
Deciphering Glioblastoma: Fundamental and Novel Insights into the Biology and Therapeutic Strategies of Gliomas
 , ,
, ,
Abstract
1. Introduction
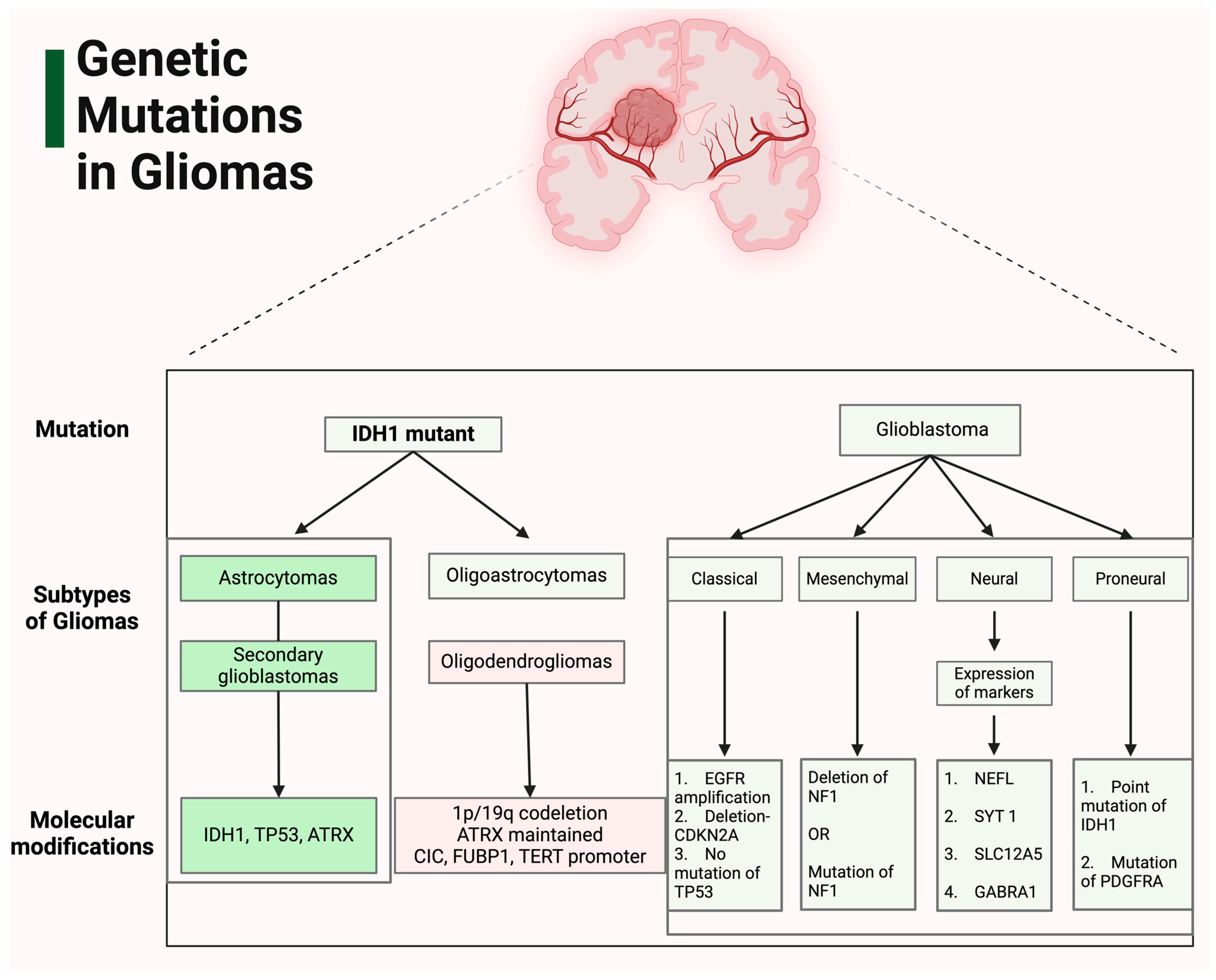
2. Genetic Landscape of Gliomas

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Physiologic Effect | Mutation | Pathogenicity | Reference |
|---|---|---|---|---|
| IDH1 |
| IDH1 R132H mutation is the most common mutation in gliomas |
| [25,26,27] |
| EGFR |
| The EGFRvIII mutation and EGFR gene amplification are the most prevalent alterations in gliomas |
| [28,29,30,31] |
| CDKN2A |
|
|
| [32,33,34] |
| TP53 (tumor protein 53) |
| Key hotspots for missense mutations include codons 175, 248, and 273 |
| [35,36,37,38,39,40,41] |
| ATRX |
| Loss of ATRX function can result from various genetic alterations, including mutations, deletions, or gene fusions. This loss is associated with a constellation of molecular changes, notably the ALT phenotype, amplification of PDGFRA, and mutations in TP53 |
| [42,43,44,45] |
| NF1 |
| A predisposing germline mutation in the NF1 gene often progresses to homozygosity, with the somatic mutation burden in NF1-associated gliomas being modulated by both age and tumor grade. High-grade tumors exhibit genetic modifications in TP53 and CDKN2A, alongside prevalent mutations in ATRX that are associated with the ALT phenotype. Furthermore, these tumors demonstrate an enrichment of genetic alterations affecting transcription/chromatin regulation and the PI3K pathways |
| [46,47,48,49] |
| PIK3CA | Cellular signaling responsible for cellular survival | The localization of PIK3CA mutations predominantly to exons 1, 9, and 20 substantiates the notion of mutational hotspots within this gene |
| [50,51,52,53] |
| PDGFRA | Transmembrane receptor that is involved in glial proliferation | Two genomic rearrangements, encompassing the starting instance of a gene fusion between the KDR VEGFR2 and the PDGFRA gene, along with six occurrences of PDGFRAΔ8,9, an intragenic deletion rearrangement. Notably, the PDGFRAΔ8,9 variant was prevalent, detected in 40% of GBM cases exhibiting PDGFRA amplification |
| [54,55,56,57] |
| PTEN | Renowned tumor suppresor gene | Deletions on 10q23. A frequently observed mutational locus resides within exon 5, which is responsible for encoding the phosphatase catalytic core motif. Additionally, recurrent mutations at CpG dinucleotides indicate the likelihood of mutations induced by deamination processes |
| [58,59,60] |
3. The Crucial Role of Glial Cells
4. Immuno-Landscape of Gliomas: Understanding the Immunological Milieu
5. The Importance of Cell Crosstalk in Gliomas
6. Transcriptomics of Gliomas
7. Emphasizing Epigenomics in Glioma Research
8. Proteomics and Metabolomics
9. Surgical Treatment
10. The Tumor Microenvironment in Gliomas: A Focal Point
11. Comparative Studies: Gliomas vs. Other Tumor Entities
12. Future Directions in Glioma Research and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| WHO | World Health Organization |
| CNS | Central nervous system |
| HGG | high-grade gliomas |
| LGGs | Low-Grade Gliomas |
| MRI | Magnetic resonance imaging |
| GBM | Glioblastom |
| IDH1 | Isocitrate dehydrogenase 1 |
| IHC | Immunohistochemistry |
| UPS | Ubiquitin proteasome system |
| AS | Astrocytoma |
| ODG | Oligodendroglioma |
| HDAC1 | Histone deacetylase 1 |
| NSCs | Neural stem cells |
| GSCs | Glioma stem cells |
| SVZ | Subventricular zone |
| OPCs | Oligodendrocyte progenitor cells |
| PUFAs | Polyunsaturated fatty acids |
| NAC | N-acetylcysteine |
| PE-PUFAs | Phosphatidylethanolamine PUFA |
| BBB | Blood-brain barrier |
| FasL | Fas ligand |
| TAAs | Tumor-associated antigens |
| MDSCs | Myeloid-Derived Suppressor Cells |
| CSF-1 | Colony Stimulating Factor-1 |
| Arg1+ | Arginase 1 positive |
| AEG1 | Astrocyte elevated gene-1 |
