
Vascular Endothelial Growth Factor Receptors in the Vascularization of Pancreatic Tumors: Implications for Prognosis and Therapy



Abstract
1. Introduction
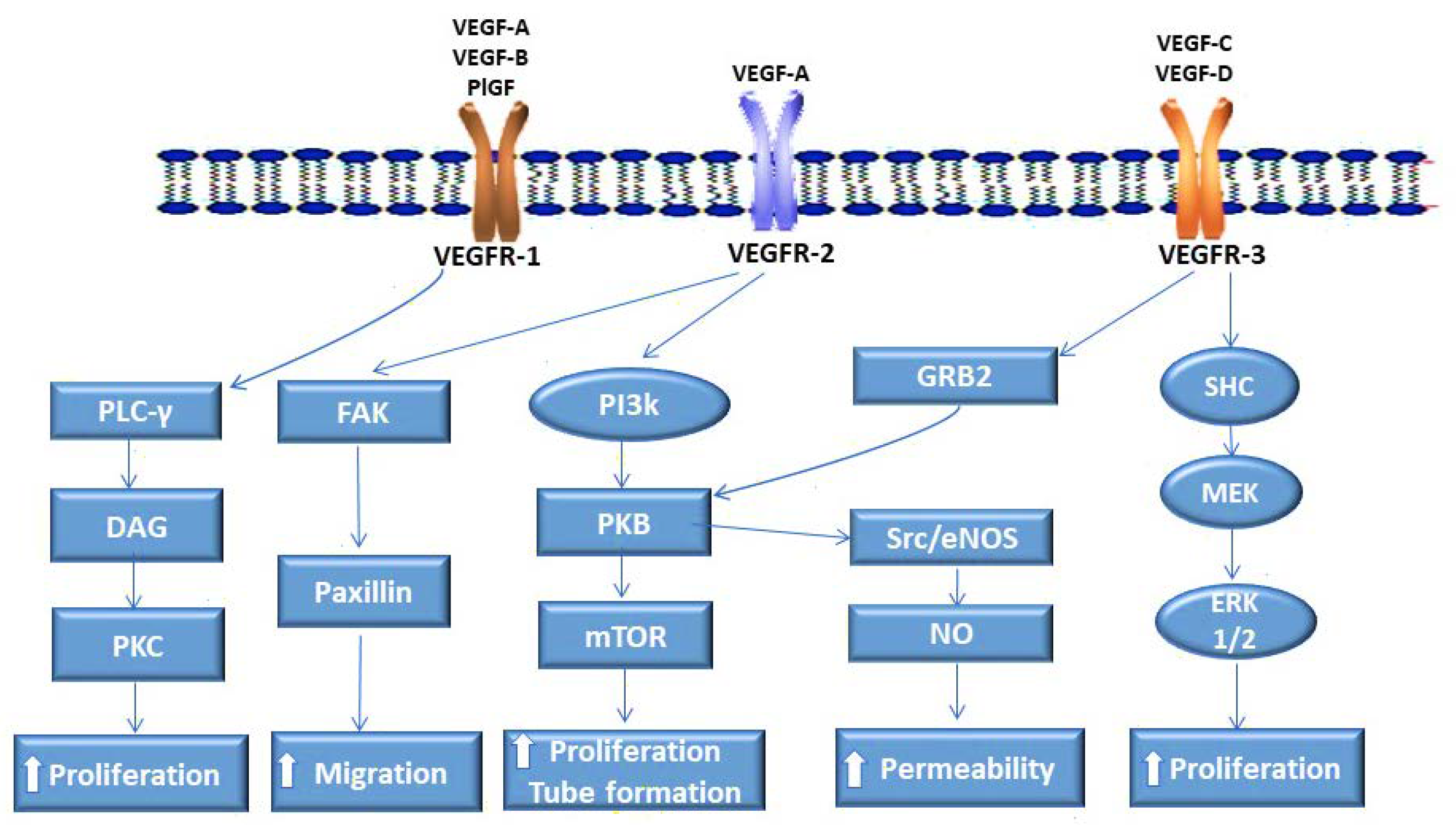
2. VEGF Receptors and Their Ligands
2.1. VEGFR-1 Enables Pancreatic Cancer Vascularization
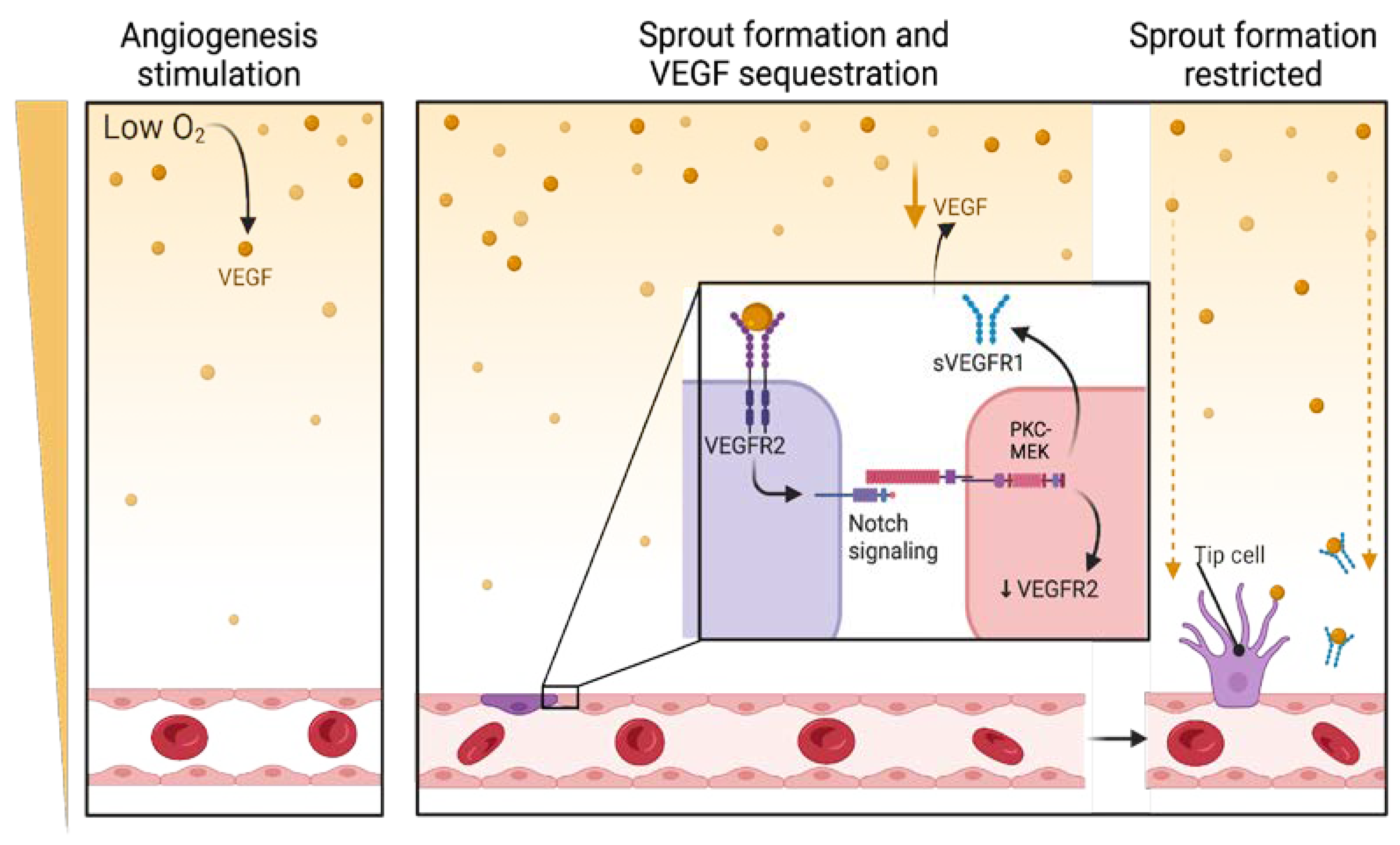
2.2. VEGFR-2 Plays a Pivotal Role in Pancreatic Cancer Angiogenesis
2.3. VEGFR-3 in the Lymphatic Dissemination of Pancreatic Cancer
3. VEGF Receptor Targeting in Pancreatic Cancer
Recent Developments in VEGFR Targeting in Pancreatic Cancer
4. VEGFR-1, -2, and -3 Variants in Pancreatic Cancer Therapy
4.1. Regulation of VEGFRs Through Alternative Splicing
4.2. Potential Role of VEGFR Splice Variants in Pancreatic Cancer Therapy
4.2.1. sVEGFR-1 Antibodies Have Potential Therapeutic Application
