
The Role of Ferroptosis and Cuproptosis in Tuberculosis Pathogenesis: Implications for Therapeutic Strategies

 ,
, 

Abstract
1. Introduction
2. Ferroptosis: Mechanistic Insights, Key Features, and Its Role in Host–Pathogen Interactions During Tuberculosis
2.1. Ferroptosis in Infectious Diseases: TB and Host–Pathogen Interactions
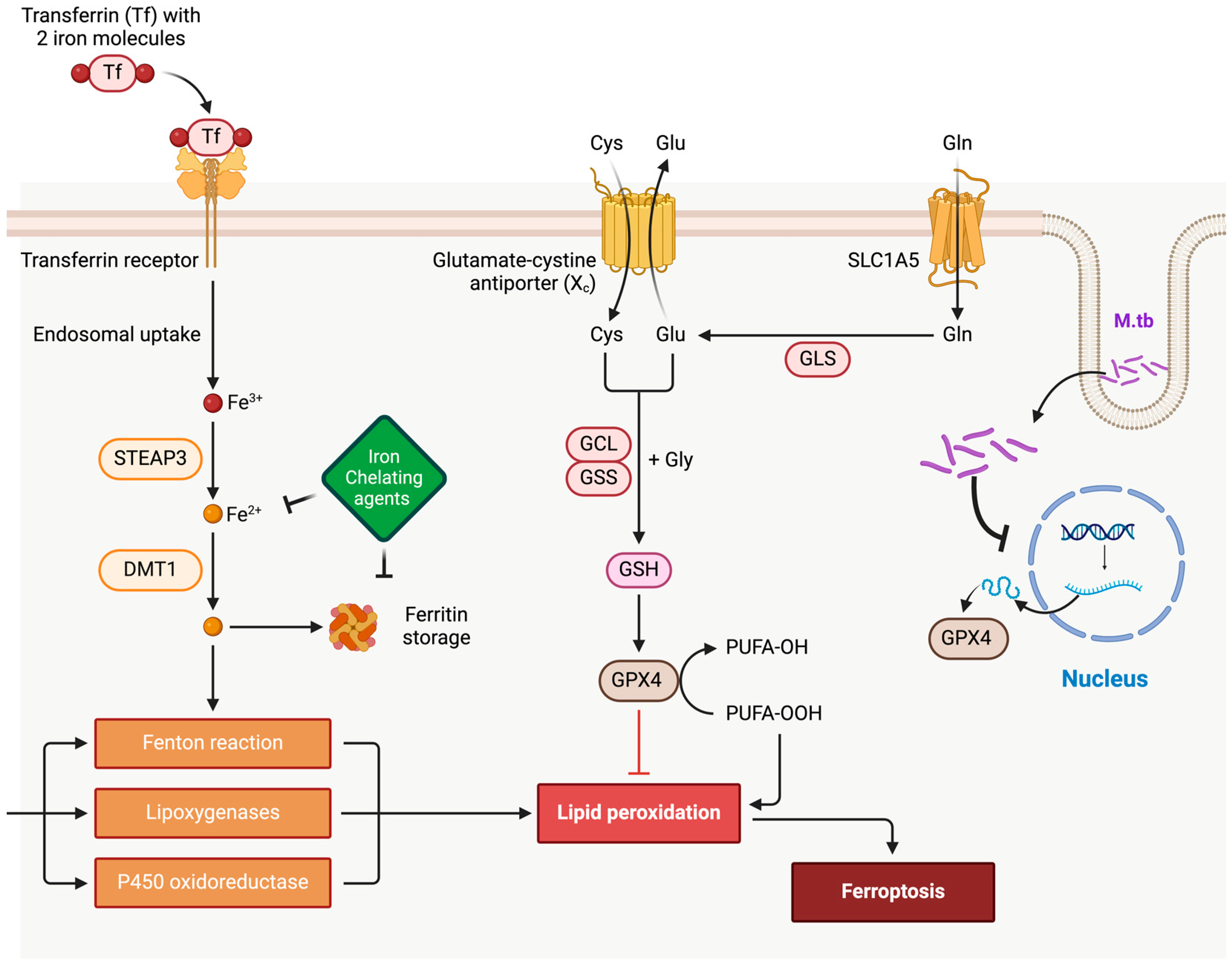
2.1.1. Host Iron Homeostasis and Ferroptosis in TB
2.1.2. Mechanisms of M.tb in Ferroptosis Modulation
- Iron Uptake and Redistribution: M.tb infection upregulates iron acquisition pathways in macrophages, including the expression of transferrin receptors and downregulation of ferroportin, which traps iron within cells. This iron accumulation promotes the Fenton reaction, generating reactive oxygen species (ROS) that can trigger lipid peroxidation—a hallmark of ferroptosis [10,24].