| TCGA | The Cancer Genome Atlas |
| GTEx | Genotype-Tissue Expression |
| CGGA | Chinese Glioma Genome Atlas |
| MIF | Macrophage migration inhibitory factor |
| PD-1 | Programmed cell death protein 1 |
| Tregs | Regulatory T cells |
| Tfr | T follicular regulatory |
| CTLA-4 | Cytotoxic T-Lymphocyte Associated protein 4 |
| PFG | Permeability Factor G |
| NK | Natural Killer |
| TGF-β | Transforming Growth Factor Beta |
| NKG2D | Natural-killer group 2, member D |
| IFN-γ | Interferon Gamma |
| CAR-T | Chimeric Antigen Receptor T |
| EGFRvIII | EGFR variant III |
| IL-13Rα | Interleukin-13 Receptor Alpha 2 |
| TME | Tumor microenviroment |
| Treg | Regulatory T cell |
| TIM-3 | T-cell immunoglobulin domain and mucin domain-3 |
| DCs | Dendritic cells |
| DIPG | Diffuse Intrinsic Pontine Glioma |
| SRS | Stereotactic Radiosurgery |
| CSCs | Cancer stem cells |
| TAMs | Tumor-associated macrophages |
| EVs | Extracellular vesicles |
| CX-43 | Connexin-43 |
| JNK | C-Jun N-terminal kinase |
| Grnd | Grindelwald |
| Egr | Eiger |
| BDNF | Brain-derived neurotrophic factor |
| GPER | G protein-coupled estrogen receptor |
| GICs | Glioma-initiating cells |
| AQPs | Aquaporins |
| EGFR | Epidermal growth factor receptor |
| ABAT | Aminobutyrate aminotransferase |
| PDGFRA | Platelet-derived growth factor receptor alpha |
| RNA | Ribonucleic acid |
| TFs | Transcription factors |
| lncRNA | Long non-coding RNA |
| DNA | Deoxyribonucleic acid |
| PTMS | Posttranslational modifications |
| SENP1 | SUMO-specific protease |
| miRNA | MicroRNA |
| DII1-4 | Delta-like family |
| PKB | Protein kinase B |
| PI3K | PI3 kinase |
| ACSS2 | Acetyl-CoA synthetase 2 |
| SREBP | Sterol regulatory element-binding protein-1 |
| EOR | Extent of resection |
| LOS | Length of hospital day |
| CSR | Complete surgical resection |
| IORT | Intraoperative radiation therapy |
| IoMRI | Intraoperative MRI |
| ECM | Extracellular matrix |
| TREM2 | Triggering receptor expressed on myeloid cells 2 |
| CAFs | Cancer-associated fibroblasts |
| FN1 | Fibronectin |
| MDSCs | Myeloid-Derived Suppressor Cells |
| VEGF | Vascular endothelial growth factor |
| vWF | Von Willebrand factor |
| B2R | Bradykinin receptor-2 |
| ICI | Immune checkpoint inhibition |
| CAR | Chimeric antigen receptor |
| oAds | Oncolytic adenovirus |
| DIPG | Diffuse Intrinsic Pontine Glioma |
| NSG | NOD scid gamma |
| EGR | Early growth response |
| ROS | Reactive oxidative stress |
| ER | Endoplasmic reticulum |
| IP3R | Inositol triphosphate receptor |
| TTFields | Tumor Treating Fields |
| CCNU | Lomustine |
| MGMT | Methyltransferase |
| DCVax-L | Dendritic cell vaccination |
| nGBM | Newly diagnosed glioblastoma |
| rGBM | Recurrent glioblastoma |
| OS | Overall survival |
| PD-1 | Programmed cell death protein |
| PD-L1 | Programmed cell death protein ligand 1 |
| ncRNAs | Non-coding RNAs |
| AI | Artificial intelligence |
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Tom, M.C.; Park, D.Y.J.; Yang, K.; Leyrer, C.M.; Wei, W.; Jia, X.; Varra, V.; Yu, J.S.; Chao, S.T.; Balagamwala, E.H.; et al. Malignant Transformation of Molecularly Classified Adult Low-Grade Glioma. Int. J. Radiat. Oncol. 2019, 105, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Hoang-Minh, L.B.; Dutra-Clarke, M.; Breunig, J.J.; Sarkisian, M.R. Glioma cell proliferation is enhanced in the presence of tumor-derived cilia vesicles. Cilia 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Larjavaara, S.; Mäntylä, R.; Salminen, T.; Haapasalo, H.; Raitanen, J.; Jääskeläinen, J.; Auvinen, A. Incidence of gliomas by anatomic location. Neuro-Oncol. 2007, 9, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, B.K.; Hansen, S.; Laursen, R.J.; Kosteljanetz, M.; Schultz, H.; Nørgård, B.M.; Guldberg, R.; Gradel, K.O. Epidemiology of glioma: Clinical characteristics, symptoms, and predictors of glioma patients grade I–IV in the the Danish Neuro-Oncology Registry. J. Neurooncol. 2017, 135, 571–579. [Google Scholar] [CrossRef]
