
GPX4 Inhibition Enhances the Pro-Oxidant and ER Stress Effects of Tempol in Colon and Gastric Cancer Cell Lines


Abstract
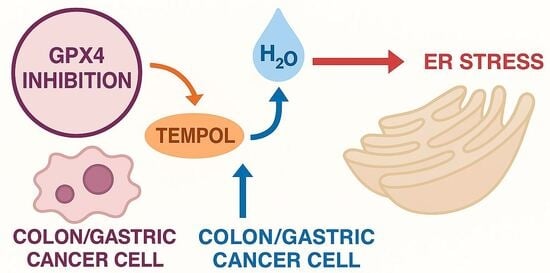
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Cell Viability and IC50 Determination (MTT Assay)
2.4. Cell Homogenization
2.5. Total Protein Quantification
2.6. Quantification of Oxidative Stress Markers
2.6.1. Intracellular Hydrogen Peroxide (H2O2) Level Quantification
2.6.2. Total Antioxidant Status and Total Oxidant Status Measurement
2.7. Quantification of ER Stress Markers (ELISA)
2.8. Statistical Analysis
3. Results
3.1. Dose–Response and Single-Agent Activity
3.2. Combination Cytotoxicity and Bliss Independence Analysis (Cell Viability)
3.3. Oxidative Stress: H2O2, TOS, TAS Indices
3.4. ER Stress Markers
3.5. Integrated Summary
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATF6 | Activating Transcription Factor 6 |
| BSA | Bovine Serum Albumin |
| CHOP | C/EBP Homologous Protein |
| CO2 | Carbon Dioxide |
| CRL | Cell Repository Line |
| DMEM | Dulbecco’s Modified Eagle Medium |
| DMSO | Dimethyl Sulfoxide |
| EGTA | Ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| ER | Endoplasmic Reticulum |
| FBS | Fetal Bovine Serum |
| FDA | Food and Drug Administration |
| GPX4 | Glutathione Peroxidase 4 |
| GRP78 | Glucose-Regulated Protein 78 |
| H2O2 | Hydrogen Peroxide |
| IC50 | Half Maximal Inhibitory Concentration |
| IRE1α | Inositol-Requiring Enzyme 1 alpha |
| JNK | c-Jun N-terminal Kinase |
| mM | millimolar |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NaCl | Sodium chloride |
| NaF | Sodium fluoride |
| nm | Nanometer |
| OD | Optical Density |
| PBS | Phosphate-Buffered Saline |
| PERK | Protein Kinase R-like ER Kinase |
| RIPA | Radioimmunoprecipitation Assay |
| ROS | Reactive Oxygen Species |
| rpm | Revolutions Per Minute |
| SD | Standard Deviation |
| Tris-HCl | Tris hydrochloride |
| UPR | Unfolded Protein Response |
| μg | Microgram |
| μL | Microliter |
| μM | Micromolar |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; Ciucis, C.D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox Homeostasis and Cellular Antioxidant Systems: Crucial Players in Cancer Growth and Therapy. Oxid. Med. Cell. Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef]
- Huang, R.; Chen, H.; Liang, J.; Li, Y.; Yang, J.; Luo, C.; Tang, Y.; Ding, Y.; Liu, X.; Yuan, Q.; et al. Dual Role of Reactive Oxygen Species and their Application in Cancer Therapy. J. Cancer 2021, 12, 5543–5561. [Google Scholar] [CrossRef]
- Kim, H.; Bhattacharya, A.; Qi, L. Endoplasmic reticulum quality control in cancer: Friend or foe. Semin. Cancer Biol. 2015, 33, 25–33. [Google Scholar] [CrossRef]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, G.; Kaplan, H.M. Tempol Induces Oxidative Stress, ER Stress and Apoptosis via MAPK/Akt/mTOR Pathway Suppression in HT29 (Colon) and CRL-1739 (Gastric) Cancer Cell Lines. Curr. Issues Mol. Biol. 2025, 47, 574. [Google Scholar] [CrossRef]
- Lewandowski, M.; Gwozdzinski, K. Nitroxides as Antioxidants and Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 2490. [Google Scholar] [CrossRef]
