
Metabolomics and Transcriptomics Jointly Explore the Mechanism of Pod Color Variation in Purple Pod Pea


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Metabolite Detection and Metabolomic Data Analysis
2.3. RNA Isolation, Sequencing and Bioinformatics Analysis
2.4. Real-Time Quantitative Polymerase Chain Reaction (RT-qPCR) Validation
2.5. Weighted Gene Co-Expression Network Analysis (WGCNA)
3. Results
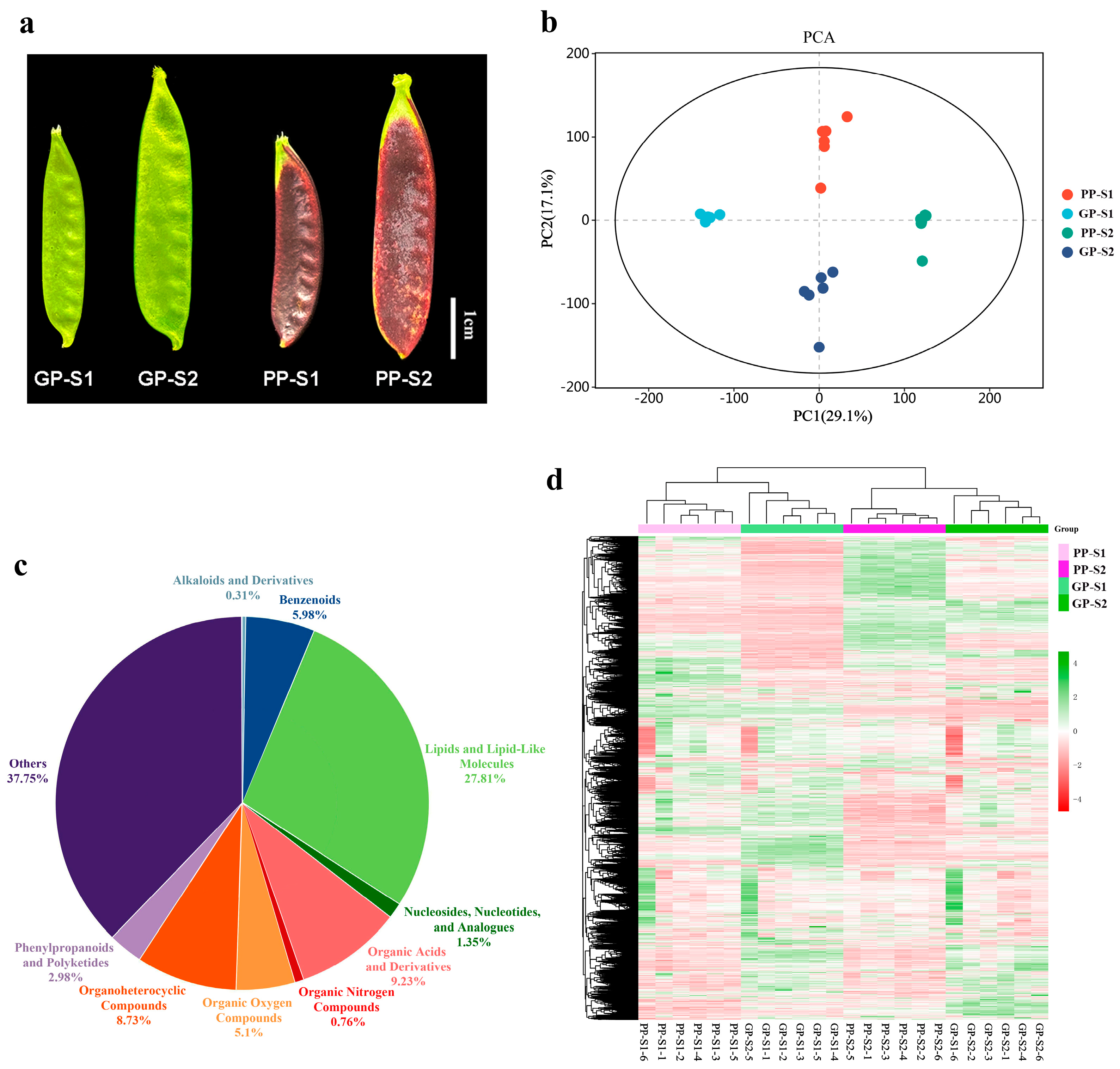
3.1. Metabolome Analysis of Green and Purple Pods
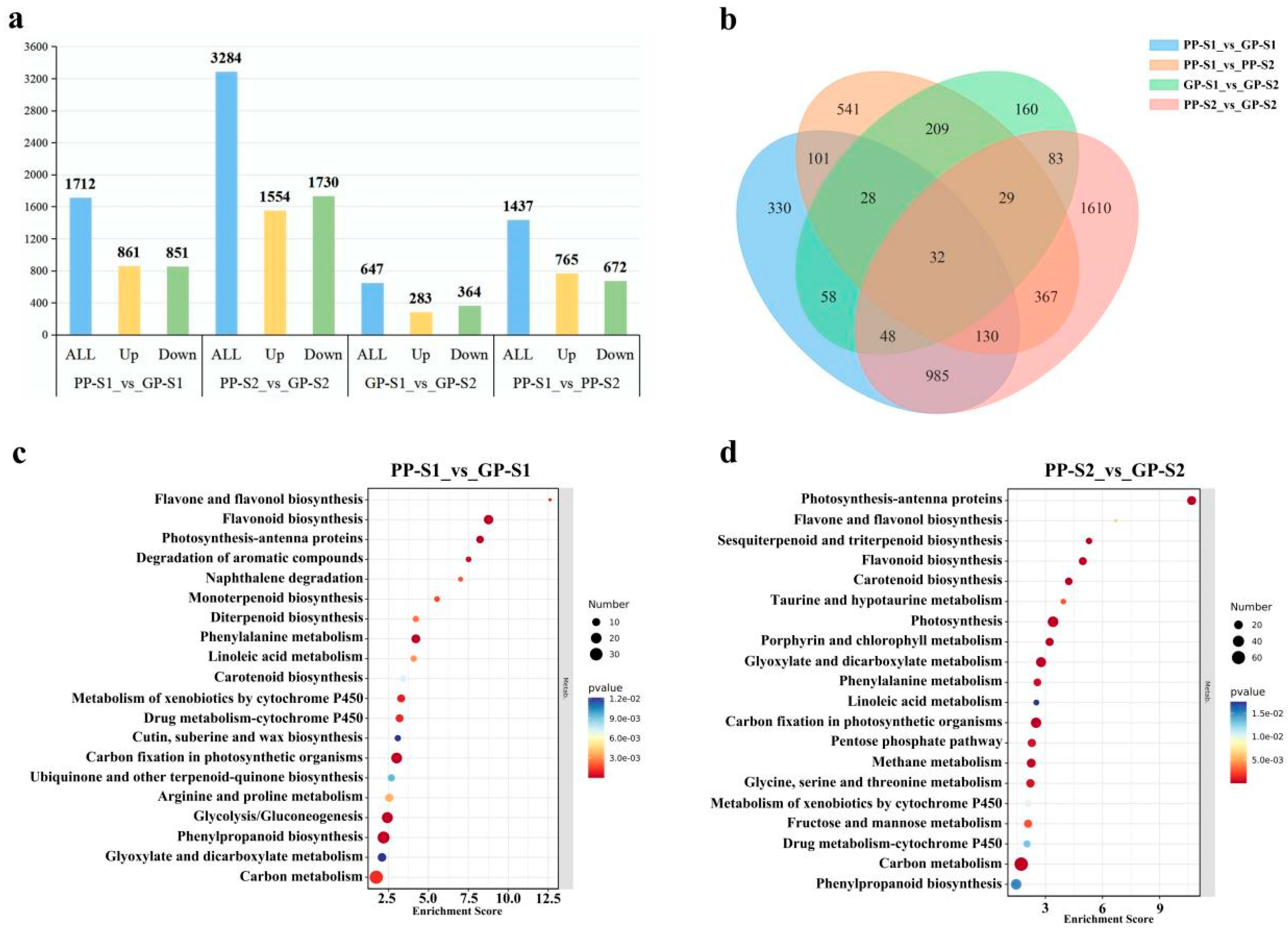
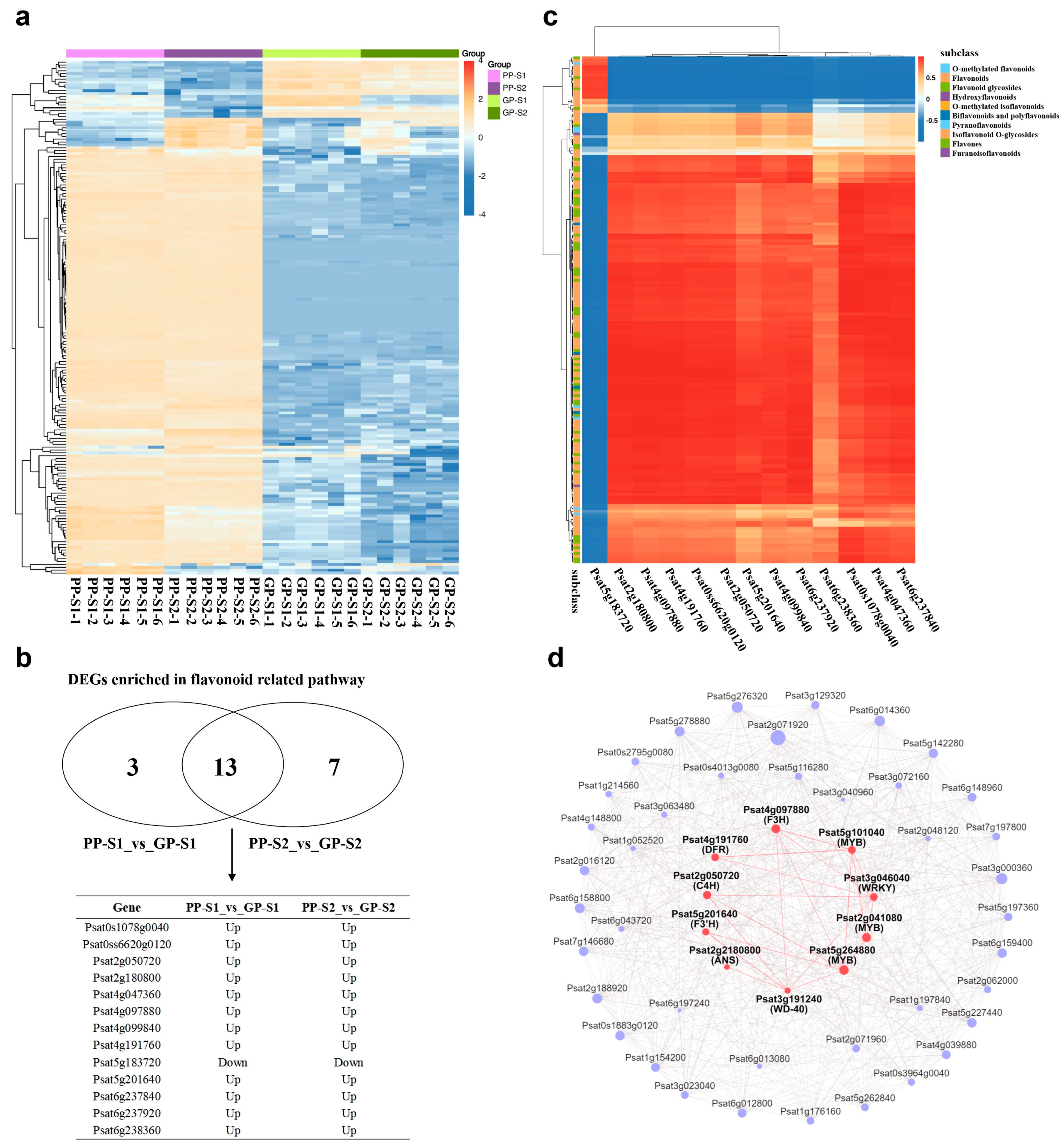
3.2. Transcriptome Analysis of Green and Purple Pods
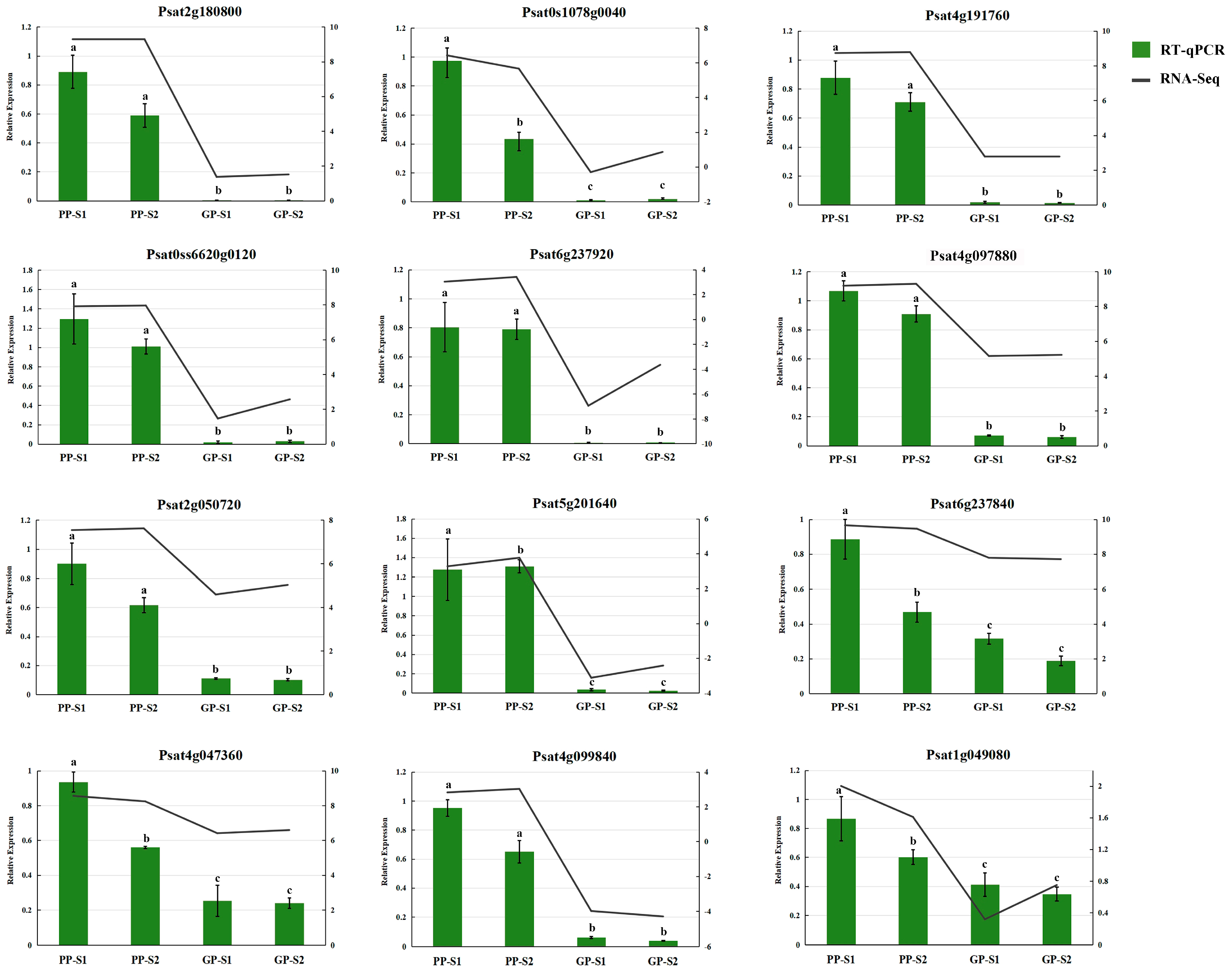
3.3. Verification of RNA-Seq Data by RT-qPCR
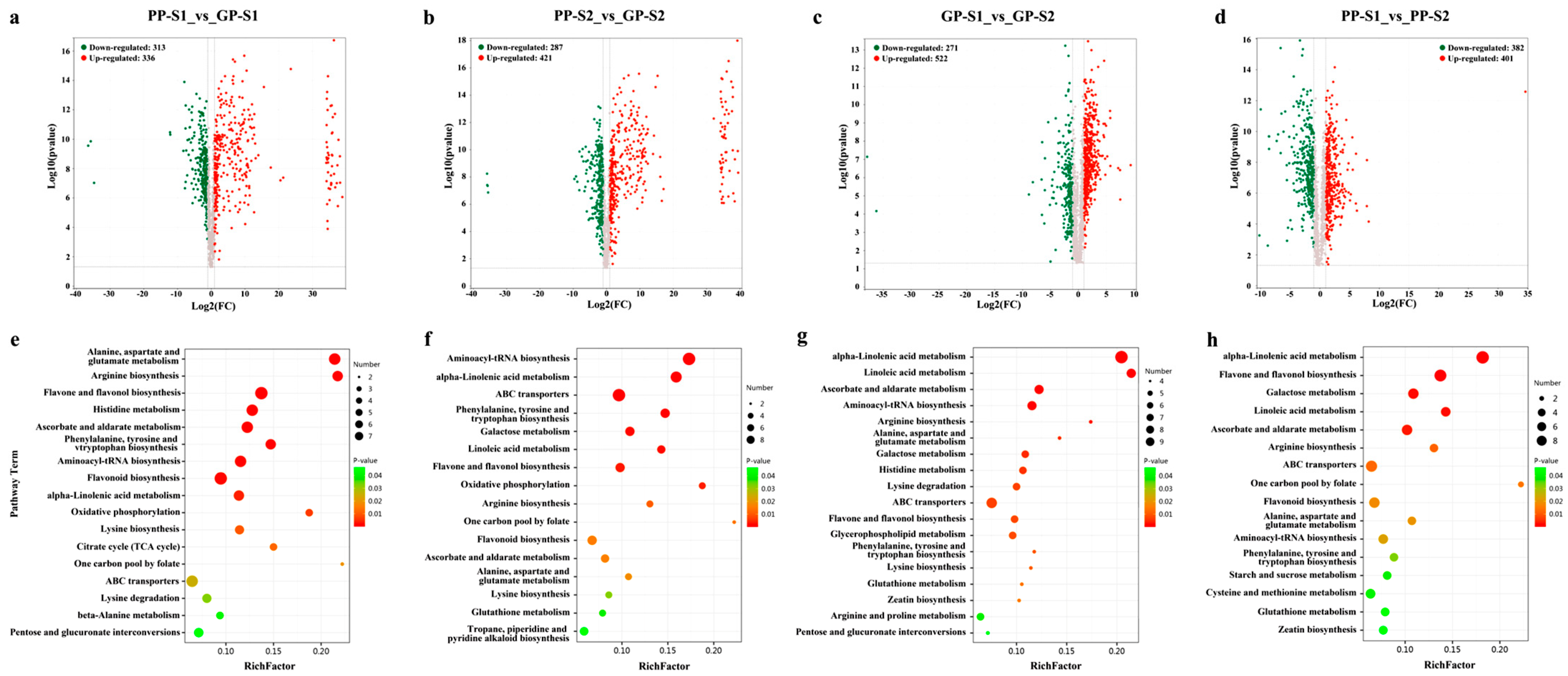
3.4. Analysis of DAMs and DEGs Involved in the Flavonoid Biosynthesis Pathway
3.5. Association Analysis of Transcriptome and Metabolome Involved in the Flavonoid Metabolic Network
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- McCrory, M.A.; Hamaker, B.R.; Lovejoy, J.C.; Eichelsdoerfer, P.E. Pulse consumption, satiety, and weight management. Adv. Nutr. 2010, 1, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Zohary, D.; Hopf, M. Domestication of pulses in the Old World: Legumes were companions of wheat and barley when agriculture began in the Near East. Science 1973, 182, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Smýkal, P.; Coyne, C.J.; Ambrose, M.J.; Maxted, N.; Schaefer, H.; Blair, M.W.; Berger, J.; Greene, S.L.; Nelson, M.N.; Besharat, N.; et al. Legume Crops Phylogeny and Genetic Diversity for Science and Breeding. Crit. Rev. Plant Sci. 2014, 34, 43–104. [Google Scholar] [CrossRef]
- Ellis, T.H.; Hofer, J.M.; Timmerman-Vaughan, G.M.; Coyne, C.J.; Hellens, R.P. Mendel, 150 years on. Trends Plant Sci. 2011, 16, 590–596. [Google Scholar] [CrossRef]
- Shirasawa, K.; Sasaki, K.; Hirakawa, H.; Isobe, S. Genomic region associated with pod color variation in pea (Pisum sativum). G3 2021, 11. [Google Scholar] [CrossRef]
- Liu, N.; Lyu, X.L.; Zhang, X.Y.; Zhang, G.W.; Zhang, Z.Q.; Guan, X.Y.; Chen, X.Y.; Yang, X.M.; Feng, Z.J.; Gao, Q.; et al. Reference genome sequence and population genomic analysis of peas provide insights into the genetic basis of Mendelian and other agronomic traits. Nat. Genet. 2024, 56, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Chen, B.Z.; Hofer, J.; Shi, Y.; Jiang, M.; Song, B.; Cheng, H.; Lu, L.; Wang, L.Y.; Howard, A.; et al. Genomic and Genetic Insights into Mendel’s Pea genes. bioRxiv 2024, 5, 31. [Google Scholar] [CrossRef]
- Yamuangmorn, S.; Prom-u-Thai, C. The Potential of High-Anthocyanin Purple Rice as a Functional Ingredient in Human Health. Antioxidants 2021, 10, 833. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Tao, J. Recent advances on the development and regulation of flower color in ornamental plants. Front. Plant Sci. 2015, 6, 261. [Google Scholar] [CrossRef]