- Antioxidant Defense Manipulation: M.tb enhances its survival by interfering with host antioxidant defenses, particularly through the inhibition of glutathione peroxidase 4 (GPX4), a critical regulator of ferroptosis. Studies show that GPX4 activity is reduced in M.tb-infected macrophages, exacerbating lipid peroxidation and ferroptotic cell death. This dynamic supports the hypothesis that M.tb exploits ferroptosis to damage surrounding tissues, facilitating bacterial dissemination [25,26].
- Lipid Metabolism Alterations: M.tb infection alters host lipid metabolism by increasing polyunsaturated fatty acid (PUFA) production, which serves as substrates for lipid peroxidation during ferroptosis. Additionally, M.tb-derived factors can modulate host lipoxygenases, further driving ferroptotic pathways [27,28].
- Immune Evasion via Ferroptosis Induction: Excessive ferroptosis in macrophages compromises their ability to contain M.tb. By inducing lipid peroxidation and membrane damage, M.tb undermines the integrity of granulomas—organized immune structures essential for bacterial containment. This disruption aids M.tb in escaping immune surveillance and spreading to new tissues [10].
2.1.3. Implications for TB Pathogenesis and Therapeutic Approaches
3. Copper-Induced Cell Death: Role of Copper in Cellular Metabolism and Stress Response
3.1. Cuproptosis: Mechanistic Features and Cellular Impact
3.2. Cuproptosis in Infectious Diseases: TB and the Host–Pathogen Interface
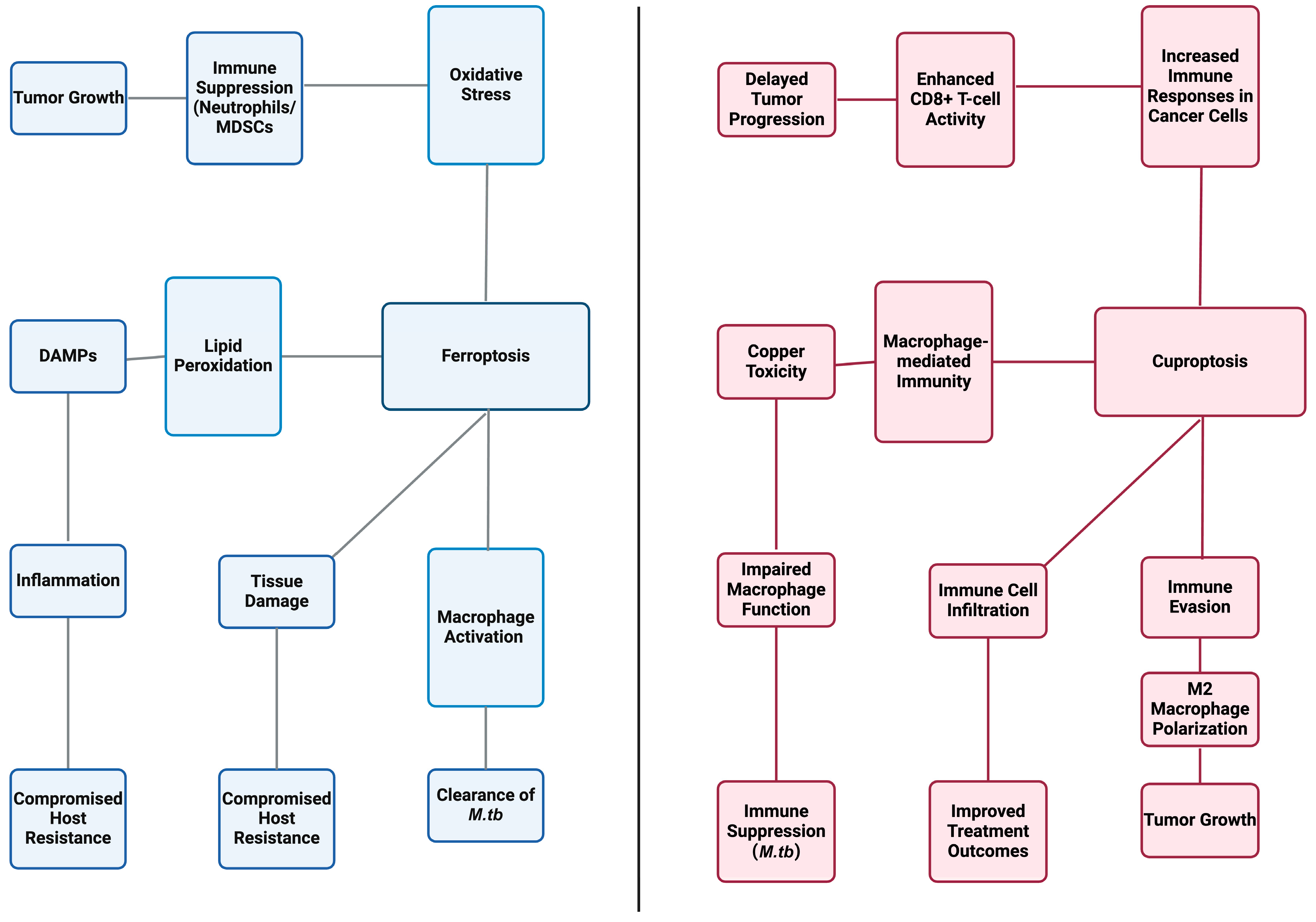
4. Synergistic Interactions of Ferroptosis and Cuproptosis in Tuberculosis Pathogenesis
4.1. Combined Effects on Host Cells
4.1.1. Impact on Macrophage Function
4.1.2. Influence of Ferroptosis and Cuproptosis on Tuberculosis Immunity
4.2. Pathogenesis Modulation by Ferroptosis and Cuproptosis in TB
4.2.1. Bacterial Survival and Replication
4.2.2. Host Resistance and Susceptibility
5. Innovative Therapeutic Opportunities in TB: Targeting Ferroptosis and Cuproptosis
5.1. Targeting Ferroptosis: Potential and Limitations in Tuberculosis Treatment
5.2. Therapeutic Exploration of Cuproptosis in Tuberculosis Management
6. Mitochondrial Dynamics in Macrophage Cell Death: Future Directions for Tuberculosis Therapies
7. Methods
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- WHO. Tuberculosis; World Health Organization: Geneva, Switzerland, 2020; Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 17 May 2020).
- Williams, P.M.; Pratt, R.H.; Walker, W.L.; Price, S.F.; Stewart, R.J.; Feng, P.-J.I. Tuberculosis—United States, 2023. MMWR—Morb. Mortal. Wkly. Rep. 2024, 73, 265–270. [Google Scholar] [CrossRef]
- Brett, K.; Dulong, C.; Severn, M. Identification of Tuberculosis: A Review of the Guidelines; Canadian Agency for Drugs and Technologies in Health: Toronto, ON, USA, 2020. [Google Scholar]
- Kiazyk, S.; Ball, T. Latent tuberculosis infection: An overview. Can. Commun. Dis. Rep. 2017, 43, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Acharya, B.; Acharya, A.; Gautam, S.; Ghimire, S.P.; Mishra, G.; Parajuli, N.; Sapkota, B. Advances in diagnosis of Tuberculosis: An update into molecular diagnosis of Mycobacterium tuberculosis. Mol. Biol. Rep. 2020, 47, 4065–4075. [Google Scholar] [CrossRef] [PubMed]