4.2.2. Role of sVEGFR-2 in Pancreatic Cancer Treatment
4.2.3. Potential of sVEGFR-3 in Cancer Treatment
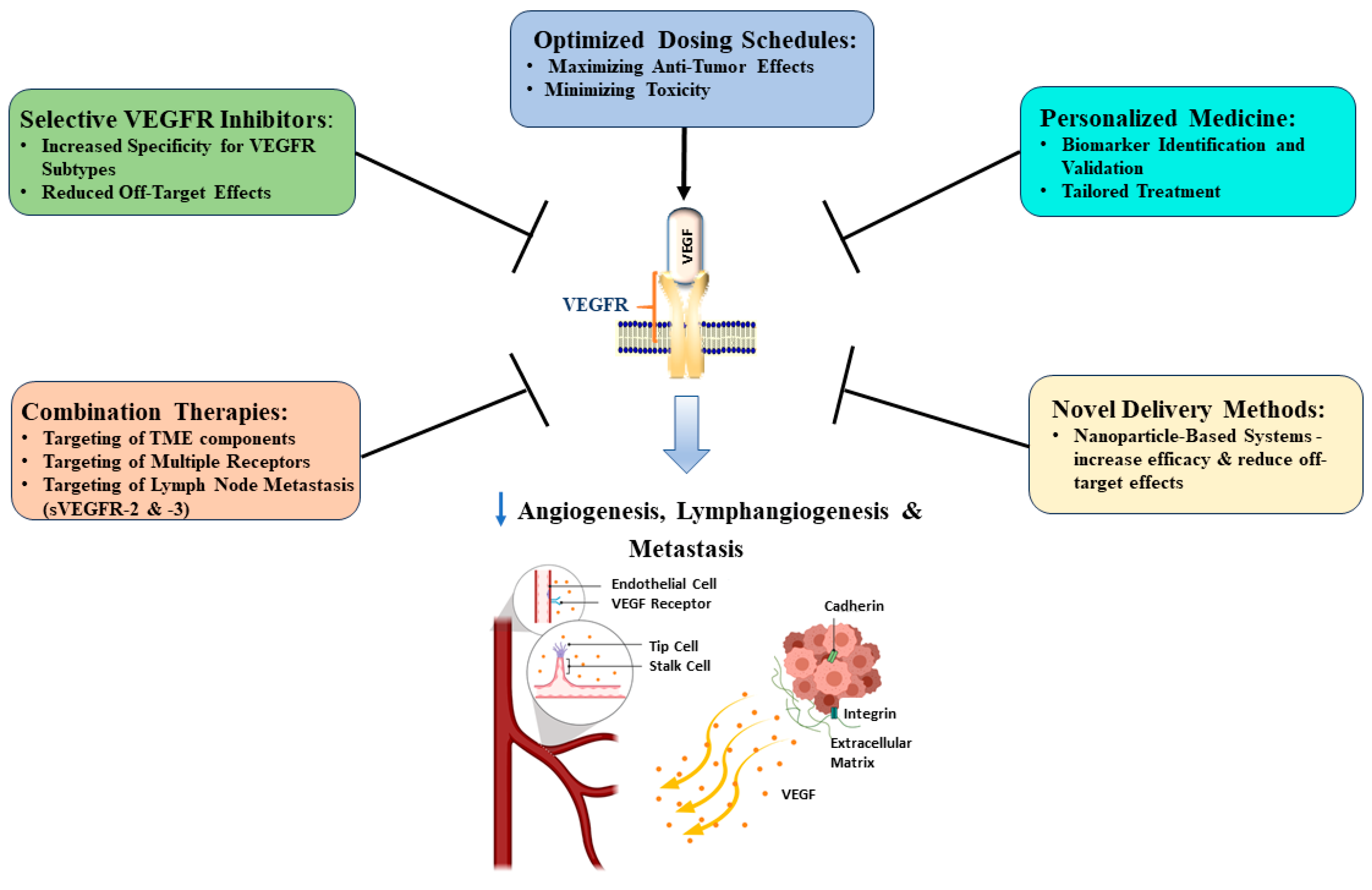
4.3. Proposed Approaches to Improve VEGFR Targeting in Pancreatic Cancer Treatment
5. VEGFR Role in Pancreatic Cancer Prognosis
Potential Biomarkers for Monitoring Treatment Effectiveness
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Yeo, D.; Giardina, C.; Saxena, P.; Rasko, J.E.J. The next wave of cellular immunotherapies in pancreatic cancer. Mol. Ther. Oncolytics 2022, 24, 561–576. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Ervik, M.; Lam, F.; Laversanne, M.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2024. Available online: https://gco.iarc.who.int/today (accessed on 29 January 2025).