- Chayut, N.; Yuan, H.; Ohali, S.; Meir, A.; Yeselson, Y.; Portnoy, V.; Zheng, Y.; Fei, Z.; Lewinsohn, E.; Katzir, N.; et al. A bulk segregant transcriptome analysis reveals metabolic and cellular processes associated with Orange allelic variation and fruit beta-carotene accumulation in melon fruit. BMC Plant Biol. 2015, 15, 274. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, G.; Xu, S.; Mao, W.; Hu, Q.; Gong, Y. Comparative Transcriptomic Analyses of Vegetable and Grain Pea (Pisum sativum L.) Seed Development. Front. Plant Sci. 2015, 6, 1039. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.Z.; Yang, M.; Zhang, X.Y.; Fan, Y.F.; Wang, X.S.; Xiang, C. Comparative analysis of transcriptome and metabolome explores the underlying mechanism of pod color variation in pea (Pisum sativum L.). J. Plant. Biochem. Biot. 2024, 33, 144–156. [Google Scholar] [CrossRef]
- He, J.; Giusti, M.M. Anthocyanins: Natural Colorants with Health-Promoting Properties. Annu. Rev. Food Sci. Technol. 2010, 1, 163–187. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Zhang, Y.; Butelli, E.; Martin, C. Engineering anthocyanin biosynthesis in plants. Curr. Opin. Plant Biol. 2014, 19, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, R.; Francioso, A.; Mosca, L.; Silva, P. Anthocyanins: A Comprehensive Review of Their Chemical Properties and Health Effects on Cardiovascular and Neurodegenerative Diseases. Molecules 2020, 25, 3809. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.Y.; Lou, Q.; Li, H.B.; Yue, J.; Liu, Y.L.; Wang, Y.J. Anatomical and biochemical studies of bicolored flower development in Muscari latifolium. Protoplasma 2013, 250, 1273–1281. [Google Scholar] [CrossRef]
- Lou, Q.; Liu, Y.L.; Qi, Y.Y.; Jiao, S.Z.; Tian, F.F.; Jiang, L.; Wang, Y.J. Transcriptome sequencing and metabolite analysis reveals the role of delphinidin metabolism in flower color in grape hyacinth. J. Exp. Bot. 2014, 65, 3157–3164. [Google Scholar] [CrossRef]
- Bloor, S.J.; Falshaw, R. Covalently linked anthocyanin-flavonol pigments from blue Agapanthus flowers. Phytochemistry 2000, 53, 575–579. [Google Scholar] [CrossRef]
- Mizuno, T.; Yabuya, T.; Kitajima, J.; Iwashina, T. Identification of novel Cglycosylflavones and their contribution to flower colour of the Dutch iris cultivars. Plant Physiol. Biochem. 2013, 72, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Kondo, T. Structure and molecular stacking of anthocyanins—Flower color variation. Angew. Chem. Int. Ed. Engl. 1991, 30, 17–33. [Google Scholar] [CrossRef]
