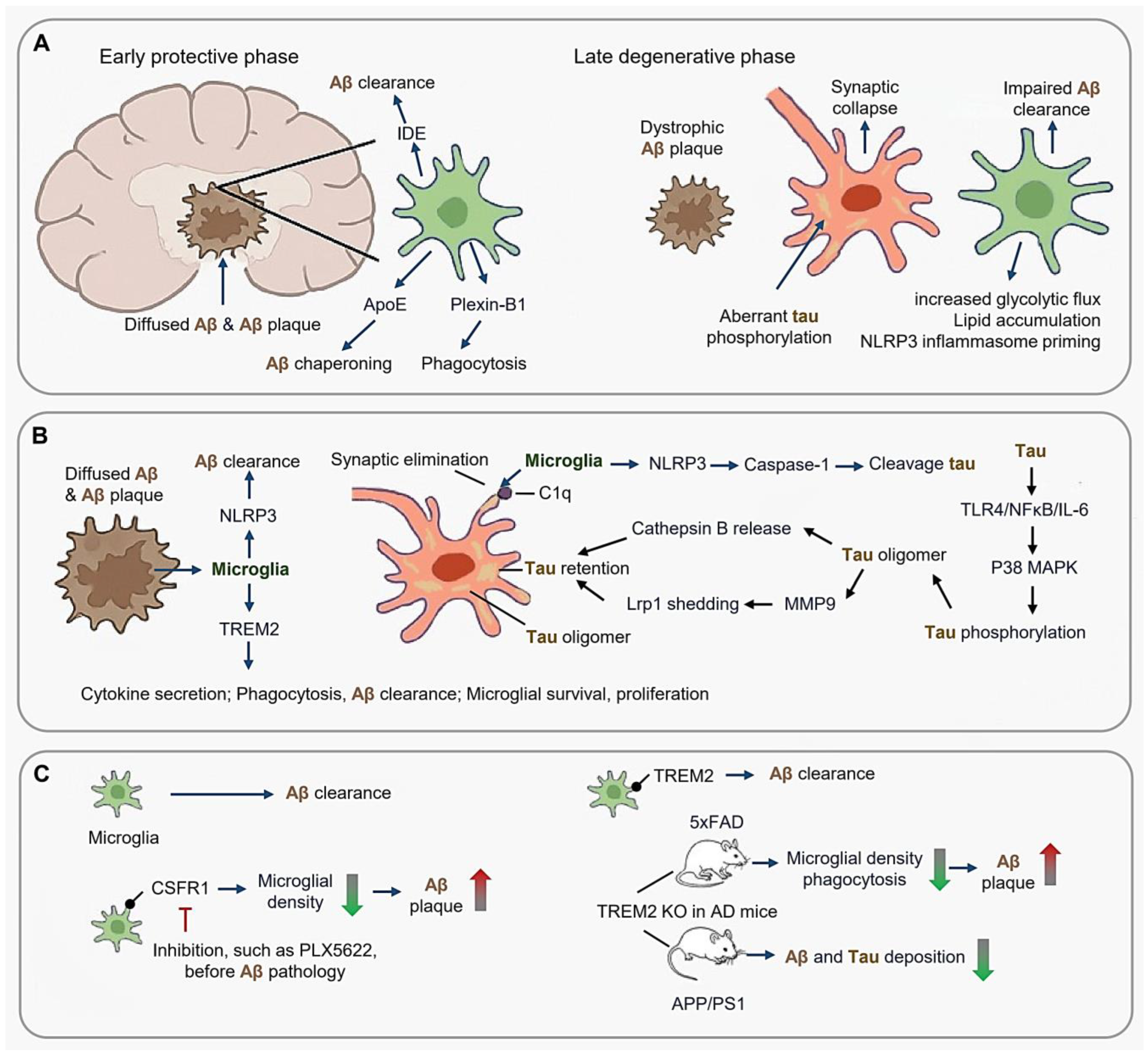
3.1. Alzheimer’s Disease: Phase-Dependent Microglial Dysregulation in Aβ-Tau Synergy
3.1.1. Microglial Response to Aβ Plaques and Tau-Microglia Crosstalk
3.1.2. Spatiotemporal Evolution of Disease-Associated Microglia (DAM)
3.1.3. Therapeutic Implications
3.2. Parkinson’s Disease: Microglial Orchestrators of α-Synucleinopathy
3.2.1. Phase-Dependent Microglial Activation in PD Pathogenesis
3.2.2. Therapeutic Modulation of Microglial Checkpoints
3.3. Huntington’s Disease: Microglial Dysregulation in Mutant Huntingtin Proteostasis
3.3.1. Tripartite Pathogenesis: mHTT Subversion of Microglial Homeostasis
3.3.2. Therapeutic Rebalancing of Microglial States
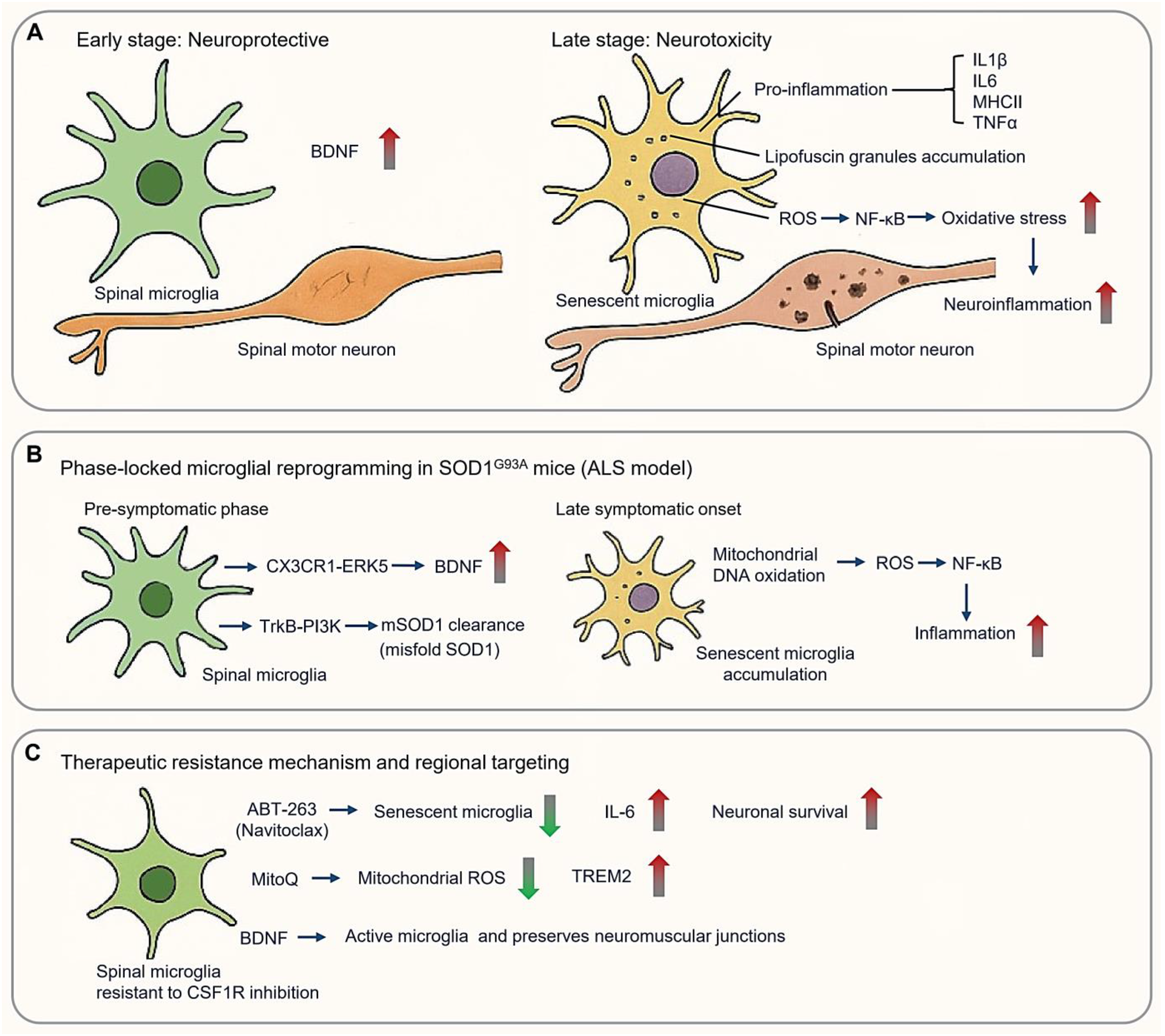
3.4. Amyotrophic Lateral Sclerosis: Spinal Microglial Dichotomy
3.4.1. Phase-Locked Microglial Reprogramming in SOD1 Pathogenesis
3.4.2. Therapeutic Resistance Mechanisms and Regional Targeting
4. Therapeutic Interventions
4.1. Targeting Microglia for Therapy
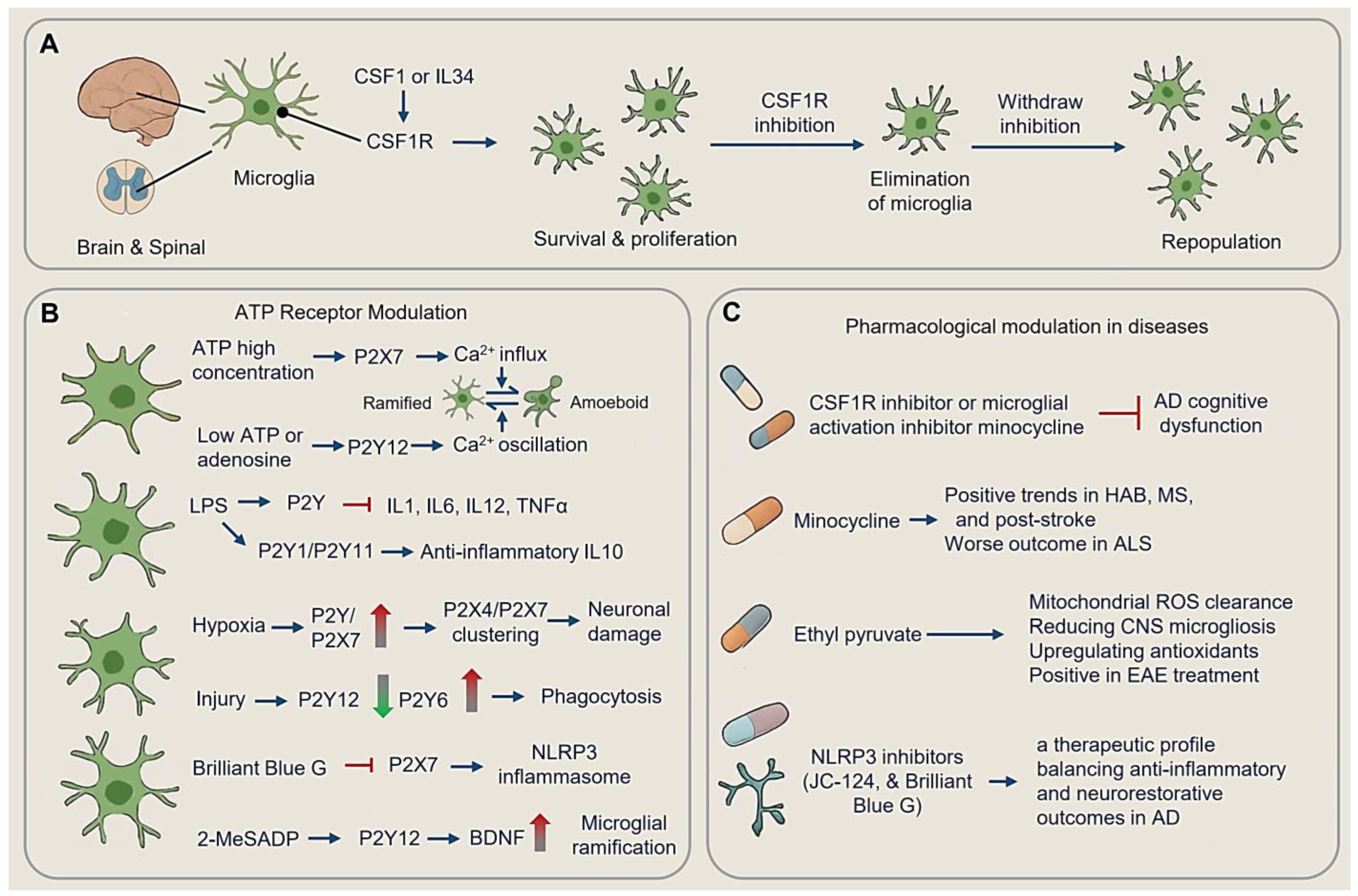
4.1.1. CSF1R and Microglial Survival
4.1.2. ATP Receptor Modulation
4.1.3. Pharmacological Modulation
4.2. Implications for Treatment Strategies
5. Lifestyle Modifications
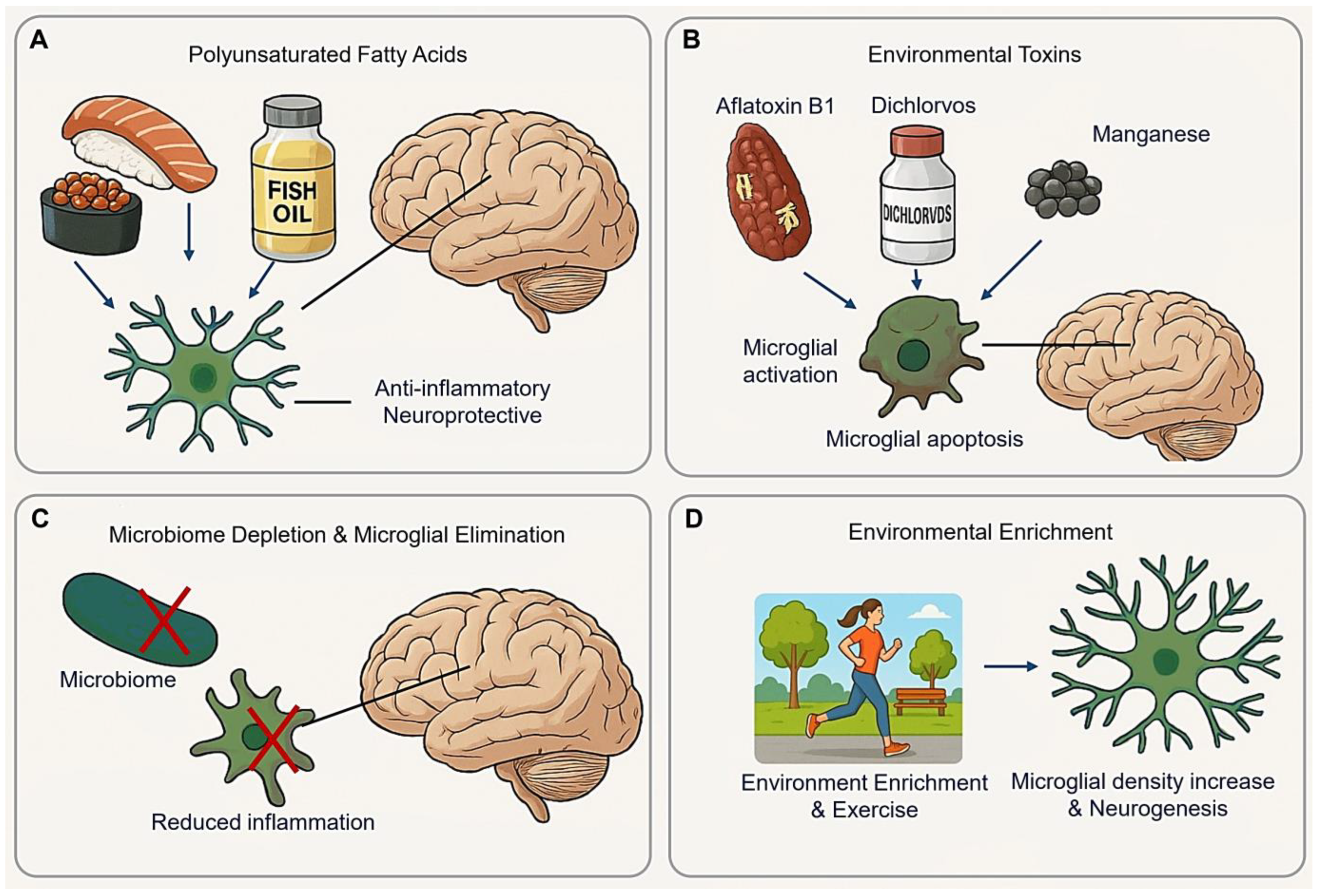
5.1. Polyunsaturated Fatty Acids (PUFAs)
5.2. Environmental Toxins
5.3. Microbiome Depletion and Microglial Elimination
5.4. Environmental Enrichment (EE)
6. Discussion
7. Conclusions
Funding
Conflicts of Interest
References
- Jia, X.; Gao, Z.; Hu, H. Microglia in depression: Current perspectives. Sci. China Life Sci. 2021, 64, 911–925. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Gunner, G.; Cheadle, L.; Johnson, K.M.; Ayata, P.; Badimon, A.; Mondo, E.; Nagy, M.A.; Liu, L.; Bemiller, S.M.; Kim, K.W.; et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 2019, 22, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Thion, M.S.; Ginhoux, F.; Garel, S. Microglia and early brain development: An intimate journey. Science 2018, 362, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef]
- Nikodemova, M.; Kimyon, R.S.; De, I.; Small, A.L.; Collier, L.S.; Watters, J.J. Microglial numbers attain adult levels after undergoing a rapid decrease in cell number in the third postnatal week. J. Neuroimmunol. 2015, 278, 280–288. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Vargas Aguilar, S.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; Zelada Gonzalez, F.; Perrin, P.; et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef]
- Ueno, M.; Fujita, Y.; Tanaka, T.; Nakamura, Y.; Kikuta, J.; Ishii, M.; Yamashita, T. Layer V cortical neurons require microglial support for survival during postnatal development. Nat. Neurosci. 2013, 16, 543–551. [Google Scholar] [CrossRef]
- Miyamoto, A.; Wake, H.; Ishikawa, A.W.; Eto, K.; Shibata, K.; Murakoshi, H.; Koizumi, S.; Moorhouse, A.J.; Yoshimura, Y.; Nabekura, J. Microglia contact induces synapse formation in developing somatosensory cortex. Nat. Commun. 2016, 7, 12540. [Google Scholar] [CrossRef]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef] [PubMed]
- Knuesel, I.; Chicha, L.; Britschgi, M.; Schobel, S.A.; Bodmer, M.; Hellings, J.A.; Toovey, S.; Prinssen, E.P. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 2014, 10, 643–660. [Google Scholar] [CrossRef]
- Tay, T.L.; Sagar; Dautzenberg, J.; Grun, D.; Prinz, M. Unique microglia recovery population revealed by single-cell RNAseq following neurodegeneration. Acta Neuropathol. Commun. 2018, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Bilbo, S.D.; Block, C.L.; Bolton, J.L.; Hanamsagar, R.; Tran, P.K. Beyond infection—Maternal immune activation by environmental factors, microglial development, and relevance for autism spectrum disorders. Exp. Neurol. 2018, 299, 241–251. [Google Scholar] [CrossRef]
- Wu, Z.; Zhou, L.; Sun, L.; Xie, Y.; Xiao, L.; Wang, H.; Wang, G. Brief postpartum separation from offspring promotes resilience to lipopolysaccharide challenge-induced anxiety and depressive-like behaviors and inhibits neuroinflammation in C57BL/6J dams. Brain Behav. Immun. 2021, 95, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Welzel, B.; Loscher, W. Effects of season, daytime, sex, and stress on the incidence, latency, frequency, severity, and duration of neonatal seizures in a rat model of birth asphyxia. Epilepsy Behav. 2023, 147, 109415. [Google Scholar] [CrossRef]
- Reuss, S.; Siebrecht, E.; Stier, U.; Buchholz, H.G.; Bausbacher, N.; Schabbach, N.; Kronfeld, A.; Dieterich, M.; Schreckenberger, M. Modeling Vestibular Compensation: Neural Plasticity Upon Thalamic Lesion. Front. Neurol. 2020, 11, 441. [Google Scholar] [CrossRef]
- Hagberg, H.; Mallard, C.; Ferriero, D.M.; Vannucci, S.J.; Levison, S.W.; Vexler, Z.S.; Gressens, P. The role of inflammation in perinatal brain injury. Nat. Rev. Neurol. 2015, 11, 192–208. [Google Scholar] [CrossRef]
- Lear, B.A.; Lear, C.A.; Dhillon, S.K.; Davidson, J.O.; Gunn, A.J.; Bennet, L. Evolution of grey matter injury over 21 days after hypoxia-ischaemia in preterm fetal sheep. Exp. Neurol. 2023, 363, 114376. [Google Scholar] [CrossRef]
- Dean, D.C., 3rd; Hurley, S.A.; Kecskemeti, S.R.; O’Grady, J.P.; Canda, C.; Davenport-Sis, N.J.; Carlsson, C.M.; Zetterberg, H.; Blennow, K.; Asthana, S.; et al. Association of Amyloid Pathology With Myelin Alteration in Preclinical Alzheimer Disease. JAMA Neurol. 2017, 74, 41–49. [Google Scholar] [CrossRef]