- Park, W.H. Tempol Inhibits the Growth of Lung Cancer and Normal Cells through Apoptosis Accompanied by Increased O2•− Levels and Glutathione Depletion. Molecules 2022, 27, 7341. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, I.M.U.; Santos, F.R.; da Silva, H.M.; Minatel, E.; Mesquitta, M.; Salvador, M.J.; Montico, F.; Cagnon, V.H.A. Tempol effect on oxidative and mitochondrial markers in preclinical models for prostate cancer. Toxicol. Res. 2024, 13, tfae056. [Google Scholar] [CrossRef]
- Ferreira, G.C.; Pinheiro, L.C.; Oliveira-Paula, G.H.; Angelis, C.D.; Portella, R.L.; Tanus-Santos, J.E. Antioxidant tempol modulates the increases in tissue nitric oxide metabolites concentrations after oral nitrite administration. Chem. Biol. Interact. 2021, 349, 109658. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Sun, S.; Johnson, T.; Qi, R.; Zhang, S.; Zhang, J.; Yang, K. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep. 2021, 35, 109235. [Google Scholar] [CrossRef] [PubMed]
- Weaver, K.; Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10, 891. [Google Scholar] [CrossRef]
- Park, W.H. Tempol differently affects cellular redox changes and antioxidant enzymes in various lung-related cells. Sci. Rep. 2021, 11, 14869. [Google Scholar] [CrossRef]
- Eaton, J.K.; Furst, L.; Ruberto, R.A.; Moosmayer, D.; Hilpmann, A.; Ryan, M.J.; Zimmermann, K.; Cai, L.L.; Niehues, M.; Badock, V.; et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 2020, 16, 497–506. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Li, B.; Zheng, C.; Liu, G.; Liu, Z.; Zhang, L.; Xu, P. Discovery of ML210-Based glutathione peroxidase 4 (GPX4) degrader inducing ferroptosis of human cancer cells. Eur. J. Med. Chem. 2023, 254, 115343. [Google Scholar] [CrossRef]
- Ai, L.; Li, R.; Wang, X.; Liu, Z.; Li, Y. Tempol alleviates acute lung injury by affecting glutathione synthesis through Nrf2 and inhibiting ferroptosis in lung epithelial cells. J. Biochem. Mol. Toxicol. 2024, 38, e23674. [Google Scholar] [CrossRef]
- Bliss, C.I. The toxicity of poisons applied jointly. Ann. Appl. Biol. 1939, 26, 585–615. [Google Scholar] [CrossRef]
- Demidenko, E.; Miller, T.W. Statistical determination of synergy based on Bliss definition of drugs independence. PLoS ONE 2019, 14, e0224137. [Google Scholar] [CrossRef]
- Monti, E.; Supino, R.; Colleoni, M.; Costa, B.; Ravizza, R.; Gariboldi, M.B. Nitroxide TEMPOL impairs mitochondrial function and induces apoptosis in HL60 cells. J. Cell. Biochem. 2001, 82, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Maiorino, M.; Conrad, M.; Ursini, F. GPx4, Lipid Peroxidation, and Cell Death: Discoveries, Rediscoveries, and Open Issues. Antioxid. Redox Signal 2018, 29, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, R.L.; Mesgarzadeh, J.S.; Hendershot, L.M. Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol. Cell 2022, 82, 1477–1491. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, D.H.; Choudry, H.A.; Bartlett, D.L.; Lee, Y.J. Ferroptosis-Induced Endoplasmic Reticulum Stress: Cross-talk between Ferroptosis and Apoptosis. Mol. Cancer Res. 2018, 16, 1073–1076. [Google Scholar] [CrossRef]
- Jordan, J.B.; Smallwood, M.J.; Smerdon, G.R.; Winyard, P.G. Cellular Pre-Adaptation to The High O2 Concentration Used in Standard Cell Culture Confers Resistance to Subsequent H2O2-Induced Cell Death. Antioxidants 2024, 13, 269. [Google Scholar] [CrossRef]