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Ferrara, N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am. J. Physiol.-Cell Physiol. 2001, 280, C1358–C1366. [Google Scholar] [CrossRef]
- Mabeta, P.; Pepper, M.S. Manipulating the tumor microenvironment: Opportunities for therapeutic targeting. Front. Anti-Cancer Drug Discov. 2017, 8, 46–71. [Google Scholar]
- Marks, E.C.; Wilkinson, T.M.; Frampton, C.M.; Skelton, L.; Pilbrow, A.P.; Yandle, T.G.; Pemberton, C.J.; Doughty, R.N.; Whalley, G.A.; Ellis, C.J.; et al. Plasma levels of soluble VEGF receptor isoforms, circulating pterins and VEGF system SNPs as prognostic biomarkers in patients with acute coronary syndromes. BMC Cardiovasc. Disord. 2018, 18, 169. [Google Scholar] [CrossRef] [PubMed]
- Stuttfeld, E.; Ballmer-Hofer, K. Structure and function of VEGF receptors. IUBMB Life 2009, 61, 915–922. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef]
- Costache, M.I.; Ioana, M.; Iordache, S.; Ene, D.; Costache, C.A.; Săftoiu, A. VEGF Expression in Pancreatic Cancer and Other Malignancies: A Review of the Literature. Romanian J. Intern. Med. 2015, 53, 199–208. [Google Scholar] [CrossRef]
- Xelwa, N.; Candy, G.P.; Devar, J.; Omoshoro-Jones, J.; Smith, M.; Nweke, E.E. Targeting Growth Factor Signaling Pathways in Pancreatic Cancer: Towards Inhibiting Chemoresistance. Front. Oncol. 2021, 11, 683788. [Google Scholar] [CrossRef]
- Craven, K.E.; Gore, J.; Korc, M. Overview of pre-clinical and clinical studies targeting angiogenesis in pancreatic ductal adenocarcinoma. Cancer Lett. 2016, 381, 201–210. [Google Scholar] [CrossRef]
- An, Y.-F.; Pu, N.; Jia, J.-B.; Wang, W.-Q.; Liu, L. Therapeutic advances targeting tumor angiogenesis in pancreatic cancer: Current dilemmas and future directions. Biochim. Biophys. Acta (BBA) Rev. Cancer 2023, 1878, 188958. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Ullrich, A. Growth factor receptor tyrosine kinases. Annu. Rev. Biochem. 1988, 57, 443–478. [Google Scholar] [CrossRef]
- Shibuya, M. Structure and dual function of vascular endothelial growth factor receptor-1 (Flt-1). Int. J. Biochem. Cell Biol. 2001, 33, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Wey, J.S.; Fan, F.; Gray, M.J.; Bauer, T.W.; McCarty, M.F.; Somcio, R.; Liu, W.; Evans, D.B.; Wu, Y.; Hicklin, D.J.; et al. Vascular endothelial growth factor receptor-1 promotes migration and invasion in pancreatic carcinoma cell lines. Cancer 2005, 104, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L. VEGF receptor signal transduction–A brief update. Vasc. Pharmacol. 2016, 86, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Bando, H.; Weich, H.A.; Brokelmann, M.; Horiguchi, S.; Funata, N.; Ogawa, T.; Toi, M. Association between intratumoral free and total VEGF, soluble VEGFR-1, VEGFR-2 and prognosis in breast cancer. Br. J. Cancer 2005, 92, 553–561. [Google Scholar] [CrossRef]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.-K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis selectively requires the p110α isoform of PI3K to control endothelial cell migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef]
- Göhrig, A.; Hilfenhaus, G.; Rosseck, F.; Welzel, M.; Moser, B.; Barbone, G.; Kunze, C.A.; Rein, J.; Wilken, G.; Böhmig, M.; et al. Placental growth factor promotes neural invasion and predicts disease prognosis in resectable pancreatic cancer. J. Exp. Clin. Cancer Res. 2024, 43, 153. [Google Scholar] [CrossRef]
- Weddell, J.C.; Chen, S.; Imoukhuede, P. VEGFR1 promotes cell migration and proliferation through PLCγ and PI3K pathways. NPJ Syst. Biol. Appl. 2017, 4, 1. [Google Scholar] [CrossRef]
- Korc, M. Pathways for aberrant angiogenesis in pancreatic cancer. Mol. Cancer 2003, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Lee, J.; Yun, J.H.; Choi, C.; Cho, S.; Kim, S.J.; Kim, J.H. Autocrine DUSP28 signaling mediates pancreatic cancer malignancy via regulation of PDGF-A. Sci. Rep. 2017, 7, 12760. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Sasaki, T.; Kuwada, Y.; Murakami, M.; Yamasaki, S.; Chayama, K. Expressions of angiogenic factors in pancreatic ductal carcinoma: A correlative study with clinicopathologic parameters and patient survival. Pancreas 2003, 26, 344–349. [Google Scholar] [CrossRef]
- Hoffmann, A.C.; Mori, R.; Vallbohmer, D.; Brabender, J.; Klein, E.; Drebber, U.; Baldus, S.E.; Cooc, J.; Azuma, M.; Metzger, R.; et al. High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia 2008, 10, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Sampedro, A.; Gaggia, G.; Ney, A.; Mahamed, I.; Acedo, P. The state-of-the-art of phase II/III clinical trials for targeted pancreatic cancer therapies. J. Clin. Med. 2021, 10, 566. [Google Scholar] [CrossRef]
- Jakobsson, L.; Franco, C.A.; Bentley, K.; Collins, R.T.; Ponsioen, B.; Aspalter, I.M.; Rosewell, I.; Busse, M.; Thurston, G.; Medvinsky, A.; et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat. Cell Biol. 2010, 12, 943–953. [Google Scholar] [CrossRef]
- Fukasawa, M.; Korc, M. Vascular endothelial growth factor-trap suppresses tumorigenicity of multiple pancreatic cancer cell lines. Clin. Cancer Res. 2004, 10, 3327–3332. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamaguchi, S.; Chida, K.; Shibuya, M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-γ and Dann synthesis in vascular endothelial cells. EMBO J. 2001, 20, 2768–2778. [Google Scholar] [CrossRef]