- Kracht, L.; Borggrewe, M.; Eskandar, S.; Brouwer, N.; Chuva de Sousa Lopes, S.M.; Laman, J.D.; Scherjon, S.A.; Prins, J.R.; Kooistra, S.M.; Eggen, B.J.L. Human fetal microglia acquire homeostatic immune-sensing properties early in development. Science 2020, 369, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Block, C.L.; Eroglu, O.; Mague, S.D.; Smith, C.J.; Ceasrine, A.M.; Sriworarat, C.; Blount, C.; Beben, K.A.; Malacon, K.E.; Ndubuizu, N.; et al. Prenatal environmental stressors impair postnatal microglia function and adult behavior in males. Cell Rep. 2022, 40, 111161. [Google Scholar] [CrossRef]
- Franceschini, A.; Capece, M.; Chiozzi, P.; Falzoni, S.; Sanz, J.M.; Sarti, A.C.; Bonora, M.; Pinton, P.; Di Virgilio, F. The P2X7 receptor directly interacts with the NLRP3 inflammasome scaffold protein. FASEB J. 2015, 29, 2450–2461. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.C.; Pastrana, E.; Tavazoie, M.; Doetsch, F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 2009, 12, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Ponomarev, E.D.; Veremeyko, T.; Barteneva, N.; Krichevsky, A.M.; Weiner, H.L. MicroRNA-124 promotes microglia quiescence and suppresses EAE by deactivating macrophages via the C/EBP-alpha-PU.1 pathway. Nat. Med. 2011, 17, 64–70. [Google Scholar] [CrossRef]
- Ponomarev, E.D.; Veremeyko, T.; Weiner, H.L. MicroRNAs are universal regulators of differentiation, activation, and polarization of microglia and macrophages in normal and diseased CNS. Glia 2013, 61, 91–103. [Google Scholar] [CrossRef]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef]
- Trang, T.; Beggs, S.; Salter, M.W. Brain-derived neurotrophic factor from microglia: A molecular substrate for neuropathic pain. Neuron Glia Biol. 2011, 7, 99–108. [Google Scholar] [CrossRef]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef]
- Zhou, Y.; Ulland, T.K.; Colonna, M. TREM2-Dependent Effects on Microglia in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, S.; Luo, H.; Xu, F.; Liang, J.; Ma, C.; Ren, L.; Wang, H.; Hou, Y. Aflatoxin B1 induces microglia cells apoptosis mediated by oxidative stress through NF-kappaB signaling pathway in mice spinal cords. Environ. Toxicol. Pharmacol. 2022, 90, 103794. [Google Scholar] [CrossRef]
- Scheller, M.; Foerster, J.; Heyworth, C.M.; Waring, J.F.; Lohler, J.; Gilmore, G.L.; Shadduck, R.K.; Dexter, T.M.; Horak, I. Altered development and cytokine responses of myeloid progenitors in the absence of transcription factor, interferon consensus sequence binding protein. Blood 1999, 94, 3764–3771. [Google Scholar] [CrossRef]
- Gabriele, L.; Phung, J.; Fukumoto, J.; Segal, D.; Wang, I.M.; Giannakakou, P.; Giese, N.A.; Ozato, K.; Morse, H.C., 3rd. Regulation of apoptosis in myeloid cells by interferon consensus sequence-binding protein. J. Exp. Med. 1999, 190, 411–421. [Google Scholar] [CrossRef]
- de Almeida, M.M.A.; Goodkey, K.; Voronova, A. Regulation of microglia function by neural stem cells. Front. Cell. Neurosci. 2023, 17, 1130205. [Google Scholar] [CrossRef] [PubMed]
- Pluchino, S.; Zanotti, L.; Rossi, B.; Brambilla, E.; Ottoboni, L.; Salani, G.; Martinello, M.; Cattalini, A.; Bergami, A.; Furlan, R.; et al. Neurosphere-derived multipotent precursors promote neuroprotection by an immunomodulatory mechanism. Nature 2005, 436, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Kokaia, Z.; Darsalia, V. Human Neural Stem Cells for Ischemic Stroke Treatment. Results Probl. Cell Differ. 2018, 66, 249–263. [Google Scholar] [CrossRef]
- Gao, M.; Dong, Q.; Yao, H.; Zhang, Y.; Yang, Y.; Dang, Y.; Zhang, H.; Yang, Z.; Xu, M.; Xu, R. Induced neural stem cells modulate microglia activation states via CXCL12/CXCR4 signaling. Brain Behav. Immun. 2017, 59, 288–299. [Google Scholar] [CrossRef]
- Brousse, B.; Mercier, O.; Magalon, K.; Daian, F.; Durbec, P.; Cayre, M. Endogenous neural stem cells modulate microglia and protect against demyelination. Stem Cell Rep. 2021, 16, 1792–1804. [Google Scholar] [CrossRef]
- Dixon, K.J.; Theus, M.H.; Nelersa, C.M.; Mier, J.; Travieso, L.G.; Yu, T.S.; Kernie, S.G.; Liebl, D.J. Endogenous neural stem/progenitor cells stabilize the cortical microenvironment after traumatic brain injury. J. Neurotrauma 2015, 32, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef]
- Neniskyte, U.; Kuliesiute, U.; Vadisiute, A.; Jevdokimenko, K.; Coletta, L.; Deivasigamani, S.; Pamedytyte, D.; Daugelaviciene, N.; Dabkeviciene, D.; Perlas, E.; et al. Phospholipid scramblase Xkr8 is required for developmental axon pruning via phosphatidylserine exposure. EMBO J. 2023, 42, e111790. [Google Scholar] [CrossRef]
- Li, E.; Noda, M.; Doi, Y.; Parajuli, B.; Kawanokuchi, J.; Sonobe, Y.; Takeuchi, H.; Mizuno, T.; Suzumura, A. The neuroprotective effects of milk fat globule-EGF factor 8 against oligomeric amyloid beta toxicity. J. Neuroinflamm. 2012, 9, 148. [Google Scholar] [CrossRef]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef]
- Greer, J.M.; Capecchi, M.R. Hoxb8 is required for normal grooming behavior in mice. Neuron 2002, 33, 23–34. [Google Scholar] [CrossRef]
- Elmore, M.R.; Najafi, A.R.; Koike, M.A.; Dagher, N.N.; Spangenberg, E.E.; Rice, R.A.; Kitazawa, M.; Matusow, B.; Nguyen, H.; West, B.L.; et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 2014, 82, 380–397. [Google Scholar] [CrossRef]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Ginhoux, F.; Lim, S.; Hoeffel, G.; Low, D.; Huber, T. Origin and differentiation of microglia. Front. Cell. Neurosci. 2013, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Badimon, A.; Strasburger, H.J.; Ayata, P.; Chen, X.; Nair, A.; Ikegami, A.; Hwang, P.; Chan, A.T.; Graves, S.M.; Uweru, J.O.; et al. Negative feedback control of neuronal activity by microglia. Nature 2020, 586, 417–423. [Google Scholar] [CrossRef]