- Saktiawati, A.M.; Putera, D.D.; Setyawan, A.; Mahendradhata, Y.; van der Werf, T.S. Diagnosis of tuberculosis through breath test: A systematic review. eBioMedicine 2019, 46, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.J.; Booty, M.G.; Rosebrock, T.R.; Nunes-Alves, C.; Desjardins, D.M.; Keren, I.; Fortune, S.M.; Remold, H.G.; Behar, S.M. Efferocytosis Is an Innate Antibacterial Mechanism. Cell Host Microbe 2012, 12, 289–300. [Google Scholar] [CrossRef]
- Lerner, T.R.; Borel, S.; Greenwood, D.J.; Repnik, U.; Russell, M.R.; Herbst, S.; Jones, M.L.; Collinson, L.M.; Griffiths, G.; Gutierrez, M.G. Mycobacterium tuberculosis replicates within necrotic human macrophages. J. Cell Biol. 2017, 216, 583–594. [Google Scholar] [CrossRef]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef]
- Amaral, E.P.; Costa, D.L.; Namasivayam, S.; Riteau, N.; Kamenyeva, O.; Mittereder, L.; Mayer-Barber, K.D.; Andrade, B.B.; Sher, A. A major role for ferroptosis in Mycobacterium tuberculosis–induced cell death and tissue necrosis. J. Exp. Med. 2019, 216, 556–570. [Google Scholar] [CrossRef]
- Boelaert, J.R.; Vandecasteele, S.J.; Appelberg, R.; Gordeuk, V.R. The Effect of the Host’s Iron Status on Tuberculosis. J. Infect. Dis. 2007, 195, 1745–1753. [Google Scholar] [CrossRef]
- Meunier, E.; Neyrolles, O. Die another way: Ferroptosis drives tuberculosis pathology. J. Exp. Med. 2019, 216, 471–473. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Long, Q.; Nong, L.; Zheng, Y.; Meng, X.; Zhu, Q. Identification of immune infiltration and cuproptosis-related molecular clusters in tuberculosis. Front. Immunol. 2023, 14, 1205741. [Google Scholar] [CrossRef]
- Tsang, T.; Davis, C.I.; Brady, D.C. Copper biology. Curr. Biol. 2021, 31, R421–R427. [Google Scholar] [CrossRef]
- Mohan, G.; Kulshreshtha, S.; Sharma, P. Zinc and Copper in Indian Patients of Tuberculosis: Impact on Antitubercular Therapy. Biol. Trace Element Res. 2006, 111, 63–70. [Google Scholar] [CrossRef]
- Bahi, G.A.; Boyvin, L.; Méité, S.; M’boh, G.M.; Yeo, K.; N’guessan, K.R.; Bidié, A.D.P.; Djaman, A.J. Assessments of serum copper and zinc concentration, and the Cu/Zn ratio determination in patients with multidrug resistant pulmonary tuberculosis (MDR-TB) in Côte d’Ivoire. BMC Infect. Dis. 2017, 17, 257. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Battistelli, C.; Sabarese, G.; Santangelo, L.; Montaldo, C.; Gonzalez, F.J.; Tripodi, M.; Cicchini, C. The lncRNA HOTAIR transcription is controlled by HNF4α-induced chromatin topology modulation. Cell Death Differ. 2018, 26, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef]
- Chao, A.; Sieminski, P.J.; Owens, C.P.; Goulding, C.W. Iron Acquisition in Mycobacterium tuberculosis. Chem. Rev. 2019, 119, 1193–1220. [Google Scholar] [CrossRef]
- Hood, M.I.; Skaar, E.P. Nutritional immunity: Transition metals at the pathogen–host interface. Nat. Rev. Microbiol. 2012, 10, 525–537. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Zhang, Y.; Lei, Z.; Lu, Z.; Tan, S.; Ge, P.; Chai, Q.; Zhao, M.; Zhang, X.; Li, B.; et al. A mycobacterial effector promotes ferroptosis-dependent pathogenicity and dissemination. Nat. Commun. 2023, 14, 1430. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, C.; Cheng, L.; Ullah, H.; Sha, S.; Kang, J.; Ma, X.; Ma, Y. Mycobacterium tuberculosis Rv1324 Protein Contributes to Mycobacterial Persistence and Causes Pathological Lung Injury in Mice by Inducing Ferroptosis. Microbiol. Spectr. 2023, 11, e0252622. [Google Scholar] [CrossRef] [PubMed]