- Mabeta, P.; Steenkamp, V. The VEGF/VEGFR Axis Revisited: Implications for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 15585. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, D.; Liu, H.; Wang, Z.; Ma, G.; Yu, M.; Zhang, Y.; Zeng, Y. Expression levels of VEGF-C and VEGFR-3 in renal cell carcinoma and their association with lymph node metastasis. Exp. Ther. Med. 2021, 21, 554. [Google Scholar] [CrossRef] [PubMed]
- Su, J.L.; Chen, P.S.; Chien, M.H.; Chen, P.B.; Chen, Y.H.; Lai, C.C.; Hung, M.C.; Kuo, M.L. Further evidence for expression and function of the VEGF-C/VEGFR-3 axis in cancer cells. Cancer Cell 2008, 13, 557–560. [Google Scholar] [CrossRef]
- Donnan, M.D.; Deb, D.K.; Onay, T.; Scott, R.P.; Ni, E.; Zhou, Y.; Quaggin, S.E. Formation of the glomerular microvasculature is regulated by VEGFR-3. Am. J. Physiol.-Renal Physiol. 2023, 324, F91–F105. [Google Scholar] [CrossRef]
- Su, J.L.; Yen, C.J.; Chen, P.S.; Chuang, S.E.; Hong, C.C.; Kuo, I.H.; Chen, H.Y.; Hung, M.C.; Kuo, M.L. The role of the VEGF-C/VEGFR-3 axis in cancer progression. Br. J. Cancer 2007, 96, 541–545. [Google Scholar] [CrossRef]
- Haiko, P.; Makinen, T.; Keskitalo, S.; Taipale, J.; Karkkainen, M.J.; Baldwin, M.E.; Stacker, S.A.; Achen, M.G.; Alitalo, K. Deletion of Vascular Endothelial Growth Factor C (VEGF-C) and VEGF-D Is Not Equivalent to VEGF Receptor 3 Deletion in Mouse Embryos. Mol. Cell. Biol. 2008, 28, 4843–4850. [Google Scholar] [CrossRef]
- Tang, R.F.; Wang, S.X.; Peng, L.; Wang, S.X.; Zhang, M.; Li, Z.F.; Zhang, Z.M.; Xiao, Y.; Zhang, F.R. Expression of vascular endothelial growth factors A and C in human pancreatic cancer. World J. Gastroenterol. 2006, 12, 280–286. [Google Scholar] [CrossRef]
- Tang, R.F.; Itakura, J.; Aikawa, T.; Matsuda, K.; Fujii, H.; Korc, M.; Matsumoto, Y. Overexpression of lymphangiogenic growth factor VEGF-C in human pancreatic cancer. Pancreas 2001, 22, 285–292. [Google Scholar] [CrossRef]
- Tammela, T.; Zarkada, G.; Wallgard, E.; Murtomäki, A.; Suchting, S.; Wirzenius, M.; Waltari, M.; Hellström, M.; Schomber, T.; Peltonen, R.; et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 2008, 454, 656–660. [Google Scholar] [CrossRef]
- Patel, S.A.; Nilsson, M.B.; Le, X.; Cascone, T.; Jain, R.K.; Heymach, J.V. Molecular mechanisms and future implications of VEGF/VEGFR in cancer therapy. Clin. Cancer Res. 2023, 29, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Hsu, M.C.; Pan, M.R.; Hung, W.C. Two Birds, One Stone: Double Hits on Tumor Growth and Lymphangiogenesis by Targeting Vascular Endothelial Growth Factor Receptor 3. Cells 2019, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Lu, Y.; Gui, M.; Guan, J.; Lin, M.; Zhao, J.; Mao, Q.; Lin, J. Qingjie Fuzheng Granule suppresses lymphangiogenesis in colorectal cancer via the VEGF-C/VEGFR-3 dependent PI3K/AKT pathway. Biomed. Pharmacother. 2021, 137, 111331. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Büchler, P.; Giese, N.; Giese, T.; Wilting, J.; Büchler, M.W.; Friess, H. Role of lymphangiogenesis and lymphangiogenic factors during pancreatic cancer progression and lymphatic spread. Int. J. Oncol. 2006, 28, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, M.J.; He, T.; Zheng, Z.; Zheng, X.L.; Qu, B.H.; Wang, Y.; Duan, X.; Zheng, Y.J. Expression of vascular endothelial growth factor C in pancreatic cancer and its effect upon lymph node metastasis. Zhonghua Yi Xue Za Zhi 2009, 89, 2386–2390. [Google Scholar] [PubMed]
- Cheng, P.; Jin, G.; Hu, X.; Shi, M.; Zhang, Y.; Liu, R.; Zhou, Y.; Shao, C.; Zheng, J.; Zhu, M. Analysis of tumor-induced lymphangiogenesis and lymphatic vessel invasion of pancreatic carcinoma in the peripheral nerve plexus. Cancer Sci. 2012, 103, 1756–1763. [Google Scholar] [CrossRef]
- Kurahara, H.; Takao, S.; Maemura, K.; Shinchi, H.; Natsugoe, S.; Aikou, T. Impact of vascular endothelial growth factor-C and -D expression in human pancreatic cancer: Its relationship to lymph node metastasis. Clin. Cancer Res. 2004, 10, 8413–8420. [Google Scholar] [CrossRef]
- Fontanella, C.; Ongaro, E.; Bolzonello, S.; Guardascione, M.; Fasola, G.; Aprile, G. Clinical advances in the development of novel VEGFR2 inhibitors. Ann. Transl. Med. 2014, 2, 1. [Google Scholar]
- Lee, P.Y.; Yeoh, Y.; Low, T.Y. A recent update on small-molecule kinase inhibitors for targeted cancer therapy and their therapeutic insights from mass spectrometry-based proteomic analysis. FEBS J. 2023, 290, 2845–2864. [Google Scholar] [CrossRef]
- Capdevila, J.; Fazio, N.; Lopez, C.; Teulé, A.; Valle, J.W.; Tafuto, S.; Custodio, A.; Reed, N.; Raderer, M.; Grande, E.; et al. Lenvatinib in Patients with Advanced Grade 1/2 Pancreatic and Gastrointestinal Neuroendocrine Tumors: Results of the Phase II TALENT Trial (GETNE1509). J. Clin. Oncol. 2021, 39, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Bozzarelli, S.; Rimassa, L.; Giordano, L.; Sala, S.; Tronconi, M.C.; Pressiani, T.; Smiroldo, V.; Prete, M.G.; Spaggiari, P.; Personeni, N.; et al. Regorafenib in Patients with Refractory Metastatic Pancreatic Cancer: A Phase II Study (RESOUND). Future Oncol. 2019, 15, 4009–4017. [Google Scholar] [CrossRef]
- Chan, J.A.; Geyer, S.; Zemla, T.; Knopp, M.V.; Behr, S.; Pulsipher, S.; Ou, F.-S.; Dueck, A.C.; Acoba, J.; Shergill, A.; et al. Phase 3 Trial of Cabozantinib to Treat Advanced Neuroendocrine Tumors. N. Engl. J. Med. 2024, 392, 653. [Google Scholar] [CrossRef]