- Kana, V.; Desland, F.A.; Casanova-Acebes, M.; Ayata, P.; Badimon, A.; Nabel, E.; Yamamuro, K.; Sneeboer, M.; Tan, I.L.; Flanigan, M.E.; et al. CSF-1 controls cerebellar microglia and is required for motor function and social interaction. J. Exp. Med. 2019, 216, 2265–2281. [Google Scholar] [CrossRef]
- Kierdorf, K.; Prinz, M. Microglia: Same same, but different. J. Exp. Med. 2019, 216, 2223–2225. [Google Scholar] [CrossRef]
- Borst, K.; Dumas, A.A.; Prinz, M. Microglia: Immune and non-immune functions. Immunity 2021, 54, 2194–2208. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Imbeault, S.; Larsson, M.K.; Oliveros, A.; Nilsson, I.A.K.; Codeluppi, S.; Orhan, F.; Bhat, M.; Tufvesson-Alm, M.; Gracias, J.; et al. GRK3 deficiency elicits brain immune activation and psychosis. Mol. Psychiatry 2021, 26, 6820–6832. [Google Scholar] [CrossRef]
- Chitu, V.; Gokhan, S.; Stanley, E.R. Modeling CSF-1 receptor deficiency diseases—How close are we? FEBS J. 2022, 289, 5049–5073. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.T.; Dorman, L.C.; Pan, S.; Vainchtein, I.D.; Han, R.T.; Nakao-Inoue, H.; Taloma, S.E.; Barron, J.J.; Molofsky, A.B.; Kheirbek, M.A.; et al. Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 2020, 182, 388–403.e15. [Google Scholar] [CrossRef]
- Vainchtein, I.D.; Alsema, A.M.; Dubbelaar, M.L.; Grit, C.; Vinet, J.; van Weering, H.R.J.; Al-Izki, S.; Biagini, G.; Brouwer, N.; Amor, S.; et al. Characterizing microglial gene expression in a model of secondary progressive multiple sclerosis. Glia 2023, 71, 588–601. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Roumier, A.; Bechade, C.; Poncer, J.C.; Smalla, K.H.; Tomasello, E.; Vivier, E.; Gundelfinger, E.D.; Triller, A.; Bessis, A. Impaired synaptic function in the microglial KARAP/DAP12-deficient mouse. J. Neurosci. 2004, 24, 11421–11428. [Google Scholar] [CrossRef] [PubMed]
- Otero, K.; Turnbull, I.R.; Poliani, P.L.; Vermi, W.; Cerutti, E.; Aoshi, T.; Tassi, I.; Takai, T.; Stanley, S.L.; Miller, M.; et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat. Immunol. 2009, 10, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Erny, D.; Goldmann, T.; Sander, V.; Schulz, C.; Perdiguero, E.G.; Wieghofer, P.; Heinrich, A.; Riemke, P.; Holscher, C.; et al. Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 2013, 16, 273–280. [Google Scholar] [CrossRef]
- Zheng, H.; Jia, L.; Liu, C.C.; Rong, Z.; Zhong, L.; Yang, L.; Chen, X.F.; Fryer, J.D.; Wang, X.; Zhang, Y.W.; et al. TREM2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J. Neurosci. 2017, 37, 1772–1784. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, C.C.; Atagi, Y.; Chen, X.F.; Jia, L.; Yang, L.; He, W.; Zhang, X.; Kang, S.S.; Rosenberry, T.L.; et al. Opposing roles of the triggering receptor expressed on myeloid cells 2 and triggering receptor expressed on myeloid cells-like transcript 2 in microglia activation. Neurobiol. Aging 2016, 42, 132–141. [Google Scholar] [CrossRef]
- Wu, K.; Byers, D.E.; Jin, X.; Agapov, E.; Alexander-Brett, J.; Patel, A.C.; Cella, M.; Gilfilan, S.; Colonna, M.; Kober, D.L.; et al. TREM-2 promotes macrophage survival and lung disease after respiratory viral infection. J. Exp. Med. 2015, 212, 681–697. [Google Scholar] [CrossRef]
- Qin, Q.; Teng, Z.; Liu, C.; Li, Q.; Yin, Y.; Tang, Y. TREM2, microglia, and Alzheimer’s disease. Mech. Ageing Dev. 2021, 195, 111438. [Google Scholar] [CrossRef]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef]
- Utz, S.G.; See, P.; Mildenberger, W.; Thion, M.S.; Silvin, A.; Lutz, M.; Ingelfinger, F.; Rayan, N.A.; Lelios, I.; Buttgereit, A.; et al. Early Fate Defines Microglia and Non-parenchymal Brain Macrophage Development. Cell 2020, 181, 557–573.e18. [Google Scholar] [CrossRef]
- Cronk, J.C.; Derecki, N.C.; Ji, E.; Xu, Y.; Lampano, A.E.; Smirnov, I.; Baker, W.; Norris, G.T.; Marin, I.; Coddington, N.; et al. Methyl-CpG Binding Protein 2 Regulates Microglia and Macrophage Gene Expression in Response to Inflammatory Stimuli. Immunity 2015, 42, 679–691. [Google Scholar] [CrossRef]
- Schafer, D.P.; Heller, C.T.; Gunner, G.; Heller, M.; Gordon, C.; Hammond, T.; Wolf, Y.; Jung, S.; Stevens, B. Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. eLife 2016, 5, e15224. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef]
- Guo, L.; Bertola, D.R.; Takanohashi, A.; Saito, A.; Segawa, Y.; Yokota, T.; Ishibashi, S.; Nishida, Y.; Yamamoto, G.L.; Franco, J.; et al. Bi-allelic CSF1R Mutations Cause Skeletal Dysplasia of Dysosteosclerosis-Pyle Disease Spectrum and Degenerative Encephalopathy with Brain Malformation. Am. J. Hum. Genet. 2019, 104, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Oosterhof, N.; Chang, I.J.; Karimiani, E.G.; Kuil, L.E.; Jensen, D.M.; Daza, R.; Young, E.; Astle, L.; van der Linde, H.C.; Shivaram, G.M.; et al. Homozygous Mutations in CSF1R Cause a Pediatric-Onset Leukoencephalopathy and Can Result in Congenital Absence of Microglia. Am. J. Hum. Genet. 2019, 104, 936–947. [Google Scholar] [CrossRef]
- Ballasch, I.; Garcia-Garcia, E.; Vila, C.; Perez-Gonzalez, A.; Sancho-Balsells, A.; Fernandez, J.; Soto, D.; Puigdellivol, M.; Gasull, X.; Alberch, J.; et al. Ikzf1 as a novel regulator of microglial homeostasis in inflammation and neurodegeneration. Brain Behav. Immun. 2023, 109, 144–161. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, K.; Rubino, S.J.; Burns, J.C.; Smith, B.A.; McCarl, C.A.; Zhu, J.; Jandreski, L.; Cullen, P.; Carlile, T.M.; Li, A.; et al. MOG autoantibodies trigger a tightly-controlled FcR and BTK-driven microglia proliferative response. Brain 2021, 144, 2361–2374. [Google Scholar] [CrossRef]