- Di Carlo, D.T.; Cagnazzo, F.; Benedetto, N.; Morganti, R.; Perrini, P. Multiple high-grade gliomas: Epidemiology, management, and outcome. A systematic review and meta-analysis. Neurosurg. Rev. 2019, 42, 263–275. [Google Scholar] [CrossRef] [PubMed]
- McNeill, K.A. Epidemiology of Brain Tumors. Neurol. Clin. 2016, 34, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.; Kelly, M.E.; Yeboa, D.N.; Buerki, R.A.; Cioffi, G.; Balaji, S.; Ostrom, Q.T.; Kruchko, C.; Barnholtz-Sloan, J.S. Epidemiology of brainstem high-grade gliomas in children and adolescents in the United States, 2000–2017. Neuro-Oncol. 2021, 23, 990–998. [Google Scholar] [CrossRef]
- Lin, D.; Wang, M.; Chen, Y.; Gong, J.; Chen, L.; Shi, X.; Lan, F.; Chen, Z.; Xiong, T.; Sun, H.; et al. Trends in Intracranial Glioma Incidence and Mortality in the United States, 1975–2018. Front. Oncol. 2021, 11, 748061. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-Oncol. 2020, 22 (Suppl. S1), iv1–iv96. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; de Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-Oncol. 2015, 16 (Suppl. S10), x1–x36. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncol. 2018, 20 (Suppl. S4), iv1–iv86. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z.; Hua, C.; Xu, Y.; Li, Y.; Zhao, G. Primary Malignant Brain Tumors following Systemic Malignancies: A Population-Based Analysis. Neuroepidemiology 2022, 56, 452–459. [Google Scholar] [CrossRef]
- Chen, R.; Smith-Cohn, M.; Cohen, A.L.; Colman, H. Glioma Subclassifications and Their Clinical Significance. Neurotherapeutics 2017, 14, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L. Cancer Statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. Available online: https://acsjournals.onlinelibrary.wiley.com/doi/10.3322/caac.21590 (accessed on 6 December 2023). [CrossRef]
- Santra, A.; Kumar, R.; Sharma, P.; Bal, C.; Julka, P.K.; Malhotra, A. F-18 FDG PET-CT for predicting survival in patients with recurrent glioma: A prospective study. Neuroradiology 2011, 53, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Tazin, F.; Kumar, H.; Israr, M.A.; Omoleye, D.; Orlang, V. Li-Fraumeni Syndrome: A Rare Genetic Disorder. Cureus 2022, 14, e29240. [Google Scholar] [CrossRef]
- Karpel-Massler, G.; Nguyen, T.T.T.; Shang, E.; Siegelin, M.D. Novel IDH1-Targeted Glioma Therapies. CNS Drugs 2019, 33, 1155–1166. [Google Scholar] [CrossRef]
- Nishikawa, T.; Watanabe, R.; Kitano, Y.; Yamamichi, A.; Motomura, K.; Ohka, F.; Aoki, K.; Hirano, M.; Kato, A.; Yamaguchi, J.; et al. Reliability of IDH1-R132H and ATRX and/or p53 immunohistochemistry for molecular subclassification of Grade 2/3 gliomas. Brain Tumor Pathol. 2022, 39, 14–24. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef]
- Vriend, J.; Klonisch, T. Genes of the Ubiquitin Proteasome System Qualify as Differential Markers in Malignant Glioma of Astrocytic and Oligodendroglial Origin. Cell. Mol. Neurobiol. 2023, 43, 1425–1452. [Google Scholar] [CrossRef]
- Zhao, B.; Xia, Y.; Yang, F.; Wang, Y.; Wang, Y.; Wang, Y.; Dai, C.; Wang, Y.; Ma, W. Molecular landscape of IDH-mutant astrocytoma and oligodendroglioma grade 2 indicate tumor purity as an underlying genomic factor. Mol. Med. 2022, 28, 34. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, X.; Qi, M.; Li, M.; Dong, M.; Xu, R.; Zhang, C. Systematic analysis identifies REST as an oncogenic and immunological biomarker in glioma. Sci. Rep. 2023, 13, 3023. [Google Scholar] [CrossRef]
- Wang, G.; Li, M.; Dong, M.; Xu, R.; Zhang, C. Systematic Pan-Cancer Analysis Identifies REST as an Oncogenic and Immunological Biomarker in Brain Low Grade Glioma. Available online: https://www.researchsquare.com (accessed on 6 December 2023).