- Hernando-Calvo, A.; Han, M.; Ayodele, O.; Wang, B.X.; Bruce, J.P.; Abbas-Aghababazadeh, F.; Vila-Casadesús, M.; Sanz-Garcia, E.; Yang, S.Y.C.; Berman, H.K.; et al. A Phase II, Open-Label, Randomized Trial of Durvalumab with Olaparib or Cediranib in Patients with Mismatch Repair—Proficient Colorectal or Pancreatic Cancer. Clin. Color. Cancer 2024, 23, 272–284.e9. [Google Scholar] [CrossRef] [PubMed]
- Grande, E.; Capdevila, J.; Castellano, D.; Teulé, A.; Durán, I.; Fuster, J.; Sevilla, I.; Escudero, P.; Sastre, J.; García-Donas, J. Pazopanib in pretreated advanced neuroendocrine tumors: A phase II, open-label trial of the Spanish Task Force Group for Neuroendocrine Tumors (GETNE). Ann. Oncol. 2015, 26, 1987–1993. [Google Scholar] [CrossRef]
- Blumenthal, G.M.; Cortazar, P.; Zhang, J.J.; Tang, S.; Sridhara, R.; Murgo, A.; Justice, R.; Pazdur, R. FDA approval summary: Sunitinib for the treatment of progressive well-differentiated locally advanced or metastatic pancreatic neuroendocrine tumors. Oncologist 2012, 17, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, A.; Gilabert, M.; François, E.; Dahan, L.; Perrier, H.; Lamy, R.; Re, D.; Largillier, R.; Gasmi, M.; Tchiknavorian, X.; et al. BAYPAN study: A double-blind phase III randomized trial comparing gemcitabine plus sorafenib and gemcitabine plus placebo in patients with advanced pancreatic cancer. Ann. Oncol. 2012, 23, 2799–2805. [Google Scholar] [CrossRef]
- Kindler, H.L.; Ioka, T.; Richel, D.J.; Bennouna, J.; Létourneau, R.; Okusaka, T.; Funakoshi, A.; Furuse, J.; Park, Y.S.; Ohkawa, S.; et al. Axitinib plus gemcitabine versus placebo plus gemcitabine in patients with advanced pancreatic adenocarcinoma: A double-blind randomised phase 3 study. Lancet Oncol. 2011, 12, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, J.; Li, N.; Huang, W.; Chen, D.; Tao, W.; Wang, H. An open-label, placebo-controlled, randomized phase II trial of camrelizumab combined with or without apatinib or capecitabine in the treatment of previously treated advanced pancreatic cancer. J. Clin. Oncol. 2024, 42, e16324. [Google Scholar] [CrossRef]
- Une, N.; Takano-Kasuya, M.; Kitamura, N.; Ohta, M.; Inose, T.; Kato, C.; Nishimura, R.; Tada, H.; Miyagi, S.; Ishida, T.; et al. The anti-angiogenic agent lenvatinib induces tumor vessel normalization and enhances radiosensitivity in hepatocellular tumors. Med. Oncol. 2021, 38, 60. [Google Scholar] [CrossRef]
- Antoniotti, C.; Marmorino, F.; Pennati, M.; Zaffaroni, N.; Rossini, D.; Borelli, B.; Zucchelli, G.; Moretto, R.; Pietrantonio, F.; Masi, G.; et al. Circulating angiogenesis-related markers as predictors of benefit from regorafenib in metastatic colorectal cancer (mCRC) patients (pts). Ann. Oncol. 2017, 28, vi4. [Google Scholar] [CrossRef]
- Wu, J.Q.; Fan, R.Y.; Zhang, S.R.; Li, C.Y.; Shen, L.Z.; Wei, P.; He, Z.H.; He, M.F. A systematical comparison of anti-angiogenesis and anti-cancer efficacy of ramucirumab, apatinib, regorafenib and cabozantinib in zebrafish model. Life Sci. 2020, 247, 117402. [Google Scholar] [CrossRef]
- Keating, G.M. Bevacizumab: A review of its use in advanced cancer. Drugs 2014, 74, 1891–1925. [Google Scholar] [CrossRef]
- Robert, N.J.; Saleh, M.N.; Paul, D.; Generali, D.; Gressot, L.; Copur, M.S.; Brufsky, A.M.; Minton, S.E.; Giguere, J.K.; Smith, J.W., II; et al. Sunitinib plus paclitaxel versus bevacizumab plus paclitaxel for first-line treatment of patients with advanced breast cancer: A phase III, randomized, open-label trial. Clin. Breast Cancer 2011, 11, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Coon, J.S.T.; Liu, Z.; Hoyle, M.; Rogers, G.; Green, C.; Moxham, T.; Welch, K.; Stein, K. Sunitinib and bevacizumab for first-line treatment of metastatic renal cell carcinoma: A systematic review and indirect comparison of clinical effectiveness. Br. J. Cancer 2009, 101, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Herrmann, J. Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis. Oncol. 2018, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Bowler, E.; Oltean, S. Alternative splicing in angiogenesis. Int. J. Mol. Sci. 2019, 20, 2067. [Google Scholar] [CrossRef]
- Wu, F.T.; Stefanini, M.O.; Gabhann, F.M.; Kontos, C.D.; Annex, B.H.; Popel, A.S. A systems biology perspective on sVEGFR1: Its biological function, pathogenic role and therapeutic use. J. Cell Mol. Med. 2010, 14, 528–552. [Google Scholar] [CrossRef]
- Saito, T.; Takeda, N.; Amiya, E.; Nakao, T.; Abe, H.; Semba, H.; Soma, K.; Koyama, K.; Hosoya, Y.; Imai, Y.; et al. VEGF-A induces its negative regulator, soluble form of VEGFR-1, by modulating alternative splicing. FEBS Lett. 2013, 587, 2179–2185. [Google Scholar] [CrossRef]
- Gerber, H.-P.; Condorelli, F.; Park, J.; Ferrara, N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes: Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J. Biol. Chem. 1997, 272, 23659–23667. [Google Scholar] [CrossRef]
- Natua, S.; Ashok, C.; Shukla, S. Hypoxia-induced alternative splicing in human diseases: The pledge, the turn, and the prestige. Cell Mol. Life Sci. 2021, 78, 2729–2747. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Wilgus, T.A. Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair. Adv. Wound Care 2014, 3, 647–661. [Google Scholar] [CrossRef]