- Madore, C.; Nadjar, A.; Delpech, J.C.; Sere, A.; Aubert, A.; Portal, C.; Joffre, C.; Laye, S. Nutritional n-3 PUFAs deficiency during perinatal periods alters brain innate immune system and neuronal plasticity-associated genes. Brain Behav. Immun. 2014, 41, 22–31. [Google Scholar] [CrossRef]
- Delpech, J.C.; Wei, L.; Hao, J.; Yu, X.; Madore, C.; Butovsky, O.; Kaffman, A. Early life stress perturbs the maturation of microglia in the developing hippocampus. Brain Behav. Immun. 2016, 57, 79–93. [Google Scholar] [CrossRef]
- Silvin, A.; Ginhoux, F. Microglia heterogeneity along a spatio-temporal axis: More questions than answers. Glia 2018, 66, 2045–2057. [Google Scholar] [CrossRef]
- Verdonk, F.; Roux, P.; Flamant, P.; Fiette, L.; Bozza, F.A.; Simard, S.; Lemaire, M.; Plaud, B.; Shorte, S.L.; Sharshar, T.; et al. Phenotypic clustering: A novel method for microglial morphology analysis. J. Neuroinflamm. 2016, 13, 153. [Google Scholar] [CrossRef] [PubMed]
- Marin-Teva, J.L.; Dusart, I.; Colin, C.; Gervais, A.; van Rooijen, N.; Mallat, M. Microglia promote the death of developing Purkinje cells. Neuron 2004, 41, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Frank, M.G.; Adhikary, S.; Sobesky, J.L.; Weber, M.D.; Watkins, L.R.; Maier, S.F. The danger-associated molecular pattern HMGB1 mediates the neuroinflammatory effects of methamphetamine. Brain Behav. Immun. 2016, 51, 99–108. [Google Scholar] [CrossRef]
- Liu, T.W.; Chen, C.M.; Chang, K.H. Biomarker of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 4148. [Google Scholar] [CrossRef]
- Dwyer, Z.; Rudyk, C.; Situt, D.; Beauchamp, S.; Abdali, J.; Dinesh, A.; Legancher, N.; Sun, H.; Schlossmacher, M.; Hayley, S.; et al. Microglia depletion prior to lipopolysaccharide and paraquat treatment differentially modulates behavioral and neuronal outcomes in wild type and G2019S LRRK2 knock-in mice. Brain Behav. Immun. -Health 2020, 5, 100079. [Google Scholar] [CrossRef]
- Ganz, T.; Fainstein, N.; Elad, A.; Lachish, M.; Goldfarb, S.; Einstein, O.; Ben-Hur, T. Microbial pathogens induce neurodegeneration in Alzheimer’s disease mice: Protection by microglial regulation. J. Neuroinflamm. 2022, 19, 5. [Google Scholar] [CrossRef]
- Song, W.M.; Colonna, M. The identity and function of microglia in neurodegeneration. Nat. Immunol. 2018, 19, 1048–1058. [Google Scholar] [CrossRef]
- Chen, Y.; Colonna, M. Microglia in Alzheimer’s disease at single-cell level. Are there common patterns in humans and mice? J. Exp. Med. 2021, 218, e20202717. [Google Scholar] [CrossRef]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Villacampa, N.; Heneka, M.T. Microglia: You’ll Never Walk Alone! Immunity 2018, 48, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Braak, H.; Markesbery, W.R. Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship. J. Neuropathol. Exp. Neurol. 2009, 68, 1–14. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Suzuki, A.; Hasebe, R.; Horiuchi, M. Flow Cytometric Detection of PrP(Sc) in Neurons and Glial Cells from Prion-Infected Mouse Brains. J. Virol. 2018, 92, e01457-17. [Google Scholar] [CrossRef]
- Smith, A.M.; Dragunow, M. The human side of microglia. Trends Neurosci. 2014, 37, 125–135. [Google Scholar] [CrossRef]
- Streit, W.J.; Xue, Q.S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef]
- Krauthausen, M.; Kummer, M.P.; Zimmermann, J.; Reyes-Irisarri, E.; Terwel, D.; Bulic, B.; Heneka, M.T.; Muller, M. CXCR3 promotes plaque formation and behavioral deficits in an Alzheimer’s disease model. J. Clin. Investig. 2015, 125, 365–378. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Jay, T.R.; von Saucken, V.E.; Landreth, G.E. TREM2 in Neurodegenerative Diseases. Mol. Neurodegener. 2017, 12, 56. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.F.; Hu, H.; Tan, L.; Yu, J.T. Microglia Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2021, 58, 3388–3404. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Kamphuis, W.; Kooijman, L.; Schetters, S.; Orre, M.; Hol, E.M. Transcriptional profiling of CD11c-positive microglia accumulating around amyloid plaques in a mouse model for Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 1847–1860. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Yin, Z.; Raj, D.; Saiepour, N.; Van Dam, D.; Brouwer, N.; Holtman, I.R.; Eggen, B.J.L.; Moller, T.; Tamm, J.A.; Abdourahman, A.; et al. Immune hyperreactivity of Abeta plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 2017, 55, 115–122. [Google Scholar] [CrossRef]
- Ising, C.; Heneka, M.T. Chronic inflammation: A potential target in tauopathies. Lancet. Neurol. 2023, 22, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Streit, W.J.; Khoshbouei, H.; Bechmann, I. The Role of Microglia in Sporadic Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 79, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Venigalla, M.; Raju, R.; Munch, G. Pharmacological considerations for treating neuroinflammation with curcumin in Alzheimer’s disease. J. Neural Transm. 2022, 129, 755–771. [Google Scholar] [CrossRef] [PubMed]
- Hellwig, S.; Masuch, A.; Nestel, S.; Katzmarski, N.; Meyer-Luehmann, M.; Biber, K. Forebrain microglia from wild-type but not adult 5xFAD mice prevent amyloid-beta plaque formation in organotypic hippocampal slice cultures. Sci. Rep. 2015, 5, 14624. [Google Scholar] [CrossRef]