- Alves, F.; Lane, D.; Nguyen, T.P.M.; Bush, A.I.; Ayton, S. In defence of ferroptosis. Signal Transduct. Target. Ther. 2025, 10, 2. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2020, 31, 107–125. [Google Scholar] [CrossRef]
- Jumabayi, W.; Reyimu, A.; Zheng, R.; Paerhati, P.; Rahman, M.; Zou, X.; Xu, A. Ferroptosis: A new way to intervene in the game between Mycobacterium tuberculosis and macrophages. Microb. Pathog. 2024, 197, 107014. [Google Scholar] [CrossRef]
- Wufuer, D.; Li, Y.; Aierken, H.; Zheng, J. Bioinformatics-led discovery of ferroptosis-associated diagnostic biomarkers and molecule subtypes for tuberculosis patients. Eur. J. Med. Res. 2023, 28, 445. [Google Scholar] [CrossRef]
- Tan, W.; Zhang, J.; Chen, L.; Wang, Y.; Chen, R.; Zhang, H.; Liang, F. Copper homeostasis and cuproptosis-related genes: Therapeutic perspectives in non-alcoholic fatty liver disease. Diabetes, Obes. Metab. 2024, 26, 4830–4845. [Google Scholar] [CrossRef]
- Fanzo, J. Understanding human water turnover in times of water scarcity. Cell Metab. 2023, 35, 231–232. [Google Scholar] [CrossRef]
- Chen, L.; Min, J.; Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Target. Ther. 2022, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Turski, M.L.; Thiele, D.J. New Roles for Copper Metabolism in Cell Proliferation, Signaling, and Disease. J. Biol. Chem. 2009, 284, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Michel, B.; Sinha, A.K. The inactivation of rfaP, rarA or sspA gene improves the viability of the Escherichia coli DNA polymerase III holD mutant. Mol. Microbiol. 2017, 104, 1008–1026. [Google Scholar] [CrossRef]
- Forman, R.; Logunova, L.; Smith, H.; Wemyss, K.; Mair, I.; Boon, L.; Allen, J.E.; Muller, W.; Pennock, J.L.; Else, K.J. Trichuris muris infection drives cell-intrinsic IL4R alpha independent colonic RELMα+ macrophages. PLoS Pathog. 2021, 17, e1009768. [Google Scholar] [CrossRef]
- Rowland, J.L.; Niederweis, M. Resistance mechanisms of Mycobacterium tuberculosis against phagosomal copper overload. Tuberculosis 2012, 92, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Terán, I.; Goh, K.G.; Ulett, G.C. Resisting death by metal: Metabolism and Cu/Zn homeostasis in bacteria. Emerg. Top. Life Sci. 2024, 8, 45–56. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, B.; Luo, J.; Yang, Y.; Weng, Q.; Fang, S.; Zhao, Z.; Tu, J.; Chen, M.; Ji, J. Cuproptosis, ferroptosis and PANoptosis in tumor immune microenvironment remodeling and immunotherapy: Culprits or new hope. Mol. Cancer 2024, 23, 255. [Google Scholar] [CrossRef]
- Luo, Z.; Lu, R.; Shi, T.; Ruan, Z.; Wang, W.; Guo, Z.; Zhan, Z.; Ma, Y.; Lian, X.; Ding, C.; et al. Enhanced Bacterial Cuproptosis-Like Death via Reversal of Hypoxia Microenvironment for Biofilm Infection Treatment. Adv. Sci. 2024, 11, e2308850. [Google Scholar] [CrossRef]
- Guo, R.; Fang, X.; Shang, K.; Wen, J.; Ding, K. Induction of ferroptosis: A new strategy for the control of bacterial infections. Microbiol. Res. 2024, 284, 127728. [Google Scholar] [CrossRef]
- Jinson, S.; Zhang, Z.; Lancaster, G.I.; Murphy, A.J.; Morgan, P.K. Iron, lipid peroxidation and ferroptosis play pathogenic roles in atherosclerosis. Cardiovasc. Res. 2024. [Google Scholar] [CrossRef]