- Lakatos, D.; Somfai, E.; Méhes, E.; Czirók, A. Soluble VEGFR-1 signaling guides vascular patterns into dense branching morphologies. J. Theor. Biol. 2018, 456, 261–278. [Google Scholar] [CrossRef]
- Mamer, S.B.; Wittenkeller, A.; Imoukhuede, P. VEGF-A splice variants bind VEGFRs with differential affinities. Sci. Rep. 2020, 10, 14413. [Google Scholar] [CrossRef] [PubMed]
- Harris, H.; Wolk, A.; Larsson, A.; Vasson, M.P.; Basu, S. Soluble vascular endothelial growth factor receptors 2 (sVEGFR-2) and 3 (sVEGFR-3) and breast cancer risk in the Swedish Mammography Cohort. Int. J. Mol. Epidemiol. Genet. 2016, 7, 81–86. [Google Scholar]
- Takahashi, K.; Mizukami, H.; Saga, Y.; Takei, Y.; Urabe, M.; Kume, A.; Machida, S.; Fujiwara, H.; Suzuki, M.; Ozawa, K. Suppression of lymph node and lung metastases of endometrial cancer by muscle-mediated expression of soluble vascular endothelial growth factor receptor-3. Cancer Sci. 2013, 104, 1107–1111. [Google Scholar] [CrossRef]
- Harding, T.C.; Lalani, A.S.; Roberts, B.N.; Yendluri, S.; Luan, B.; Koprivnikar, K.E.; Gonzalez-Edick, M.; Huan-Tu, G.; Musterer, R.; VanRoey, M.J.; et al. AAV serotype 8-mediated gene delivery of a soluble VEGF receptor to the CNS for the treatment of glioblastoma. Mol. Ther. 2006, 13, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Kumasawa, K.; Sato, N.; Takiuchi, T.; Nakamura, H.; Kimura, T. Soluble VEGF receptor 1 (sFLT1) induces non-apoptotic death in ovarian and colorectal cancer cells. Sci. Rep. 2016, 6, 24853. [Google Scholar] [CrossRef]
- Sato, N.; Kumasawa, K.; Yamashita, M.; Miyake, T.; Nakamura, H.; Kimura, T. Therapeutic potential of combination therapy of soluble VEGF receptor 1 and conventional chemotherapy for ovarian cancer growth. J. Obstet. Gynaecol. 2020, 46, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lalani, A.S.; Harding, T.C.; Gonzalez, M.; Wu, W.W.; Luan, B.; Tu, G.H.; Koprivnikar, K.; VanRoey, M.J.; He, Y.; et al. Inhibition of lymphogenous metastasis using adeno-associated virus-mediated gene transfer of a soluble VEGFR-3. Cancer Res. 2005, 65, 6901–6909. [Google Scholar] [CrossRef]
- Kujala, A.; Valkonen, E.; Sallinen, H.; Tuppurainen, L.; Laakso, H.; Ylä-Herttuala, E.; Liimatainen, T.; Kujala, J.; Jokelainen, O.; Sironen, R.; et al. AAV8-mediated sVEGFR2 and sVEGFR3 gene therapy combined with chemotherapy reduces the growth and microvasculature of human ovarian cancer and prolongs the survival in mice. Front. Med. 2022, 9, 1018208. [Google Scholar] [CrossRef]
- Sallinen, H.; Anttila, M.; Narvainen, J.; Koponen, J.; Hamalainen, K.; Kholova, I.; Heikura, T.; Toivanen, P.; Kosma, V.M.; Heinonen, S.; et al. Antiangiogenic gene therapy with soluble VEGFR-1,-2, and-3 reduces the growth of solid human ovarian carcinoma in mice. Mol. Ther. 2009, 17, 278–284. [Google Scholar] [CrossRef]
- Shibata, M.A.; Shibata, E.; Tanaka, Y.; Shiraoka, C.; Kondo, Y. Soluble VEGFR3 gene therapy suppresses multi-organ metastasis in a mouse mammary cancer model. Cancer Sci. 2020, 111, 2837–2849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, B.; Shi, J.; Zhao, L.; Zhang, X.; Wang, C.; Hou, S.; Qian, W.; Kou, G.; Wang, H.; et al. Suppression of tumor growth and metastasis by simultaneously blocking vascular endothelial growth factor (VEGF)–A and VEGF-C with a receptor-immunoglobulin fusion protein. Cancer Res. 2010, 70, 2495–2503. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.T.; Chang, M.C.; Wei, S.C.; Tien, Y.W.; Hsu, C.; Liang, P.C.; Tsao, P.N.; Jan, I.S.; Wong, J.M. Serum vascular endothelial growth factor/soluble vascular endothelial growth factor receptor 1 ratio is an independent prognostic marker in pancreatic cancer. Pancreas 2008, 37, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Obata, Y.; Yagyu, K.; Lin, Y.; Nakajima, T.; Kobayashi, O.; Kikuichi, M.; Ushijima, R.; Kurosawa, M.; Ueda, J. Reduced serum vascular endothelial growth factor receptor-2 (sVEGFR-2) and sVEGFR-1 levels in gastric cancer patients. Cancer Sci. 2011, 102, 866–869. [Google Scholar] [CrossRef]
- Shaik, F.; Cuthbert, G.A.; Homer-Vanniasinkam, S.; Muench, S.P.; Ponnambalam, S.; Harrison, M.A. Structural Basis for Vascular Endothelial Growth Factor Receptor Activation and Implications for Disease Therapy. Biomolecules 2020, 10, 1673. [Google Scholar] [CrossRef]
- Tang, Y.; Nakada, M.T.; Kesavan, P.; McCabe, F.; Millar, H.; Rafferty, P.; Bugelski, P.; Yan, L. Extracellular Matrix Metalloproteinase Inducer Stimulates Tumor Angiogenesis by Elevating Vascular Endothelial Cell Growth Factor and Matrix Metalloproteinases. Cancer Res. 2005, 65, 3193–3199. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.D.; Camp, E.R.; Fan, F.; Shen, L.; Gray, M.J.; Liu, W.; Somcio, R.; Bauer, T.W.; Wu, Y.; Hicklin, D.J.; et al. Vascular Endothelial Growth Factor Receptor-1 Activation Mediates Epithelial to Mesenchymal Transition in Human Pancreatic Carcinoma Cells. Cancer Res. 2006, 66, 46–51. [Google Scholar] [CrossRef]
- Khan, A.; Khan, H.; Hughes, G.K.; Ladd, C.; McIntire, R.; Gardner, B.; Peña, A.M.; Schoutko, A.; Tuia, J.; Minley, K.; et al. Assessing patient risk, benefit, and outcomes in drug development: A decade of ramucirumab clinical trials. Cancer Med. 2024, 13, e7130. [Google Scholar] [CrossRef]
- Thomas, A.G.; Awasthi, N. Targeted therapy for pancreatic cancer: Lessons learned and future opportunities. Dig. Med. Res. 2021, 4, 32. [Google Scholar] [CrossRef]