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.M.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, A.; Walter, D.M.; Gudiel, A.A.; Schek, N.; Feldser, D.M.; Witze, E.S. Blocking EGFR palmitoylation suppresses PI3K signaling and mutant KRAS lung tumorigenesis. Sci. Signal. 2020, 13, eaax2364. [Google Scholar] [CrossRef]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B.; et al. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef]
- Brito, C.; Azevedo, A.; Esteves, S.; Marques, A.R.; Martins, C.; Costa, I.; Mafra, M.; Bravo Marques, J.M.; Roque, L.; Pojo, M. Clinical insights gained by refining the 2016 WHO classification of diffuse gliomas with: EGFR amplification, TERT mutations, PTEN deletion and MGMT methylation. BMC Cancer 2019, 19, 968. [Google Scholar] [CrossRef]
- Lopez-Gines, C.; Cerda-Nicolas, M.; Gil-Benso, R.; Pellin, A.; Lopez-Guerrero, J.A.; Callaghan, R.; Benito, R.; Roldan, P.; Piquer, J.; Llacer, J.; et al. Association of chromosome 7, chromosome 10 and EGFR gene amplification in glioblastoma multiforme. Clin. Neuropathol. 2005, 24, 209–218. [Google Scholar]
- Dębniak, T.; Scott, R.J.; Huzarski, T.; Byrski, T.; Rozmiarek, A.; Dębniak, B.; Górski, B.; Cybulski, C.; Mędrek, K.; Mierzejewski, M.; et al. CDKN2A common variant and multi-organ cancer risk—A population-based study. Int. J. Cancer 2006, 118, 3180–3182. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Poi, M.J.; Tsai, M.-D. The Regulatory Mechanisms of Tumor Suppressor P16INK4A and Relevance to Cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef] [PubMed]
- Danishevich, A.; Bilyalov, A.; Nikolaev, S.; Khalikov, N.; Isaeva, D.; Levina, Y.; Makarova, M.; Nemtsova, M.; Chernevskiy, D.; Sagaydak, O.; et al. CDKN2A Gene Mutations: Implications for Hereditary Cancer Syndromes. Biomedicines 2023, 11, 3343. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, B.K.A.; McLendon, R.E.; Herndon, J.E.; Friedman, H.S.; Friedman, A.H.; Bigner, D.D.; Bigner, S.H. Alterations of the TP53 Gene in Human Gliomas. Cancer Res. 1994, 54, 1324–1330. [Google Scholar]
- Noor, H.; Briggs, N.E.; McDonald, K.L.; Holst, J.; Vittorio, O. TP53 Mutation Is a Prognostic Factor in Lower Grade Glioma and May Influence Chemotherapy Efficacy. Cancers 2021, 13, 5362. [Google Scholar] [CrossRef]
- Amatya, V.J.; Naumann, U.; Weller, M.; Ohgaki, H. TP53 promoter methylation in human gliomas. Acta Neuropathol. 2005, 110, 178–184. [Google Scholar] [CrossRef]
- Gillet, E.; Alentorn, A.; Doukouré, B.; Mundwiller, E.; van Thuij, H.; Reijneveld, J.C.; Medina, J.A.M.; Liou, A.; Marie, Y.; Mokhtari, K.; et al. TP53 and p53 statuses and their clinical impact in diffuse low grade gliomas. J. Neurooncol. 2014, 118, 131–139. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.; Liu, J.; You, C.; Liu, Y.; Mao, Q. Gain of Function of Mutant TP53 in Glioblastoma: Prognosis and Response to Temozolomide. Ann. Surg. Oncol. 2014, 21, 1337–1344. [Google Scholar] [CrossRef]
- Pollack, I.F.; Finkelstein, S.D.; Burnham, J.; Holmes, E.J.; Hamilton, R.L.; Yates, A.J.; Finlay, J.L.; Sposto, R.; for the Children’s Cancer Group. Age and TP53 Mutation Frequency in Childhood Malignant Gliomas: Results in a Multi-institutional Cohort. Cancer Res. 2001, 61, 7404–7407. [Google Scholar]
- Ham, S.W.; Jeon, H.-Y.; Jin, X.; Kim, E.-J.; Kim, J.-K.; Shin, Y.J.; Lee, Y.; Kim, S.H.; Lee, S.Y.; Seo, S.; et al. TP53 gain-of-function mutation promotes inflammation in glioblastoma. Cell Death Differ. 2019, 26, 409–425. [Google Scholar] [CrossRef]