- Zhao, R.; Sukocheva, O.; Tse, E.; Neganova, M.; Aleksandrova, Y.; Zheng, Y.; Gu, H.; Zhao, D.; Madhunapantula, S.V.; Zhu, X.; et al. Cuproptosis, the novel type of oxidation-induced cell death in thoracic cancers: Can it enhance the success of immunotherapy? Cell Commun. Signal. 2024, 22, 379. [Google Scholar] [CrossRef] [PubMed]
- Lou, Q.-M.; Lai, F.-F.; Li, J.-W.; Mao, K.-J.; Wan, H.-T.; He, Y. Mechanisms of cuproptosis and its relevance to distinct diseases. Apoptosis 2024, 29, 981–1006. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, L.; Zhang, Z.; Chen, P.; Shu, H.; Yang, C.; Chu, Y.; Liu, J. Ferroptosis: An important player in the inflammatory response in diabetic nephropathy. Front. Immunol. 2023, 14, 1294317. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ma, J.-Q.; Wang, C.-C.; Zhou, J.; Sun, Y.-D.; Wei, X.-L.; Zhao, Z.-Q. Ferroptosis: A potential target of macrophages in plaque vulnerability. Open Life Sci. 2023, 18, 20220722. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Wang, J.; Gao, G.F.; Liu, C.H. Insights into battles between Mycobacterium tuberculosis and macrophages. Protein Cell 2014, 5, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Zangiabadi, S.; Chamoun, K.P.; Nguyen, K.; Tang, Y.; Sweeney, G.; Abdul-Sater, A.A. Copper infused fabric attenuates inflammation in macrophages. PLoS ONE 2023, 18, e0287741. [Google Scholar] [CrossRef]
- Chen, Y.; Fang, Z.-M.; Yi, X.; Wei, X.; Jiang, D.-S. The interaction between ferroptosis and inflammatory signaling pathways. Cell Death Dis. 2023, 14, 205. [Google Scholar] [CrossRef]
- Dou, J.; Liu, X.; Yang, L.; Huang, D.; Tan, X. Ferroptosis interaction with inflammatory microenvironments: Mechanism, biology, and treatment. Biomed. Pharmacother. 2022, 155, 113711. [Google Scholar] [CrossRef]
- Scriba, T.J.; Maseeme, M.; Young, C.; Taylor, L.; Leslie, A.J. Immunopathology in human tuberculosis. Sci. Immunol. 2024, 9, eado5951. [Google Scholar] [CrossRef]
- Marinho, F.V.; Benmerzoug, S.; Rose, S.; Campos, P.C.; Marques, J.T.; Báfica, A.; Barber, G.; Ryffel, B.; Oliveira, S.C.; Quesniaux, V.F. The cGAS/STING Pathway Is Important for Dendritic Cell Activation but Is Not Essential to Induce Protective Immunity against Mycobacterium tuberculosis Infection. J. Innate Immun. 2018, 10, 239–252. [Google Scholar] [CrossRef]
- Feng, Q.; Huo, C.; Wang, M.; Huang, H.; Zheng, X.; Xie, M. Research progress on cuproptosis in cancer. Front. Pharmacol. 2024, 15, 1290592. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Yang, Y.; Gao, Y.; He, J. Cuproptosis: Mechanisms and links with cancers. Mol. Cancer 2023, 22, 46. [Google Scholar] [CrossRef] [PubMed]
- Amaral, E.P.; Namasivayam, S.; Queiroz, A.T.L.; Fukutani, E.; Hilligan, K.L.; Aberman, K.; Fisher, L.; Bomfim, C.C.B.; Kauffman, K.; Buchanan, J.; et al. BACH1 promotes tissue necrosis and Mycobacterium tuberculosis susceptibility. Nat. Microbiol. 2023, 9, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Darwin, K.H. Copper homeostasis in Mycobacterium tuberculosis. Metallomics 2015, 7, 929–934. [Google Scholar] [CrossRef]
- Ansari, M.Y.; Batra, S.D.; Ojha, H.; Dhiman, K.; Ganguly, A.; Tyagi, J.S.; Mande, S.C. A novel function of Mycobacterium tuberculosis chaperonin paralog GroEL1 in copper homeostasis. FEBS Lett. 2020, 594, 3305–3323. [Google Scholar] [CrossRef]
- Marcus, S.A.; Sidiropoulos, S.W.; Steinberg, H.; Talaat, A.M. CsoR Is Essential for Maintaining Copper Homeostasis in Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0151816. [Google Scholar] [CrossRef]