- Kim, D.K.; Jeong, J.; Lee, D.S.; Hyeon, D.Y.; Park, G.W.; Jeon, S.; Lee, K.B.; Jang, J.-Y.; Hwang, D.; Kim, H.M.; et al. PD-L1-directed PlGF/VEGF blockade synergizes with chemotherapy by targeting CD141+ cancer-associated fibroblasts in pancreatic cancer. Nat. Commun. 2022, 13, 6292. [Google Scholar] [CrossRef]
- Manzari, M.T.; Shamay, Y.; Kiguchi, H.; Rosen, N.; Scaltriti, M.; Heller, D.A. Targeted drug delivery strategies for precision medicines. Nat. Rev. Mater. 2021, 6, 351–370. [Google Scholar] [CrossRef]
- Ahn, H.; Choi, J.; Kim, K.; Kim, H.; Choi, S.; Park, S.; Park, J.; Lim, H.; Kang, W.; Lee, J. Phase II study of pazopanib monotherapy in metastatic gastroenteropancreatic neuroendocrine tumours. Br. J. Cancer. 2013, 109, 1414–1419. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-Based Drug Delivery in Cancer Therapy and Its Role in Overcoming Drug Resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef]
- Zanjanchi, P.; Asghari, S.M.; Mohabatkar, H.; Shourian, M.; Shafiee Ardestani, M. Conjugation of VEGFR1/R2-targeting peptide with gold nanoparticles to enhance antiangiogenic and antitumoral activity. J. Nanobiotechnol. 2022, 20, 7. [Google Scholar] [CrossRef] [PubMed]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Murukesh, N.; Dive, C.; Jayson, G.C. Biomarkers of angiogenesis and their role in the development of VEGF inhibitors. Br. J. Cancer 2010, 102, 8–18. [Google Scholar] [CrossRef]
- Hansson, E.K.; Amantea, M.A.; Westwood, P.; Milligan, P.A.; Houk, B.E.; French, J.; Karlsson, M.O.; Friberg, L.E. PKPD modeling of VEGF, sVEGFR-2, sVEGFR-3, and sKIT as predictors of tumor dynamics and overall survival following sunitinib treatment in GIST. CPT Pharmacomet. Syst. Pharmacol. 2013, 2, e84. [Google Scholar] [CrossRef]
- Yuki, S.; Yamazaki, K.; Sunakawa, Y.; Taniguchi, H.; Masuishi, T.; Shiozawa, M.; Bando, H.; Nishina, T.; Yasui, H.; Ohta, T.; et al. Analysis of plasma angiogenesis factors on the efficacy of 2nd-line (2L) chemotherapy (chemo) combined with angiogenesis inhibitors (AIs) in metastatic colorectal cancer (mCRC): Results from GI-SCREEN CRC Ukit study. Ann. Oncol. 2022, 33, S695–S696. [Google Scholar] [CrossRef]
- Shaib, W.L.; Manali, R.; Liu, Y.; El-Rayes, B.; Loehrer, P.; O’Neil, B.; Cohen, S.; Khair, T.; Robin, E.; Huyck, T.; et al. Phase II randomised, double-blind study of mFOLFIRINOX plus ramucirumab versus mFOLFIRINOX plus placebo in advanced pancreatic cancer patients (HCRN GI14-198). Eur. J. Cancer 2023, 189, 112847. [Google Scholar] [CrossRef]
- Rougier, P.; Riess, H.; Manges, R.; Karasek, P.; Humblet, Y.; Barone, C.; Santoro, A.; Assadourian, S.; Hatteville, L.; Philip, P.A. Randomised, placebo-controlled, double-blind, parallel-group phase III study evaluating aflibercept in patients receiving first-line treatment with gemcitabine for metastatic pancreatic cancer. Eur. J. Cancer 2013, 49, 2633–2642. [Google Scholar] [CrossRef]
- Fang, Y.T.; Yang, W.W.; Niu, Y.R.; Sun, Y.K. Recent advances in targeted therapy for pancreatic adenocarcinoma. World J. Gastrointest. Oncol. 2023, 15, 571–595. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Chen, X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr. Drug Targets 2010, 11, 1000–1017. [Google Scholar] [CrossRef] [PubMed]
- Sennino, B.; Ishiguro-Oonuma, T.; Wei, Y.; Naylor, R.M.; Williamson, C.W.; Bhagwandin, V.; Tabruyn, S.P.; You, W.K.; Chapman, H.A.; Christensen, J.G.; et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov. 2012, 2, 270–287. [Google Scholar] [CrossRef]
- Geindreau, M.; Ghiringhelli, F.; Bruchard, M. Vascular Endothelial Growth Factor, a Key Modulator of the Anti-Tumor Immune Response. Int. J. Mol. Sci. 2021, 22, 4871. [Google Scholar] [CrossRef]
- Hao, S.; Han, W.; Ji, Y.; Sun, H.; Shi, H.; Ma, J.; Yip, J.; Ding, Y. BANCR positively regulates the HIF-1α/VEGF-C/VEGFR-3 pathway in a hypoxic microenvironment to promote lymphangiogenesis in pancreatic cancer cells. Oncol. Lett. 2022, 24, 422. [Google Scholar] [CrossRef]
- Qin, J.; Chen, X.; Yu-Lee, L.-y.; Tsai, M.-J.; Tsai, S.Y. Nuclear Receptor COUP-TFII Controls Pancreatic Islet Tumor Angiogenesis by Regulating Vascular Endothelial Growth Factor/Vascular Endothelial Growth Factor Receptor-2 Signaling. Cancer Res. 2010, 70, 8812–8821. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.H.; Dito, E.; Schillinger, B.; Venook, A.P.; Xu, Z.; Bergsland, E.K.; Wong, D.; Scott, J.; Hwang, J.; Tempero, M.A. A phase II study evaluating bevacizumab in combination with fixed-dose rate gemcitabine and low-dose cisplatin for metastatic pancreatic cancer: Is an anti-VEGF strategy still applicable? Investig. New Drugs 2008, 26, 463–471. [Google Scholar] [CrossRef]
- Kallergi, G.; Markomanolaki, H.; Giannoukaraki, V.; Papadaki, M.A.; Strati, A.; Lianidou, E.S.; Georgoulias, V.; Mavroudis, D.; Agelaki, S. Hypoxia-inducible factor-1α and vascular endothelial growth factor expression in circulating tumor cells of breast cancer patients. Breast Cancer Res. 2009, 11, R84. [Google Scholar] [CrossRef]
- Chen, H.; Li, L.; Wang, S.; Lei, Y.; Ge, Q.; Lv, N.; Zhou, X.; Chen, C. Reduced miR-126 expression facilitates angiogenesis of gastric cancer through its regulation on VEGF-A. Oncotarget 2014, 5, 11873–11885. [Google Scholar] [CrossRef]
- Sebestyén, A.; Kopper, L.; Dankó, T.; Tímár, J. Hypoxia Signaling in Cancer: From Basics to Clinical Practice. Pathol. Oncol. Res. 2021, 27, 1609802. [Google Scholar] [CrossRef] [PubMed]