- Xue, Y.; Gibbons, R.; Yan, Z.; Yang, D.; McDowell, T.L.; Sechi, S.; Qin, J.; Zhou, S.; Higgs, D.; Wang, W. The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc. Natl. Acad. Sci. USA 2003, 100, 10635–10640. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef]
- Anastasaki, C.; Gutmann, D.H. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum. Mol. Genet. 2014, 23, 6712–6721. [Google Scholar] [CrossRef]
- Anastasaki, C.; Wegscheid, M.L.; Hartigan, K.; Papke, J.B.; Kopp, N.D.; Chen, J.; Cobb, O.; Dougherty, J.D.; Gutmann, D.H. Human iPSC-Derived Neurons and Cerebral Organoids Establish Differential Effects of Germline NF1 Gene Mutations. Stem Cell Rep. 2020, 14, 541–550. [Google Scholar] [CrossRef]
- Costa, R.M.; Yang, T.; Huynh, D.P.; Pulst, S.M.; Viskochil, D.H.; Silva, A.J.; Brannan, C.I. Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat. Genet. 2001, 27, 399–405. [Google Scholar] [CrossRef]
- Danglot, G.; Régnier, V.; Fauvet, D.; Vassal, G.; Kujas, M.; Bernheim, A. Neurofibromatosis 1 (NF1) mRNAs expressed in the central nervous system are differentially spliced in the 5′ part of the gene. Hum. Mol. Genet. 1995, 4, 915–920. [Google Scholar] [CrossRef]
- Hartmann, C.; Bartels, G.; Gehlhaar, C.; Holtkamp, N.; von Deimling, A. PIK3CA mutations in glioblastoma multiforme. Acta Neuropathol. 2005, 109, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Batchelor, T.T.; Iafrate, A.J.; Dias-Santagata, D.; Borger, D.R.; Ellisen, L.W.; Yang, D.; Louis, D.N.; Cahill, D.P.; Chi, A.S. PIK3CA activating mutations are associated with more disseminated disease at presentation and earlier recurrence in glioblastoma. Acta Neuropathol. Commun. 2019, 7, 66. [Google Scholar] [CrossRef]
- Broderick, D.K.; Di, C.; Parrett, T.J.; Samuels, Y.R.; Cummins, J.M.; McLendon, R.E.; Fults, D.W.; Velculescu, V.E.; Bigner, D.D.; Yan, H. Mutations of PIK3CA in Anaplastic Oligodendrogliomas, High-Grade Astrocytomas, and Medulloblastomas. Cancer Res. 2004, 64, 5048–5050. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Clarke, I.D.; Dirks, P.B. A human brain tumor-derived PDGFR-α deletion mutant is transforming. Oncogene 2003, 22, 722–733. [Google Scholar] [CrossRef]
- Hu, W.; Duan, H.; Zhong, S.; Zeng, J.; Mou, Y. High frequency of PDGFRA and MUC family gene mutations in diffuse hemispheric glioma, H3 G34-mutant: A glimmer of hope? J. Transl. Med. 2022, 20, 64. [Google Scholar] [CrossRef] [PubMed]
- Koschmann, C.; Zamler, D.; MacKay, A.; Robinson, D.; Wu, Y.-M.; Doherty, R.; Marini, B.; Tran, D.; Garton, H.; Muraszko, K.; et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget 2016, 7, 65696–65706. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the human PTEN gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Huse, J.T.; Brennan, C.; Hambardzumyan, D.; Wee, B.; Pena, J.; Rouhanifard, S.H.; Sohn-Lee, C.; le Sage, C.; Agami, R.; Tuschl, T.; et al. The PTEN-regulating microRNA miR-26a is amplified in high-grade glioma and facilitates gliomagenesis in vivo. Genes Dev. 2009, 23, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shao, N.; Luo, G.; Li, L.; Zheng, L.; Nilsson-Ehle, P.; Xu, N. Mutations of PTEN gene in gliomas correlate to tumor differentiation and short-term survival rate. Anticancer Res. 2010, 30, 981–985. [Google Scholar] [PubMed]
- Barres, B.A. The Mystery and Magic of Glia: A Perspective on Their Roles in Health and Disease. Neuron 2008, 60, 430–440. [Google Scholar] [CrossRef]
- Hyun, K.-A.; Koo, G.-B.; Han, H.; Sohn, J.; Choi, W.; Kim, S.-I.; Jung, H.-I.; Kim, Y.-S. Epithelial-to-mesenchymal transition leads to loss of EpCAM and different physical properties in circulating tumor cells from metastatic breast cancer. Oncotarget 2016, 7, 24677–24687. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Qian, J.; Yang, L.; Sun, Z.; Hu, C.; Wang, X.; Hu, S.; Xie, Y. Multiparametric MRI radiomics for the differentiation of brain glial cell hyperplasia from low-grade glioma. BMC Med. Imaging 2023, 23, 116. [Google Scholar] [CrossRef] [PubMed]