- Ulrich, J.D.; Finn, M.B.; Wang, Y.; Shen, A.; Mahan, T.E.; Jiang, H.; Stewart, F.R.; Piccio, L.; Colonna, M.; Holtzman, D.M. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol. Neurodegener. 2014, 9, 20. [Google Scholar] [CrossRef]
- Jay, T.R.; Miller, C.M.; Cheng, P.J.; Graham, L.C.; Bemiller, S.; Broihier, M.L.; Xu, G.; Margevicius, D.; Karlo, J.C.; Sousa, G.L.; et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J. Exp. Med. 2015, 212, 287–295. [Google Scholar] [CrossRef]
- Badanjak, K.; Fixemer, S.; Smajic, S.; Skupin, A.; Grunewald, A. The Contribution of Microglia to Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 4676. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-kappaB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed]
- Mouton-Liger, F.; Rosazza, T.; Sepulveda-Diaz, J.; Ieang, A.; Hassoun, S.M.; Claire, E.; Mangone, G.; Brice, A.; Michel, P.P.; Corvol, J.C.; et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 2018, 66, 1736–1751. [Google Scholar] [CrossRef]
- Wang, W.; Lv, Z.; Gao, J.; Liu, M.; Wang, Y.; Tang, C.; Xiang, J. Treadmill exercise alleviates neuronal damage by suppressing NLRP3 inflammasome and microglial activation in the MPTP mouse model of Parkinson’s disease. Brain Res. Bull. 2021, 174, 349–358. [Google Scholar] [CrossRef]
- Jewell, S.; Herath, A.M.; Gordon, R. Inflammasome Activation in Parkinson’s Disease. J. Park. Dis. 2022, 12, S113–S128. [Google Scholar] [CrossRef] [PubMed]
- Tu, H.Y.; Yuan, B.S.; Hou, X.O.; Zhang, X.J.; Pei, C.S.; Ma, Y.T.; Yang, Y.P.; Fan, Y.; Qin, Z.H.; Liu, C.F.; et al. alpha-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell 2021, 20, e13522. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Liu, J.; Wang, B.; Sun, M.; Yang, H. Microglia in the Neuroinflammatory Pathogenesis of Alzheimer’s Disease and Related Therapeutic Targets. Front. Immunol. 2022, 13, 856376. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Bell, S.; Wang, J.; Wen, S.; Baloh, R.H.; Appel, S.H. TDP-43 activates microglia through NF-kappaB and NLRP3 inflammasome. Exp. Neurol. 2015, 273, 24–35. [Google Scholar] [CrossRef]
- Borhani-Haghighi, M.; Mohamadi, Y.; Kashani, I.R. In utero transplantation of neural stem cells ameliorates maternal inflammation-induced prenatal white matter injury. J. Cell. Biochem. 2019, 120, 12785–12795. [Google Scholar] [CrossRef]
- Koutsoudaki, P.N.; Papastefanaki, F.; Stamatakis, A.; Kouroupi, G.; Xingi, E.; Stylianopoulou, F.; Matsas, R. Neural stem/progenitor cells differentiate into oligodendrocytes, reduce inflammation, and ameliorate learning deficits after transplantation in a mouse model of traumatic brain injury. Glia 2016, 64, 763–779. [Google Scholar] [CrossRef]
- Gagne, J.J.; Power, M.C. Anti-inflammatory drugs and risk of Parkinson disease: A meta-analysis. Neurology 2010, 74, 995–1002. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Raymond, L.A.; Andre, V.M.; Cepeda, C.; Gladding, C.M.; Milnerwood, A.J.; Levine, M.S. Pathophysiology of Huntington’s disease: Time-dependent alterations in synaptic and receptor function. Neuroscience 2011, 198, 252–273. [Google Scholar] [CrossRef]
- Sigfridsson, E.; Marangoni, M.; Hardingham, G.E.; Horsburgh, K.; Fowler, J.H. Deficiency of Nrf2 exacerbates white matter damage and microglia/macrophage levels in a mouse model of vascular cognitive impairment. J. Neuroinflamm. 2020, 17, 367. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.; Kim, H.S.; Jeon, I.; Noh, J.E.; Park, H.J.; Lee, S.; Park, I.H.; Stevanato, L.; Hicks, C.; Corteling, R.; et al. Implantation of the clinical-grade human neural stem cell line, CTX0E03, rescues the behavioral and pathological deficits in the quinolinic acid-lesioned rodent model of Huntington’s disease. Stem Cells 2020, 38, 936–947. [Google Scholar] [CrossRef]
- Ma, L.; Morton, A.J.; Nicholson, L.F. Microglia density decreases with age in a mouse model of Huntington’s disease. Glia 2003, 43, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Li, J.; Du, P.; Jin, W.; Gao, G.; Cui, D. Senile plaques in Alzheimer’s disease arise from Abeta- and Cathepsin D-enriched mixtures leaking out during intravascular haemolysis and microaneurysm rupture. FEBS Lett. 2023, 597, 1007–1040. [Google Scholar] [CrossRef]
- Li, D.; Si, J.; Guo, Y.; Liu, B.; Chen, X.; Qi, K.; Yang, S.; Ji, E. Danggui-Buxue decoction alleviated vascular senescence in mice exposed to chronic intermittent hypoxia through activating the Nrf2/HO-1 pathway. Pharm. Biol. 2023, 61, 1041–1053. [Google Scholar] [CrossRef]
- Kim, B.W.; Koppula, S.; Kumar, H.; Park, J.Y.; Kim, I.W.; More, S.V.; Kim, I.S.; Han, S.D.; Kim, S.K.; Yoon, S.H.; et al. alpha-Asarone attenuates microglia-mediated neuroinflammation by inhibiting NF kappa B activation and mitigates MPTP-induced behavioral deficits in a mouse model of Parkinson’s disease. Neuropharmacology 2015, 97, 46–57. [Google Scholar] [CrossRef]
- Yang, W.; Chen, Y.H.; Liu, H.; Qu, H.D. Neuroprotective effects of piperine on the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease mouse model. Int. J. Mol. Med. 2015, 36, 1369–1376. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef] [PubMed]
- Minter, M.R.; Taylor, J.M.; Crack, P.J. The contribution of neuroinflammation to amyloid toxicity in Alzheimer’s disease. J. Neurochem. 2016, 136, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Yanpallewar, S.; Fulgenzi, G.; Tomassoni-Ardori, F.; Barrick, C.; Tessarollo, L. Delayed onset of inherited ALS by deletion of the BDNF receptor TrkB.T1 is non-cell autonomous. Exp. Neurol. 2021, 337, 113576. [Google Scholar] [CrossRef] [PubMed]