- Kim, R.; Hashimoto, A.; Markosyan, N.; Tyurin, V.A.; Tyurina, Y.Y.; Kar, G.; Gabrilovich, D.I. Ferroptosis of tumour neu-trophils causes immune suppression in cancer. Nature 2022, 612, 338–346. [Google Scholar] [CrossRef]
- Mao, H.; Zhao, Y.; Li, H.; Lei, L. Ferroptosis as an emerging target in inflammatory diseases. Prog. Biophys. Mol. Biol. 2020, 155, 20–28. [Google Scholar] [CrossRef]
- Cai, Z.; He, Y.E.; Yu, Z.; Hu, J.; Xiao, Z.; Zu, X.; Li, H. Cuproptosis-related modification patterns depict the tumor microen-vironment, precision immunotherapy, and prognosis of kidney renal clear cell carcinoma. Front. Immunol. 2022, 13, 933241. [Google Scholar] [CrossRef]
- Zeng, J.; Chen, H.; Liu, X.; Xia, H.; Chen, L.; Lin, D.; Wang, N.; Weng, C.; Guan, G.; Zheng, Y. Cuproptosis in microsatellite stable colon cancer cells affects the cytotoxicity of CD8+T through the WNT signaling pathway. Chem. Interact. 2024, 403, 111239. [Google Scholar] [CrossRef]
- Feng, S.; Zhang, Y.; Zhu, H.; Jian, Z.; Zeng, Z.; Ye, Y.; Xiong, X. Cuproptosis facilitates immune activation but promotes immune escape, and a machine learning–based cuproptosis-related signature is identified for predicting prognosis and immunotherapy response of gliomas. CNS Neurosci. Ther. 2024, 30, e14380. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Chen, G.; Yu, K.N.; Feng, Y.; Zhao, L.; Yang, M.; Cao, W.; Almahi, W.A.A.; Sun, M.; Xu, Y.; et al. Synergism of non-thermal plasma and low concentration RSL3 triggers ferroptosis via promoting xCT lysosomal degradation through ROS/AMPK/mTOR axis in lung cancer cells. Cell Commun. Signal. 2024, 22, 112. [Google Scholar] [CrossRef] [PubMed]
- Dirik, H.; Taşkıran, A.Ş.; Joha, Z. Ferroptosis inhibitor ferrostatin-1 attenuates morphine tolerance development in male rats by inhibiting dorsal root ganglion neuronal ferroptosis. Korean J. Pain 2024, 37, 233–246. [Google Scholar] [CrossRef]
- Tang, D.; Kroemer, G.; Kang, R. Ferroptosis in immunostimulation and immunosuppression. Immunol. Rev. 2023, 321, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting ferroptosis opens new avenues for the development of novel therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.-A.; Hsu, C.-H.; Lin, M.-C.; Chu, Y.-H.; Hung, Y.-M.; Wei, J.C.-C. Association of iron deficiency anemia with tuberculosis in Taiwan: A nationwide population-based study. PLoS ONE 2019, 14, e0221908. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, Y.; Li, H.; Han, L. FIN56, a novel ferroptosis inducer, triggers lysosomal membrane permeabilization in a TFEB-dependent manner in glioblastoma. J. Cancer 2021, 12, 6610–6619. [Google Scholar] [CrossRef]
- Ngwane, A.H.; Petersen, R.; Baker, B.; Wiid, I.; Wong, H.N.; Haynes, R.K. The evaluation of the anti-cancer drug elesclomol that forms a redox-active copper chelate as a potential anti-tubercular drug. IUBMB Life 2019, 71, 532–538. [Google Scholar] [CrossRef]
- Tarin, M.; Babaie, M.; Eshghi, H.; Matin, M.M.; Saljooghi, A.S. Elesclomol, a copper-transporting therapeutic agent targeting mitochondria: From discovery to its novel applications. J. Transl. Med. 2023, 21, 745. [Google Scholar] [CrossRef]
- Cobine, P.A.; Brady, D.C. Cuproptosis: Cellular and molecular mechanisms underlying copper-induced cell death. Mol. Cell 2022, 82, 1786–1787. [Google Scholar] [CrossRef] [PubMed]