- Friedmann-Morvinski, D.; Bushong, E.A.; Ke, E.; Soda, Y.; Marumoto, T.; Singer, O.; Ellisman, M.H.; Verma, I.M. Dedifferentiation of Neurons and Astrocytes by Oncogenes Can Induce Gliomas in Mice. Science 2012, 338, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- de Almeida Sassi, F.; Lunardi Brunetto, A.; Schwartsmann, G.; Roesler, R.; Abujamra, A.L. Glioma Revisited: From Neurogenesis and Cancer Stem Cells to the Epigenetic Regulation of the Niche. J. Oncol. 2012, 2012, 537861. Available online: https://www.hindawi.com/journals/jo/2012/537861/ (accessed on 6 December 2023). [CrossRef] [PubMed]
- Lee, J.-S.; Lee, H.J.; Moon, B.-H.; Song, S.-H.; Lee, M.-O.; Shim, S.H.; Kim, H.S.; Lee, M.C.; Kwon, J.T.; Fornace, A.J.; et al. Generation of Cancerous Neural Stem Cells Forming Glial Tumor by Oncogenic Stimulation. Stem Cell Rev. Rep. 2012, 8, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Fares, J.; Ahmed, A.U.; Ulasov, I.V.; Sonabend, A.M.; Miska, J.; Lee-Chang, C.; Balyasnikova, I.V.; Chandler, J.P.; Portnow, J.; Tate, M.C.; et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: A first-in-human, phase 1, dose-escalation trial. Lancet Oncol. 2021, 22, 1103–1114. [Google Scholar] [CrossRef]
- Petronek, M.S.; Teferi, N.; Caster, J.M.; Stolwijk, J.M.; Zaher, A.; Buatti, J.M.; Hasan, D.; Wafa, E.I.; Salem, A.K.; Gillan, E.G.; et al. Magnetite nanoparticles as a kinetically favorable source of iron to enhance GBM response to chemoradiosensitization with pharmacological ascorbate. Redox Biol. 2023, 62, 102651. [Google Scholar] [CrossRef]
- Hänninen, M.M.; Haapasalo, J.; Haapasalo, H.; Fleming, R.E.; Britton, R.S.; Bacon, B.R.; Parkkila, S. Expression of iron-related genes in human brain and brain tumors. BMC Neurosci. 2009, 10, 36. [Google Scholar] [CrossRef]
- Calzolari, A.; Larocca, L.M.; Deaglio, S.; Finisguerra, V.; Boe, A.; Raggi, C.; Ricci-Vitani, L.; Pierconti, F.; Malavasi, F.; De Maria, R.; et al. Transferrin Receptor 2 Is Frequently and Highly Expressed in Glioblastomas. Transl. Oncol. 2010, 3, 123–134. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef]
- Yee, P.P.; Wei, Y.; Kim, S.-Y.; Lu, T.; Chih, S.Y.; Lawson, C.; Tang, M.; Liu, Z.; Anderson, B.; Thamburaj, K.; et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat. Commun. 2020, 11, 5424. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; McDonald, A.; Fleming, M.D. The Steap proteins are metalloreductases. Blood 2006, 108, 1388–1394. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Zhou, Y.; Wang, X.; Zhao, H.; Nie, C.; Jiang, X. A Ferroptosis-Related Prognostic Risk Score Model to Predict Clinical Significance and Immunogenic Characteristics in Glioblastoma Multiforme. Oxid. Med. Cell. Longev. 2021, 2021, 9107857. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xu, C.; Yu, Q.; Zhong, C.; Peng, Y.; Chen, J.; Chen, G. Comprehensive landscape of STEAP family functions and prognostic prediction value in glioblastoma. J. Cell. Physiol. 2021, 236, 2988–3000. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhu, W.; Pei, D. System Xc−: A key regulatory target of ferroptosis in cancer. Investig. New Drugs 2021, 39, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Noch, E.K.; Palma, L.; Yim, I.; Bullen, N.; Barnett, D.; Walsh, A.; Bhinder, B.; Benedetti, E.; Krumsiek, J.; Gurvitch, J.; et al. Cysteine induces mitochondrial reductive stress in glioblastoma through hydrogen peroxide production. Proc. Natl. Acad. Sci. USA 2024, 121, e2317343121. [Google Scholar] [CrossRef]