- Xin, X.; Gong, T.; Hong, Y. Hydrogen peroxide initiates oxidative stress and proteomic alterations in meningothelial cells. Sci. Rep. 2022, 12, 14519. [Google Scholar] [CrossRef]
- Halliwell, B.; Clement, M.V.; Ramalingam, J.; Long, L.H. Hydrogen peroxide. Ubiquitous in cell culture and in vivo? IUBMB Life 2000, 50, 251–257. [Google Scholar] [CrossRef]
- Siao, Y.J.; Peng, C.C.; Tung, Y.C.; Chen, Y.F. Comparison of Hydrogen Peroxide Secretion From Living Cells Cultured in Different Formats Using Hydrogel-Based LSPR Substrates. Front. Bioeng. Biotechnol. 2022, 10, 869184. [Google Scholar] [CrossRef]
- Whittemore, E.R.; Loo, D.T.; Watt, J.A.; Cotman, C.W. A detailed analysis of hydrogen peroxide-induced cell death in primary neuronal culture. Neuroscience 1995, 67, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Gülden, M.; Jess, A.; Kammann, J.; Maser, E.; Seibert, H. Cytotoxic potency of H2O2 in cell cultures: Impact of cell concentration and exposure time. Free Radic. Biol. Med. 2010, 49, 1298–1305. [Google Scholar] [CrossRef]
- Ong, H.L.; Subedi, K.P.; Son, G.Y.; Liu, X.; Ambudkar, I.S. Unfolding the Interactions between Endoplasmic Reticulum Stress and Oxidative Stress. Antioxidants 2023, 12, 981. [Google Scholar] [CrossRef]
- Paxman, R.; Plate, L.; Blackwood, E.A.; Glembotski, C.; Powers, E.T.; Wiseman, R.L.; Kelly, J.W. Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins. eLife 2018, 7, e37168. [Google Scholar] [CrossRef]
- Metz, J.M.; Smith, D.; Mick, R.; Lustig, R.; Mitchell, J.; Cherakuri, M.; Glatstein, E.; Hahn, S.M. A phase I study of topical Tempol for the prevention of alopecia induced by whole brain radiotherapy. Clin. Cancer Res. 2004, 10, 6411–6417. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ELISA-Based Protein Concentrations | Cell Line | Control Mean ± SD | Tempol Alone [Tempol (2 mM)] Mean ± SD | ML210 | Combination Treatment [Tempol (2 mM) + ML210 (0.05 μM)] (Mean ± SD) | p-Value *, 95%CI of Diff (Lower to Upper) | p-Value **, 95%CI of Diff (Lower to Upper) |
|---|---|---|---|---|---|---|---|
| IRE1α (pg/mL) | CRL-1739 | 110.00 ± 6.96 | 144.2 ± 9.99 | 20.5 ± 2.36 | 224.2 ± 17.44 | 0.0004, −51.46 to −16.87 | <0.0001, −131.5 to −96.87 |
| HT29 | 100.8 ± 8.28 | 132 ± 10.86 | 19 ± 2.36 | 238 ± 12 | 0.0002, −45.95 to −16.38 | <0.0001, −152.0 to −122.4 | |
| ATF6 (pg/mL) | CRL-1739 | 0.22 ± 0.05 | 0.36 ± 0.08 | 0.48 ± 0.16 | 0.98 ± 0.14 | 0.0549, −0.28 to 0.003 | <0.0001, −0.91 to −0.62 |
| HT29 | 0.21 ± 0.08 | 0.42 ± 0.09 | 0.44 ± 0.21 | 0.99 ± 0.16 | 0.0147, −0.37 to −0.04 | <0.0001, −0.94 to −0.61 | |
| GRP78 (pg/mL) | CRL-1739 | 0.35 ± 0.06 | 0.68 ± 0.15 | 0.78 ± 0.37 | 1.57 ± 0.28 | 0.0171, −0.59 to −0.06 | <0.0001, −1.48 to −0.95 |
| HT29 | 0.43 ± 0.10 | 0.79 ± 0.10 | 0.92 ± 0.25 | 1.34 ± 0.63 | 0.2109, −0.88 to 0.18 | 0.0015, −1.44 to −0.38 | |
| H2O2 level μM/106 Cell | CRL-1739 | 6.23 ± 0.38 | 6.62 ± 1.02 | 13.66 ± 3.07 | 28.50 ± 3.01 | 0.9113, −2.99 to 2.23 | <0.0001, −24.87 to −19.66 |
| HT29 | 4.68 ± 0.40 | 4.32 ± 0.62 | 13.17 ± 3.06 | 21.33 ± 2.58 | 0.8862, −1.81 to 2.55 | <0.0001, −18.83 to −14.47 | |
| TOS (μmol H2O2 Eq./g) | CRL-1739 | 9.67 ± 1.2 | 15.17 ± 1.32 | 21 ± 2.36 | 24.67 ± 3.2 | 0.0012, −8.68 to −2.31 | <0.0001, −18.18 to −11.82 |