- Kobashi, S.; Terashima, T.; Katagi, M.; Nakae, Y.; Okano, J.; Suzuki, Y.; Urushitani, M.; Kojima, H. Transplantation of M2-Deviated Microglia Promotes Recovery of Motor Function after Spinal Cord Injury in Mice. Mol. Ther. 2020, 28, 254–265. [Google Scholar] [CrossRef]
- Drake, S.S.; Zaman, A.; Gianfelice, C.; Hua, E.M.; Heale, K.; Afanasiev, E.; Klement, W.; Stratton, J.A.; Prat, A.; Zandee, S.; et al. Senolytic treatment diminishes microglia and decreases severity of experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2024, 21, 283. [Google Scholar] [CrossRef]
- Chen, W.; Guo, C.; Huang, S.; Jia, Z.; Wang, J.; Zhong, J.; Ge, H.; Yuan, J.; Chen, T.; Liu, X.; et al. MitoQ attenuates brain damage by polarizing microglia towards the M2 phenotype through inhibition of the NLRP3 inflammasome after ICH. Pharmacol. Res. 2020, 161, 105122. [Google Scholar] [CrossRef]
- Zeng, F.; Li, Y.; Li, X.; Gu, X.; Cao, Y.; Cheng, S.; Tian, H.; Mei, R.; Mei, X. Microglia overexpressing brain-derived neurotrophic factor promote vascular repair and functional recovery in mice after spinal cord injury. Neural Regen. Res. 2024, 21, 365–376. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Easley-Neal, C.; Foreman, O.; Sharma, N.; Zarrin, A.A.; Weimer, R.M. CSF1R Ligands IL-34 and CSF1 Are Differentially Required for Microglia Development and Maintenance in White and Gray Matter Brain Regions. Front. Immunol. 2019, 10, 2199. [Google Scholar] [CrossRef]
- De, I.; Maklakova, V.; Litscher, S.; Boyd, M.M.; Klemm, L.C.; Wang, Z.; Kendziorski, C.; Collier, L.S. Microglial responses to CSF1 overexpression do not promote the expansion of other glial lineages. J. Neuroinflamm. 2021, 18, 162. [Google Scholar] [CrossRef]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. Microglia Are Critical in Host Defense against Prion Disease. J. Virol. 2018, 92, e00549-18. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, W.; Langosch, J.M.; Gebicke-Haerter, P.J.; Illes, P. Characterization and possible function of adenosine 5’-triphosphate receptors in activated rat microglia. Br. J. Pharmacol. 1994, 111, 942–950. [Google Scholar] [CrossRef]
- Illes, P.; Norenberg, W.; Gebicke-Haerter, P.J. Molecular mechanisms of microglial activation. B. Voltage- and purinoceptor-operated channels in microglia. Neurochem. Int. 1996, 29, 13–24. [Google Scholar] [CrossRef]
- McLarnon, J.G.; Zhang, L.; Goghari, V.; Lee, Y.B.; Walz, W.; Krieger, C.; Kim, S.U. Effects of ATP and elevated K+ on K+ currents and intracellular Ca2+ in human microglia. Neuroscience 1999, 91, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Z.; Chen, M.; Ping, J.; Dunn, P.; Lv, J.; Jiao, B.; Burnstock, G. Microglial morphology and its transformation after challenge by extracellular ATP in vitro. J. Neurosci. Res. 2006, 83, 91–101. [Google Scholar] [CrossRef]
- Wollmer, M.A.; Lucius, R.; Wilms, H.; Held-Feindt, J.; Sievers, J.; Mentlein, R. ATP and adenosine induce ramification of microglia in vitro. J. Neuroimmunol. 2001, 115, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Boucsein, C.; Zacharias, R.; Farber, K.; Pavlovic, S.; Hanisch, U.K.; Kettenmann, H. Purinergic receptors on microglial cells: Functional expression in acute brain slices and modulation of microglial activation in vitro. Eur. J. Neurosci. 2003, 17, 2267–2276. [Google Scholar] [CrossRef]
- Ogata, T.; Chuai, M.; Morino, T.; Yamamoto, H.; Nakamura, Y.; Schubert, P. Adenosine triphosphate inhibits cytokine release from lipopolysaccharide-activated microglia via P2y receptors. Brain Res. 2003, 981, 174–183. [Google Scholar] [CrossRef]
- Seo, D.R.; Kim, S.Y.; Kim, K.Y.; Lee, H.G.; Moon, J.H.; Lee, J.S.; Lee, S.H.; Kim, S.U.; Lee, Y.B. Cross talk between P2 purinergic receptors modulates extracellular ATP-mediated interleukin-10 production in rat microglial cells. Exp. Mol. Med. 2008, 40, 19–26. [Google Scholar] [CrossRef]
- Bianco, F.; Fumagalli, M.; Pravettoni, E.; D’Ambrosi, N.; Volonte, C.; Matteoli, M.; Abbracchio, M.P.; Verderio, C. Pathophysiological roles of extracellular nucleotides in glial cells: Differential expression of purinergic receptors in resting and activated microglia. Brain Res. Brain Res. Rev. 2005, 48, 144–156. [Google Scholar] [CrossRef]
- Morigiwa, K.; Quan, M.; Murakami, M.; Yamashita, M.; Fukuda, Y. P2 Purinoceptor expression and functional changes of hypoxia-activated cultured rat retinal microglia. Neurosci. Lett. 2000, 282, 153–156. [Google Scholar] [CrossRef]
- Cavaliere, F.; Dinkel, K.; Reymann, K. Microglia response and P2 receptor participation in oxygen/glucose deprivation-induced cortical damage. Neuroscience 2005, 136, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Pan, W.; Xu, Y.; Zhang, J.; Wan, J.; Jiang, H. Microglia-Mediated Neuroinflammation: A Potential Target for the Treatment of Cardiovascular Diseases. J. Inflamm. Res. 2022, 15, 3083–3094. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, J.; Wang, C.; Hu, H.; Li, X.; Xue, M.; Liu, J.; Cheng, W.; Wang, Y.; Li, Y.; et al. Microglial Mincle receptor in the PVN contributes to sympathetic hyperactivity in acute myocardial infarction rat. J. Cell. Mol. Med. 2019, 23, 112–125. [Google Scholar] [CrossRef]
- Metz, L.M.; Eliasziw, M. Trial of Minocycline in Clinically Isolated Syndrome of Multiple Sclerosis. N. Engl. J. Med. 2017, 377, 789. [Google Scholar] [CrossRef]