- Xin, S.; Schick, J.A. PUFAs dictate the balance of power in ferroptosis. Cell Calcium 2023, 110, 102703. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Akagi, S.; Kono, N.; Ariyama, H.; Shindou, H.; Shimizu, T.; Arai, H. Lysophosphatidylcholine acyltransferase 1 protects against cytotoxicity induced by polyunsaturated fatty acids. FASEB J. 2016, 30, 2027–2039. [Google Scholar] [CrossRef]
- Lagrost, L.; Masson, D. The expanding role of lyso-phosphatidylcholine acyltransferase-3 (LPCAT3), a phospholipid remodeling enzyme, in health and disease. Curr. Opin. Lipidol. 2022, 33, 193–198. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641.e26. [Google Scholar] [CrossRef]
- Yang, X.; Yin, F.; Liu, Q.; Ma, Y.; Zhang, H.; Guo, P.; Wen, W.; Guo, X.; Wu, Y.; Yang, Z.; et al. Ferroptosis-related genes identify tumor immune microenvironment characterization for the prediction of prognosis in cervical cancer. Ann. Transl. Med. 2022, 10, 123. [Google Scholar] [CrossRef]
- Zhou, X.; Liao, X.; Zhang, B.; He, H.; Shui, Y.; Xu, W.; Jiang, C.; Shen, L.; Wei, Q. Recurrence patterns in patients with high-grade glioma following temozolomide-based chemoradiotherapy. Mol. Clin. Oncol. 2016, 5, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, P.R.; Castro, M.G. Pushing the Limits of Glioma Resection Using Electrophysiologic Brain Mapping. J. Clin. Oncol. 2012, 30, 2437–2440. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Mao, L.; Xiong, J.; Wen, J.; Wang, Y.; Geng, D.; Liu, Y. TAZ Expression on Endothelial Cells Is Closely Related to Blood Vascular Density and VEGFR2 Expression in Astrocytomas. J. Neuropathol. Exp. Neurol. 2019, 78, 172–180. [Google Scholar] [CrossRef]
- Maraqah, H.H.; Abu-Asab, M.S.; Lee, H.S.; Aboud, O. Astrocytoma and glioblastoma IDH1-wildtype cells colonize tumor vessels and deploy vascular mimicry. Ultrastruct. Pathol. 2023, 47, 253–260. [Google Scholar] [CrossRef]
- Ho, I.A.; Ng, W.H.; Lam, P.Y. FasL and FADD delivery by a glioma-specific and cell cycle-dependent HSV-1 amplicon virus enhanced apoptosis in primary human brain tumors. Mol. Cancer 2010, 9, 270. [Google Scholar] [CrossRef]
- Ryken, T.; Frankel, B.; Longo, S.; Sibenaller, Z. Interaction of fas and fas ligand in a rat 36b10 glioma model. Neurosurg. Focus 2000, 8, 1–6. [Google Scholar] [CrossRef]
- Gratas, C.; Tohma, Y.; Meir, E.G.V.; Klein, M.; Tenan, M.; Ishii, N.; Tachibana, O.; Kleihues, P.; Ohgaki, H. Fas Ligand Expression in Glioblastoma Cell Lines and Primary Astrocytic Brain Tumors. Brain Pathol. 1997, 7, 863–869. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
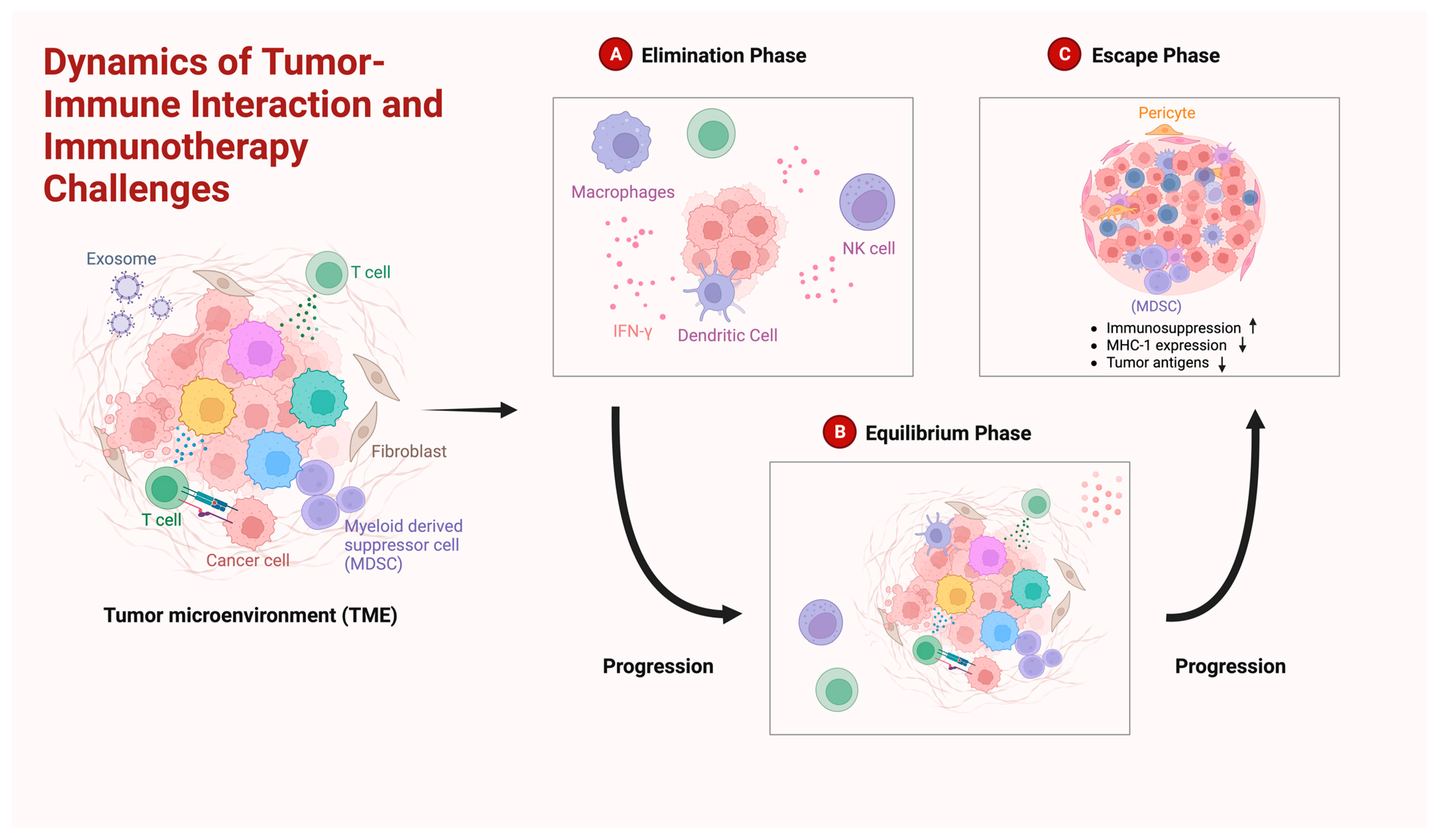
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
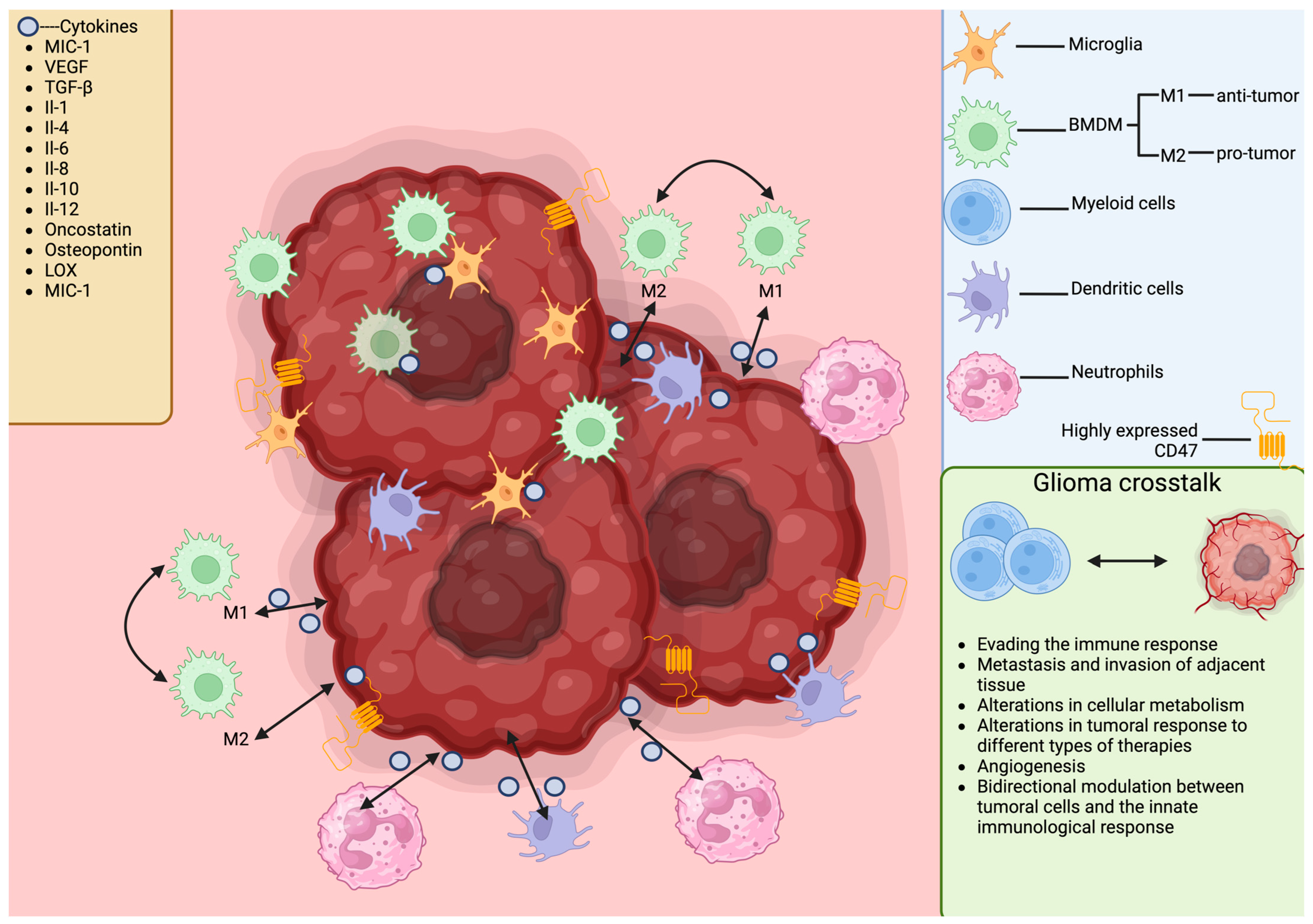
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Guo, X.; Xue, H.; Shao, Q.; Wang, J.; Guo, X.; Chen, X.; Zhang, J.; Xu, S.; Li, T.; Zhang, P.; et al. Hypoxia promotes glioma-associated macrophage infiltration via periostin and subsequent M2 polarization by upregulating TGF-beta and M-CSFR. Oncotarget 2016, 7, 80521–80542. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.S.; Braganhol, E.; Whiteside, T.L. Arginase-1+ Exosomes from Reprogrammed Macrophages Promote Glioblastoma Progression. Int. J. Mol. Sci. 2020, 21, 3990. [Google Scholar] [CrossRef]
- Li, J.; Sun, Y.; Sun, X.; Zhao, X.; Ma, Y.; Wang, Y.; Zhang, X. AEG-1 silencing attenuates M2-polarization of glioma-associated microglia/macrophages and sensitizes glioma cells to temozolomide. Sci. Rep. 2021, 11, 17348. [Google Scholar] [CrossRef]