| HT29 | 9.50 ± 1.04 | 13.67 ± 1.97 | 19 ± 2.36 | 23.17 ± 3.43 | 0.0206, −7.71 to −0.62 | <0.0001, −17.21 to −10.12 | |
| TAS (mmol Trolox Eq./g) | CRL-1739 | 1.85 ± 0.10 | 1.38 ± 0.15 | 1.19 ± 0.12 | 0.58 ± 0.20 | 0.0003, 0.23 to 0.70 | <0.0001, 1.03 to 1.50 |
| HT29 | 1.87 ± 0.15 | 1.37 ± 0.20 | 1.19 ± 0.13 | 0.90 ± 0.25 | 0.0017, 0.20 to 0.80 | <0.0001, 0.66 to 1.27 |
| Cell Line | Tempol IC50 (mM, Mean ± SD) | ML210 IC50 (µM, Mean ± SD) |
|---|---|---|
| CRL-1739 | 3.44 ± 0.50 | 0.49 ± 0.10 |
| HT29 | 3.75 ± 0.30 | 0.44 ± 0.15 |
| Cell Line | Tempol (2 mM) | ML210 (0.05 µM) | Combination | Statistical Comparison |
|---|---|---|---|---|
| CRL-1739 | 81.14 ± 4.87% | 85.71 ± 3.49% | 44.28 ± 7.06% | Combo vs. Tempol: p < 0.0001; Combo vs. ML210: p < 0.0001 |
| HT29 | 84.42 ± 3.78% | 85.00 ± 3.05% | 41.28 ± 4.95% | Combo vs. Tempol: p < 0.0001; Combo vs. ML210: p < 0.0001 |
| Cell Line | Δ (Mean ± SEM) | Interpretation |
|---|---|---|
| HT29 | +0.147 ± 0.013 | Slight synergy |
| CRL-1739 | +0.257 ± 0.038 | Moderate synergy |
| Parameter | Cell Line | Observed Effect (Mean ± SEM) | Bliss Expected | Δ (Obs–Exp) | Interpretation |
|---|---|---|---|---|---|
| H2O2 | CRL-1739 | 3.45 ± 0.16 | 1.56 ± 0.25 | +2.23 ± 0.36 | Strong synergy |
| HT29 | 3.56 ± 0.20 | 1.65 ± 0.26 | +1.92 ± 0.16 | Strong synergy | |
| TOS | CRL-1739 | 1.55 ± 0.14 | 2.42 ± 0.25 | −0.87 ± 0.18 | Strong Antagonism |
| HT29 | 1.44 ± 0.14 | 1.66 ± 0.18 | −0.22 ± 0.32 | Moderate antagonism | |
| TAS | CRL-1739 | −0.69 ± 0.05 | −0.54 ± 0.03 | −0.14 ± 0.06 | Slight/antagonism |
| HT29 | −0.52 ± 0.05 | −0.56 ± 0.04 | +0.04 ± 0.03 | Slight synergy |
| Marker | Cell Line | Observed Effect (Mean ± SEM) | Bliss Expected | Δ (Obs–Exp) | Interpretation |
|---|---|---|---|---|---|
| ATF6 | CRL-1739 | 3.53 ± 0.27 | 2.67 ± 0.27 | +0.86 ± 0.12 | Strong synergy |
| HT29 | 3.66 ± 0.33 | 2.96 ± 0.55 | +1.38 ± 0.74 | Strong synergy | |
| GRP78 | CRL-1739 | 3.45 ± 0.32 | 2.85 ± 0.84 | +0.94 ± 1.01 | Strong synergy |
| HT29 | 2.10 ± 0.64 | 2.38 ± 0.39 | −0.29 ± 0.69 | Moderate antagonism | |
| IRE1α | CRL-1739 | 1.04 ± 0.06 | 0.90 ± 0.09 | +0.13 ± 0.07 | Slight synergy |
| HT29 | 1.36 ± 0.04 | 1.23 ± 0.13 | +0.13 ± 0.12 | Slight synergy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ozdemir, G.; Kaplan, H.M. GPX4 Inhibition Enhances the Pro-Oxidant and ER Stress Effects of Tempol in Colon and Gastric Cancer Cell Lines. Curr. Issues Mol. Biol. 2025, 47, 856. https://doi.org/10.3390/cimb47100856
Ozdemir G, Kaplan HM. GPX4 Inhibition Enhances the Pro-Oxidant and ER Stress Effects of Tempol in Colon and Gastric Cancer Cell Lines. Current Issues in Molecular Biology. 2025; 47(10):856. https://doi.org/10.3390/cimb47100856
Chicago/Turabian StyleOzdemir, Gorkem, and Halil Mahir Kaplan. 2025. "GPX4 Inhibition Enhances the Pro-Oxidant and ER Stress Effects of Tempol in Colon and Gastric Cancer Cell Lines" Current Issues in Molecular Biology 47, no. 10: 856. https://doi.org/10.3390/cimb47100856
APA StyleOzdemir, G., & Kaplan, H. M. (2025). GPX4 Inhibition Enhances the Pro-Oxidant and ER Stress Effects of Tempol in Colon and Gastric Cancer Cell Lines. Current Issues in Molecular Biology, 47(10), 856. https://doi.org/10.3390/cimb47100856