- Ballester, A.R.; Molthoff, J.; de Vos, R.; Hekkert, B.; Orzaez, D.; Fernandez-Moreno, J.P.; Tripodi, P.; Grandillo, S.; Martin, C.; Heldens, J.; et al. Biochemical and molecular analysis of pink tomatoes: Deregulated expression of the gene encoding transcription factor SlMYB12 leads to pink tomato fruit color. Plant Physiol. 2010, 152, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of plant pigments: Anthocyanins, betalains and carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Springob, K.; Nakajima, J.I.; Yamazaki, M.; Saito, K. Recent advances in the biosynthesis and accumulation of anthocyanins. Nat. Prod. Rep. 2003, 20, 288–303. [Google Scholar] [CrossRef] [PubMed]
- Koes, R.; Verweij, W.; Quattrocchio, F. Flavonoids: A colorful model for the regulation and evolution of biochemical pathways. Trends Plant Sci. 2005, 10, 236–242. [Google Scholar] [CrossRef]
- Jaakola, L. New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci. 2013, 18, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Van Nocker, S.; Ludwig, P. The WD-repeat protein superfamily in Arabidopsis: Conservation and divergence in structure and function. BMC Genom. 2003, 4, 1–11. [Google Scholar] [CrossRef]
- Espley, R.V.; Brendolise, C.; Chagne, D.; Kutty-Amma, S.; Green, S.; Volz, R.; Putterill, J.; Schouten, H.J.; Gardiner, S.E.; Hellens, R.P. Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell 2009, 21, 168–183. [Google Scholar] [CrossRef]
- Espley, R.V.; Hellens, R.P.; Putterill, J.; Stevenson, D.E.; Kutty-Amma, S.; Allan, A.C. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2007, 49, 414–427. [Google Scholar] [CrossRef]