- Gordon, P.H.; Moore, D.H.; Miller, R.G.; Florence, J.M.; Verheijde, J.L.; Doorish, C.; Hilton, J.F.; Spitalny, G.M.; MacArthur, R.B.; Mitsumoto, H.; et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: A phase III randomised trial. Lancet Neurol. 2007, 6, 1045–1053. [Google Scholar] [CrossRef]
- Vaughn, A.C.; Cooper, E.M.; DiLorenzo, P.M.; O’Loughlin, L.J.; Konkel, M.E.; Peters, J.H.; Hajnal, A.; Sen, T.; Lee, S.H.; de La Serre, C.B.; et al. Energy-dense diet triggers changes in gut microbiota, reorganization of gut-brain vagal communication and increases body fat accumulation. Acta Neurobiol. Exp. 2017, 77, 18–30. [Google Scholar] [CrossRef]
- Yew, W.P.; Djukic, N.D.; Jayaseelan, J.S.P.; Walker, F.R.; Roos, K.A.A.; Chataway, T.K.; Muyderman, H.; Sims, N.R. Early treatment with minocycline following stroke in rats improves functional recovery and differentially modifies responses of peri-infarct microglia and astrocytes. J. Neuroinflamm. 2019, 16, 6. [Google Scholar] [CrossRef]
- Rooney, S.; Sah, A.; Unger, M.S.; Kharitonova, M.; Sartori, S.B.; Schwarzer, C.; Aigner, L.; Kettenmann, H.; Wolf, S.A.; Singewald, N. Neuroinflammatory alterations in trait anxiety: Modulatory effects of minocycline. Transl. Psychiatry 2020, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Sah, A.; Rooney, S.; Kharitonova, M.; Sartori, S.B.; Wolf, S.A.; Singewald, N. Enriched Environment Attenuates Enhanced Trait Anxiety in Association with Normalization of Aberrant Neuro-Inflammatory Events. Int. J. Mol. Sci. 2022, 23, 13052. [Google Scholar] [CrossRef] [PubMed]
- Leone, C.; Le Pavec, G.; Meme, W.; Porcheray, F.; Samah, B.; Dormont, D.; Gras, G. Characterization of human monocyte-derived microglia-like cells. Glia 2006, 54, 183–192. [Google Scholar] [CrossRef]
- Tufekci, K.U.; Ercan, I.; Isci, K.B.; Olcum, M.; Tastan, B.; Gonul, C.P.; Genc, K.; Genc, S. Sulforaphane inhibits NLRP3 inflammasome activation in microglia through Nrf2-mediated miRNA alteration. Immunol. Lett. 2021, 233, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Althafar, Z.M. Targeting Microglia in Alzheimer’s Disease: From Molecular Mechanisms to Potential Therapeutic Targets for Small Molecules. Molecules 2022, 27, 4124. [Google Scholar] [CrossRef]
- Gehrmann, J.; Banati, R.B. Microglial turnover in the injured CNS: Activated microglia undergo delayed DNA fragmentation following peripheral nerve injury. J. Neuropathol. Exp. Neurol. 1995, 54, 680–688. [Google Scholar] [CrossRef]
- Ogata, T.; Schubert, P. Programmed cell death in rat microglia is controlled by extracellular adenosine. Neurosci. Lett. 1996, 218, 91–94. [Google Scholar] [CrossRef]
- Petkovic, F.; Campbell, I.L.; Gonzalez, B.; Castellano, B. Astrocyte-targeted production of interleukin-6 reduces astroglial and microglial activation in the cuprizone demyelination model: Implications for myelin clearance and oligodendrocyte maturation. Glia 2016, 64, 2104–2119. [Google Scholar] [CrossRef]
- Petkovic, F.; Campbell, I.L.; Gonzalez, B.; Castellano, B. Reduced cuprizone-induced cerebellar demyelination in mice with astrocyte-targeted production of IL-6 is associated with chronically activated, but less responsive microglia. J. Neuroimmunol. 2017, 310, 97–102. [Google Scholar] [CrossRef]
- Desai, A.; Park, T.; Barnes, J.; Kevala, K.; Chen, H.; Kim, H.Y. Reduced acute neuroinflammation and improved functional recovery after traumatic brain injury by alpha-linolenic acid supplementation in mice. J. Neuroinflamm. 2016, 13, 253. [Google Scholar] [CrossRef] [PubMed]
- Souto, N.S.; Claudia Monteiro Braga, A.; Lutchemeyer de Freitas, M.; Rechia Fighera, M.; Royes, L.F.F.; Schneider Oliveira, M.; Furian, A.F. Aflatoxin B1 reduces non-enzymatic antioxidant defenses and increases protein kinase C activation in the cerebral cortex of young rats. Nutr. Neurosci. 2018, 21, 268–275. [Google Scholar] [CrossRef]
- Baldissera, M.D.; Souza, C.F.; Zeppenfeld, C.C.; Descovi, S.N.; Moreira, K.L.S.; da Rocha, M.; da Veiga, M.L.; da Silva, A.S.; Baldisserotto, B. Aflatoxin B(1)-contaminated diet disrupts the blood-brain barrier and affects fish behavior: Involvement of neurotransmitters in brain synaptosomes. Environ. Toxicol. Pharmacol. 2018, 60, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Alsayyah, A.; ElMazoudy, R.; Al-Namshan, M.; Al-Jafary, M.; Alaqeel, N. Chronic neurodegeneration by aflatoxin B1 depends on alterations of brain enzyme activity and immunoexpression of astrocyte in male rats. Ecotoxicol. Environ. Saf. 2019, 182, 109407. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, J.Y.; You, S.; Song, G.; Lim, W. Neurotoxic effects of aflatoxin B1 on human astrocytes in vitro and on glial cell development in zebrafish in vivo. J. Hazard. Mater. 2020, 386, 121639. [Google Scholar] [CrossRef]
- Mehrzad, J.; Malvandi, A.M.; Alipour, M.; Hosseinkhani, S. Environmentally relevant level of aflatoxin B(1) elicits toxic pro-inflammatory response in murine CNS-derived cells. Toxicol. Lett. 2017, 279, 96–106. [Google Scholar] [CrossRef]
- Mehrzad, J.; Hosseinkhani, S.; Malvandi, A.M. Human Microglial Cells Undergo Proapoptotic Induction and Inflammatory Activation upon in vitro Exposure to a Naturally Occurring Level of Aflatoxin B1. Neuroimmunomodulation 2018, 25, 176–183. [Google Scholar] [CrossRef]
- Sunkaria, A.; Wani, W.Y.; Sharma, D.R.; Gill, K.D. Dichlorvos exposure results in activation induced apoptotic cell death in primary rat microglia. Chem. Res. Toxicol. 2012, 25, 1762–1770. [Google Scholar] [CrossRef]
- Porte Alcon, S.; Gorojod, R.M.; Kotler, M.L. Regulated Necrosis Orchestrates Microglial Cell Death in Manganese-Induced Toxicity. Neuroscience 2018, 393, 206–225. [Google Scholar] [CrossRef]