- Smith, I. Mycobacterium tuberculosis Pathogenesis and Molecular Determinants of Virulence. Clin. Microbiol. Rev. 2003, 16, 463–496. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Virmani, T.; Kumar, G.; Deshmukh, R.; Sharma, A.; Duarte, S.; Brandão, P.; Fonte, P. Nanocarriers in Tuberculosis Treatment: Challenges and Delivery Strategies. Pharmaceuticals 2023, 16, 1360. [Google Scholar] [CrossRef]
- Garhyan, J.; Mohan, S.; Rajendran, V.; Bhatnagar, R. Preclinical Evidence of Nanomedicine Formulation to Target Mycobacterium tuberculosis at Its Bone Marrow Niche. Pathogens 2020, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Briehl, M.M.; Mazar, A.P.; Batinic-Haberle, I.; Reboucas, J.S.; Glinsmann-Gibson, B.; Rimsza, L.M.; Tome, M.E. The copper chelator ATN-224 induces peroxynitrite-dependent cell death in hematological malignancies. Free. Radic. Biol. Med. 2013, 60, 157–167. [Google Scholar] [CrossRef]
- Doñate, F.; Juarez, J.C.; Burnett, M.E.; Manuia, M.M.; Guan, X.; Shaw, D.E.; Smith, E.L.P.; Timucin, C.; Braunstein, M.J.; Batuman, O.A.; et al. Identification of biomarkers for the antiangiogenic and antitumour activity of the superoxide dismutase 1 (SOD1) inhibitor tetrathiomolybdate (ATN-224). Br. J. Cancer 2008, 98, 776–783. [Google Scholar] [CrossRef]
- Maphasa, R.E.; Meyer, M.; Dube, A. The Macrophage Response to Mycobacterium tuberculosis and Opportunities for Autophagy Inducing Nanomedicines for Tuberculosis Therapy. Front. Cell. Infect. Microbiol. 2021, 10, 618414. [Google Scholar] [CrossRef]
- Anes, E.; Pires, D.; Mandal, M.; Azevedo-Pereira, J.M. ESAT-6 a Major Virulence Factor of Mycobacterium tuberculo-sis. Biomolecules 2023, 13, 968. [Google Scholar] [CrossRef]
- Mohareer, K.; Medikonda, J.; Vadankula, G.R.; Banerjee, S. Mycobacterial Control of Host Mitochondria: Bioenergetic and Metabolic Changes Shaping Cell Fate and Infection Outcome. Front. Cell. Infect. Microbiol. 2020, 10. [Google Scholar] [CrossRef]
- Rytter, H.; Roger, K.; Chhuon, C.; Ding, X.; Coureuil, M.; Jamet, A.; Henry, T.; Guerrera, I.C.; Charbit, A. Dual proteomics of infected macrophages reveal bacterial and host players involved in the Francisella intracellular life cycle and cell to cell dissemination by merocytophagy. Sci. Rep. 2024, 14, 7797. [Google Scholar] [CrossRef]
- Allam, R.; E Lawlor, K.; Yu, E.C.; Mildenhall, A.L.; Moujalled, D.M.; Lewis, R.S.; Ke, F.; Mason, K.D.; White, M.J.; Stacey, K.J.; et al. Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. Embo Rep. 2014, 15, 982–990. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Therapeutic Strategy | Compound | Dose | Mechanism | Potential Challenges |
|---|---|---|---|---|
| Ferroptosis Inducers | Erastin | 5–10 μM | Increases iron and ROS for lipid peroxidation and cell death | Ensuring specificity to infected cells to prevent healthy cell damage |
| RSL3 | 0.25–0.5 μM | Triggers ferroptosis in infected cells | Dose monitoring to avoid excessive ROS and tissue inflammation | |
| FIN56 | 0.1–1 μM | Enhances lipid peroxidation by targeting GPX4 | Long-term toxicity with chronic iron manipulation | |
| Lipid Peroxidation Inhibitors | Ferrostatin-1 | 1–5 μM | Balances ROS to protect uninfected cells | Potential systemic oxidative stress |
| Iron Chelators | Deferoxamine | 500–1000 mg/day (lower in TB) | Reduces iron to control ROS and ferroptosis | Risk of iron deficiency or worsening anemia |