- Mahmoudi, E.; MOHAMMAD, S.B.; Yadollahi, A.; Hosseini, E. Independence of color intensity variation in red flesh apples from the number of repeat units in promoter region of the MdMYB10 gene as an allele to MdMYB1 and MdMYBA. Iran. J. Biotechnol. 2012, 2012, 153–160. [Google Scholar]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814–827. [Google Scholar] [CrossRef]
- Jiang, W.; Liu, T.; Nan, W.; Jeewani, D.C.; Niu, Y.; Li, C.; Wang, Y.; Shi, X.; Wang, C.; Wang, J. Two transcription factors TaPpm1 and TaPpb1 co-regulate anthocyanin biosynthesis in purple pericarps of wheat. J. Exp. Bot. 2018, 69, 2555–2567. [Google Scholar] [CrossRef]
- Spelt, C.; Quattrocchio, F.; Mol, J.; Koes, R. ANTHOCYANIN1 of petunia controls pigment synthesis, vacuolar pH, and seed coat development by genetically distinct mechanisms. Plant Cell 2002, 14, 2121–2135. [Google Scholar] [CrossRef] [PubMed]
- Schaart, J.G.; Dubos, C.; Romero De La Fuente, I.; van Houwelingen, A.M.; de Vos, R.C.; Jonker, H.H.; Xu, W.; Routaboul, M.J.; Lepiniec, L.; Bovy, A.G. Identification and characterization of MYB-bHLH-WD40 regulatory complexes controlling proanthocyanidin biosynthesis in strawberry (Fragaria x ananassa) fruits. New Phytol. 