- Zhan, J.; Liu, M.; Su, X.; Zhan, K.; Zhang, C.; Zhao, G. Effects of alfalfa flavonoids on the production performance, immune system, and ruminal fermentation of dairy cows. Asian-Australas. J. Anim. Sci. 2017, 30, 1416–1424. [Google Scholar] [CrossRef]
- Dodiya, H.B.; Kuntz, T.; Shaik, S.M.; Baufeld, C.; Leibowitz, J.; Zhang, X.; Gottel, N.; Zhang, X.; Butovsky, O.; Gilbert, J.A.; et al. Sex-specific effects of microbiome perturbations on cerebral Abeta amyloidosis and microglia phenotypes. J. Exp. Med. 2019, 216, 1542–1560. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Ruiz, A.; Mosley, M.; George, A.J.; Mussaji, L.F.; Fullerton, E.F.; Ruszkowski, E.M.; Jacobs, A.J.; Gewirtz, A.T.; Chassaing, B.; Forger, N.G. The microbiota influences cell death and microglial colonization in the perinatal mouse brain. Brain Behav. Immun. 2018, 67, 218–229. [Google Scholar] [CrossRef]
- Lee, J.H.; Chen, T.W.; Hsu, C.H.; Yen, Y.H.; Yang, J.C.; Cheng, A.L.; Sasaki, S.I.; Chiu, L.L.; Sugihara, M.; Ishizuka, T.; et al. A phase I study of pexidartinib, a colony-stimulating factor 1 receptor inhibitor, in Asian patients with advanced solid tumors. Investig. New Drugs 2020, 38, 99–110. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Gluck, L.; Martin, L.P.; Olszanski, A.J.; Tolcher, A.W.; Ngarmchamnanrith, G.; Rasmussen, E.; Amore, B.M.; Nagorsen, D.; Hill, J.S.; et al. First-in-Human Study of AMG 820, a Monoclonal Anti-Colony-Stimulating Factor 1 Receptor Antibody, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5703–5710. [Google Scholar] [CrossRef] [PubMed]
- Rao, Y.; Du, S.; Yang, B.; Wang, Y.; Li, Y.; Li, R.; Zhou, T.; Du, X.; He, Y.; Wang, Y.; et al. NeuroD1 induces microglial apoptosis and cannot induce microglia-to-neuron cross-lineage reprogramming. Neuron 2021, 109, 4094–4108.e5. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Waldkirch, S.; d’Errico, P.; Sauer, J.F.; Erny, D.; Savanthrapadian, S.; Loreth, D.; Katzmarski, N.; Blank, T.; Bartos, M.; Prinz, M.; et al. Seed-induced Abeta deposition is modulated by microglia under environmental enrichment in a mouse model of Alzheimer’s disease. EMBO J. 2018, 37, 167–182. [Google Scholar] [CrossRef]
- Xu, H.; Gelyana, E.; Rajsombath, M.; Yang, T.; Li, S.; Selkoe, D. Environmental Enrichment Potently Prevents Microglia-Mediated Neuroinflammation by Human Amyloid beta-Protein Oligomers. J. Neurosci. 2016, 36, 9041–9056. [Google Scholar] [CrossRef]
- Choi, S.H.; Veeraraghavalu, K.; Lazarov, O.; Marler, S.; Ransohoff, R.M.; Ramirez, J.M.; Sisodia, S.S. Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron 2008, 59, 568–580. [Google Scholar] [CrossRef]
- Jurgens, H.A.; Johnson, R.W. Environmental enrichment attenuates hippocampal neuroinflammation and improves cognitive function during influenza infection. Brain Behav. Immun. 2012, 26, 1006–1016. [Google Scholar] [CrossRef]
- Ziv, Y.; Ron, N.; Butovsky, O.; Landa, G.; Sudai, E.; Greenberg, N.; Cohen, H.; Kipnis, J.; Schwartz, M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 2006, 9, 268–275. [Google Scholar] [CrossRef]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef]
- DiPatre, P.L.; Gelman, B.B. Microglial cell activation in aging and Alzheimer disease: Partial linkage with neurofibrillary tangle burden in the hippocampus. J. Neuropathol. Exp. Neurol. 1997, 56, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Edler, M.K.; Mhatre-Winters, I.; Richardson, J.R. Microglia in Aging and Alzheimer’s Disease: A Comparative Species Review. Cells 2021, 10, 1138. [Google Scholar] [CrossRef]
- Edler, M.K.; Sherwood, C.C.; Meindl, R.S.; Hopkins, W.D.; Ely, J.J.; Erwin, J.M.; Mufson, E.J.; Hof, P.R.; Raghanti, M.A. Aged chimpanzees exhibit pathologic hallmarks of Alzheimer’s disease. Neurobiol. Aging 2017, 59, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Robillard, K.N.; Lee, K.M.; Chiu, K.B.; MacLean, A.G. Glial cell morphological and density changes through the lifespan of rhesus macaques. Brain Behav. Immun. 2016, 55, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Shobin, E.; Bowley, M.P.; Estrada, L.I.; Heyworth, N.C.; Orczykowski, M.E.; Eldridge, S.A.; Calderazzo, S.M.; Mortazavi, F.; Moore, T.L.; Rosene, D.L. Microglia activation and phagocytosis: Relationship with aging and cognitive impairment in the rhesus monkey. GeroScience 2017, 39, 199–220. [Google Scholar] [CrossRef]
- Sharaf, A.; Krieglstein, K.; Spittau, B. Distribution of microglia in the postnatal murine nigrostriatal system. Cell Tissue Res. 2013, 351, 373–382. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Pan, L.; Sun, C.; Ma, C.; Pan, H. Balancing Microglial Density and Activation in Central Nervous System Development and Disease. Curr. Issues Mol. Biol. 2025, 47, 344. https://doi.org/10.3390/cimb47050344
Wang S, Pan L, Sun C, Ma C, Pan H. Balancing Microglial Density and Activation in Central Nervous System Development and Disease. Current Issues in Molecular Biology. 2025; 47(5):344. https://doi.org/10.3390/cimb47050344
Chicago/Turabian StyleWang, Shunqi, Liangjing Pan, Chong Sun, Chaolin Ma, and Haili Pan. 2025. "Balancing Microglial Density and Activation in Central Nervous System Development and Disease" Current Issues in Molecular Biology 47, no. 5: 344. https://doi.org/10.3390/cimb47050344
APA StyleWang, S., Pan, L., Sun, C., Ma, C., & Pan, H. (2025). Balancing Microglial Density and Activation in Central Nervous System Development and Disease. Current Issues in Molecular Biology, 47(5), 344. https://doi.org/10.3390/cimb47050344