- Duran, C.L.; Borriello, L.; Karagiannis, G.S.; Entenberg, D.; Oktay, M.H.; Condeelis, J.S. Targeting Tie2 in the Tumor Microenvironment: From Angiogenesis to Dissemination. Cancers 2021, 13, 5730. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Cancer | Status | Reference |
|---|---|---|---|---|
| Lenvatinib | VEGFR-1, -2, -3 | Advanced-grade 1–2 PNETs | Phase II | [49] |
| Regorafenib | VEGFR-1, -2, -3; BRAF, cKIT, PDGFR-B | Refractory metastatic pancreatic cancer | Phase II | [50] |
| Cabozantinib | VEGFR-2 | Advanced Pancreatic Neuroendocrine Tumors (PNETs) | Phase III | [51] |
| Cediranib | VEGFR-1 -,2, -3 | PDAC | Phase II | [52] |
| Pazopanib | VEGFR-1, -2, -3, PDGFR, and c-Kit | PNET | Phase II | [53] |
| Sunitinib | VEGFR-1, -2, -3, PDGFR-α/β, c-KIT, FLT3 and RET | Locally Advanced or Metastatic PNETs | Approved 2011 | [54] |
| Sorafenib | VEGFR-2, -3, PDGFR, c-Kit and RET | Advanced PDAC | Phase II | [55] |
| Axitinib | VEGFR-1, -2, -3 | Advanced PDAC | Phase III | [56] |
| Apatinib | VEGFR-2 | PDAC | Phase II | [57] |
| Drug | Cancer | Effects | Reference |
|---|---|---|---|
| sVEGFR-1 | Breast cancer PDAC | rAAV-sVEGFR1/R2 vectorVEGFR-1 binding and angiogenesis, suppresses macrophage infiltration, has anti-proliferative effect on cancer cells | [22,66] |
| sVEGFR-1-AAV 8 | GBM | Inhibits angiogenesis and tumor growth | [75] |
| Recombinant sVEGFR-1 (rsVEGFR-1) + Carboplatin | Ovarian cancer | Inhibits ovarian cancer cell proliferation | [76] |
| rsVEGFR-1 | Ovarian cancer, CRC | Has anti-proliferative effects on ovarian and colorectal cancer cells | [77] |
| sVEGFR-2 + sVEGFR-3—AAV8 gene therapy | Ovarian cancer | Reduces cancer metastasis, inhibits lymphangiogenesis | [78,79] |
| sVEGFR-3 | Endometrial cancer | Reduces tumor growth, lymph node metastasis | [74] |
| sVEGFR-3 vector | Ovarian cancer | Decoy for VEGFR-3 binding to VEGF-C and -D | [80] |
| sVEGFR-3 gene therapy | Breast cancer | Inhibits lymphangiogenesis and multi-organ metastasis | [81] |
| sVEGFR-3-Ig Fusion protein | HCC | Inhibits tumor angiogenesis and lymphangiogenesis, suppresses primary tumor growth and lymph node metastasis | [82] |
| Biomarker | Advantages | Limitations | References |
|---|---|---|---|
| Circulating VEGF levels | Easily measurable in blood samples; consistent drug-induced increases in plasma VEGF-A levels across multiple studies | Lack of consistent prognostic or predictive value across studies; potential confounding factors in measurement, such as platelet activation during sample handling | [96] |
| Soluble VEGFR-1 and VEGFR-2 | Reflects VEGFR inhibition; may indicate drug efficacy | Requires standardization; inconsistent correlation with outcomes | [83] |
| Circulating Endothelial Cells (CECs) | Can reflect vascular damage or active angiogenesis; some studies show correlations with clinical outcomes | Methodological problems in enumeration and characterization; lack of consensus on specific markers for CECs | [96] |
| Circulating Tumor Cells (CTCs) | Linked to metastatic potential and disease progression | Limited studies in pancreatic cancer; variability in VEGFR expression | [108] |
| MicroRNAs (miRNAs) (e.g., miR-126, miR-200) | Non-invasive; involved in VEGF regulation | Complex regulation; conflicting findings across cancer types | [109] |
| DCE-MRI (Imaging biomarker) | Non-invasive and sensitive detection method; shows consistent findings across multiple studies; Demonstrates dose-level response relationships; correlates with clinical outcomes in some studies | More complex to incorporate into multi-site studies compared to CT; requires standardization across different centers | [96] |
| Tumor Hypoxia Markers (HIF-1α, CAIX) | Enable indirect assessment of anti-angiogenic treatment effectiveness | May not be specific to VEGFR inhibition | [110] |
| Tie2-Expressing Monocytes (TEMs) | Reflect anti-angiogenic therapy effectiveness | Requires further validation in clinical settings | [111] |
| Tissue-Based Biomarkers (e.g., Microvessel density, Pericyte coverage) | Enable direct assessment of VEGFR-targeted therapy impact | Invasive, requiring tumor biopsies; lack of consistent predictive value for VEGF inhibitors; may not represent the entire tumor due to sampling limitations | [96] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grobbelaar, C.; Steenkamp, V.; Mabeta, P. Vascular Endothelial Growth Factor Receptors in the Vascularization of Pancreatic Tumors: Implications for Prognosis and Therapy. Curr. Issues Mol. Biol. 2025, 47, 179. https://doi.org/10.3390/cimb47030179
Grobbelaar C, Steenkamp V, Mabeta P. Vascular Endothelial Growth Factor Receptors in the Vascularization of Pancreatic Tumors: Implications for Prognosis and Therapy. Current Issues in Molecular Biology. 2025; 47(3):179. https://doi.org/10.3390/cimb47030179
Chicago/Turabian StyleGrobbelaar, Craig, Vanessa Steenkamp, and Peace Mabeta. 2025. "Vascular Endothelial Growth Factor Receptors in the Vascularization of Pancreatic Tumors: Implications for Prognosis and Therapy" Current Issues in Molecular Biology 47, no. 3: 179. https://doi.org/10.3390/cimb47030179
APA StyleGrobbelaar, C., Steenkamp, V., & Mabeta, P. (2025). Vascular Endothelial Growth Factor Receptors in the Vascularization of Pancreatic Tumors: Implications for Prognosis and Therapy. Current Issues in Molecular Biology, 47(3), 179. https://doi.org/10.3390/cimb47030179