2013, 197, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Grain, D.; Le Gourrierec, J.; Harscoët, E.; Berger, A.; Jauvion, V.; Scagnelli, A.; Berger, N.; Bidzinski, P.; Kelemen, Z.; et al. Regulation of flavonoid biosynthesis involves an unexpected complex transcriptional regulation of TT8 expression, in Arabidopsis. New Phytol. 2013, 198, 59–70. [Google Scholar] [CrossRef]
- Li, H.; He, K.; Zhang, Z.; Hu, Y. Molecular mechanism of phosphorous signaling inducing anthocyanin accumulation in Arabidopsis. Plant Physiol. Biochem. 2023, 196, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lu, W.; Ran, L.; Dou, L.; Yao, S.; Hu, J.; Fan, D.; Li, C.; Luo, K. R2R3-MYB transcription factor MYB6 promotes anthocyanin and proanthocyanidin biosynthesis but inhibits secondary cell wall formation in Populus tomentosa. Plant J. 2019, 99, 733–751. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Mendenhall, J.; Huo, Y.; Lloyd, A. TTG1 complex MYBs, MYB5 and TT2, control outer seed coat differentiation. Dev. Biol. 2009, 325, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Jun, J.H.; Dixon, R.A. MYB5 and MYB14 play pivotal roles in seed coat polymer biosynthesis in Medicago truncatula. Plant Physiol. 2014, 165, 1424–1439. [Google Scholar] [CrossRef] [PubMed]
- Akagi, T.; Ikegami, A.; Tsujimoto, T.; Kobayashi, S.; Sato, A.; Kono, A.; Yonemori, K. DkMyb4 Is a Myb transcription factor involved in proanthocyanidin biosynthesis in Persimmon fruit. Plant Physiol. 2009, 151, 2028–2045. [Google Scholar] [CrossRef] [PubMed]