| Cuproptosis Inducers | Disulfiram | 250–500 mg/day | Enhances copper uptake for cell death in infected cells | Copper toxicity risks |
| Elesclomol | 5–10 mg/m2 | Facilitates copper ion uptake | Liver and kidney toxicity from excess copper | |
| Copper Chelators | Tetrathiomolybdate | 20 mg/day | Manages copper toxicity | Need for dose adjustment and monitoring |
| SOD1 Inhibitor | ATN-224 | 50–100 mg/day | Enhances oxidative stress in infected cells | Monitoring to minimize non-infected |
| Compound Name | Chemical Structure (IUPAC Name) | Type | Clinical Usage |
|---|---|---|---|
| Erastin | 2-[1-[4-[2-(Dimethylamino)ethoxy]phenyl]ethylidene]indolin-3-one | Small molecule, ferroptosis inducer | Not used clinically |
| RSL3 | 1S,3R-RSL3: [(1S,3R)-2-chloro-3-[[(2,4-dichlorobenzyl)sulfanyl]methyl]cyclohexyl]methyl sulfide | Small molecule, ferroptosis inducer | Not used clinically |
| FIN56 | N-[4-[[4-(1,3-Benzothiazol-2-yl)piperidin-1-yl]methyl]phenyl]quinolin-4-amine | Small molecule targeting GPX4 | Not used clinically |
| Ferrostatin-1 | 3-[4-(Phenylamino)cyclohexyl]propanoic acid | Small molecule, lipid peroxidation inhibitor | Not used clinically |
| Deferoxamine | N-[5-[[4-[5-(Acetylhydroxyamino)pentylamino]-4-oxobutanoyl]amino]pentyl]-N-hydroxyacetamide | Iron chelator | Used clinically for iron-overload conditions |
| Disulfiram | N,N-Bis(diethylthiocarbamoyl)disulfide | Alcohol deterrent and cuproptosis inducer | Used clinically for alcohol dependency |
| Elesclomol | 2-(4-Chlorophenyl)-1-[3-(dimethylamino)propyl]-1,3-dihydroimidazol-3-one | Anticancer agent and cuproptosis inducer | Investigated in clinical trials; not widely used clinically |
| Tetrathiomolybdate | MoS42− | Copper chelator | Investigated in clinical trials; limited clinical use |
| ATN-224 | Ammonium tetrathiomolybdate | Small molecule, SOD1 inhibitor | Investigated in clinical trials; not used clinically |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dawi, J.; Affa, S.; Kafaja, K.; Misakyan, Y.; Kades, S.; Dayal, S.; Fardeheb, S.; Narasimhan, A.; Tumanyan, K.; Venketaraman, V. The Role of Ferroptosis and Cuproptosis in Tuberculosis Pathogenesis: Implications for Therapeutic Strategies. Curr. Issues Mol. Biol. 2025, 47, 99. https://doi.org/10.3390/cimb47020099
Dawi J, Affa S, Kafaja K, Misakyan Y, Kades S, Dayal S, Fardeheb S, Narasimhan A, Tumanyan K, Venketaraman V. The Role of Ferroptosis and Cuproptosis in Tuberculosis Pathogenesis: Implications for Therapeutic Strategies. Current Issues in Molecular Biology. 2025; 47(2):99. https://doi.org/10.3390/cimb47020099
Chicago/Turabian StyleDawi, John, Stephen Affa, Kevin Kafaja, Yura Misakyan, Samuel Kades, Surbi Dayal, Sabrina Fardeheb, Ananya Narasimhan, Kevin Tumanyan, and Vishwanath Venketaraman. 2025. "The Role of Ferroptosis and Cuproptosis in Tuberculosis Pathogenesis: Implications for Therapeutic Strategies" Current Issues in Molecular Biology 47, no. 2: 99. https://doi.org/10.3390/cimb47020099
APA StyleDawi, J., Affa, S., Kafaja, K., Misakyan, Y., Kades, S., Dayal, S., Fardeheb, S., Narasimhan, A., Tumanyan, K., & Venketaraman, V. (2025). The Role of Ferroptosis and Cuproptosis in Tuberculosis Pathogenesis: Implications for Therapeutic Strategies. Current Issues in Molecular Biology, 47(2), 99. https://doi.org/10.3390/cimb47020099