- Deluc, L.; Barrieu, F.; Marchive, C.; Lauvergeat, V.; Decendit, A.; Richard, T.; Carde, J.P.; Merillon, J.M.; Hamdi, S. Characterization of a grapevine R2R3-MYB transcription factor that regulates the phenylpropanoid pathway. Plant Physiol. 2006, 140, 499–511. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, X.; Yang, M.; Zhang, X.; Fan, Y.; Wang, X.; Xiang, C. Metabolomics and Transcriptomics Jointly Explore the Mechanism of Pod Color Variation in Purple Pod Pea. Curr. Issues Mol. Biol. 2025, 47, 94. https://doi.org/10.3390/cimb47020094
Zhong X, Yang M, Zhang X, Fan Y, Wang X, Xiang C. Metabolomics and Transcriptomics Jointly Explore the Mechanism of Pod Color Variation in Purple Pod Pea. Current Issues in Molecular Biology. 2025; 47(2):94. https://doi.org/10.3390/cimb47020094
Chicago/Turabian StyleZhong, Xiaojuan, Mei Yang, Xiaoyan Zhang, Yuanfang Fan, Xianshu Wang, and Chao Xiang. 2025. "Metabolomics and Transcriptomics Jointly Explore the Mechanism of Pod Color Variation in Purple Pod Pea" Current Issues in Molecular Biology 47, no. 2: 94. https://doi.org/10.3390/cimb47020094
APA StyleZhong, X., Yang, M., Zhang, X., Fan, Y., Wang, X., & Xiang, C. (2025). Metabolomics and Transcriptomics Jointly Explore the Mechanism of Pod Color Variation in Purple Pod Pea. Current Issues in Molecular Biology, 47(2), 94. https://doi.org/10.3390/cimb47020094