- Raychaudhuri, B.; Rayman, P.; Huang, P.; Grabowski, M.; Hambardzumyan, D.; Finke, J.H.; Vogelbaum, M.A. Myeloid derived suppressor cell infiltration of murine and human gliomas is associated with reduction of tumor infiltrating lymphocytes. J. Neurooncol. 2015, 122, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front. Immunol. 2020, 11, 1191. [Google Scholar] [CrossRef]
- Xue, S.; Song, G.; Yu, J. The prognostic significance of PD-L1 expression in patients with glioma: A meta-analysis. Sci. Rep. 2017, 7, 4231. [Google Scholar] [CrossRef] [PubMed]
- Eschweiler, S.; Clarke, J.; Ramírez-Suástegui, C.; Panwar, B.; Madrigal, A.; Chee, S.J.; Karydis, I.; Woo, E.; Alzetani, A.; Elsheikh, S.; et al. Intratumoral follicular regulatory T cells curtail anti-PD-1 treatment efficacy. Nat. Immunol. 2021, 22, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Sun, J.; Su, H.; Luo, S.; Chen, J.; Qiu, S.; Chi, Y.; Lin, J.; Xu, X.; Zheng, D. Antitumor CD8 T cell responses in glioma patients are effectively suppressed by T follicular regulatory cells. Exp. Cell Res. 2021, 407, 112808. [Google Scholar] [CrossRef]
- Han, S.; Feng, S.; Ren, M.; Ma, E.; Wang, X.; Xu, L.; Xu, M. Glioma cell-derived placental growth factor induces regulatory B cells. Int. J. Biochem. Cell Biol. 2014, 57, 63–68. [Google Scholar] [CrossRef]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro-Oncol. 2010, 12, 7–13. [Google Scholar] [CrossRef]
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-Cell Atlas Reveals Complexity of the Immunosuppressive Microenvironment of Initial and Recurrent Glioblastoma. Front. Immunol. 2020, 11, 835. [Google Scholar] [CrossRef]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F.; et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef]
- Weenink, B.; French, P.J.; Sillevis Smitt, P.A.E.; Debets, R.; Geurts, M. Immunotherapy in Glioblastoma: Current Shortcomings and Future Perspectives. Cancers 2020, 12, 751. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Gokhale, P.C.; Klein, S.R.; Ligon, K.L.; Rodig, S.J.; Ramkissoon, S.H.; Jones, K.L.; Conway, A.S.; Liao, X.; Zhou, J.; et al. Glioblastoma Eradication Following Immune Checkpoint Blockade in an Orthotopic, Immunocompetent Model. Cancer Immunol. Res. 2016, 4, 124–135. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Esteve, J.; Giagounidis, A.; Kim, H.-J.; Miyazaki, Y.; Platzbecker, U.; Schuh, A.C.; Sekeres, M.A.; Westermann, J.; Xiao, Z.; et al. The STIMULUS Program: Clinical Trials Evaluating Sabatolimab (MBG453) Combination Therapy in Patients (Pts) with Higher-Risk Myelodysplastic Syndromes (HR-MDS) or Acute Myeloid Leukemia (AML). Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, N.; Rinne, M.L.; Sun, H.; Stein, A.M. Sabatolimab (MBG453) model-informed drug development for dose selection in patients with myelodysplastic syndrome/acute myeloid leukemia and solid tumors. CPT Pharmacomet. Syst. Pharmacol. 2023, 12, 1653–1665. [Google Scholar] [CrossRef]
- Kim, J.E.; Patel, M.A.; Mangraviti, A.; Kim, E.S.; Theodros, D.; Velarde, E.; Liu, A.; Sankey, E.W.; Tam, A.; Xu, H.; et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin. Cancer Res. 2017, 23, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Ausejo-Mauleon, I.; Labiano, S.; De La Nava, D.; Laspidea, V.; Zalacain, M.; Marrodán, L.; García-Moure, M.; González-Huarriz, M.; Hervás-Corpión, I.; Dhandapani, L.; et al. TIM-3 blockade in diffuse intrinsic pontine glioma models promotes tumor regression and antitumor immune memory. Cancer Cell 2023, 41, 1911–1926.e8. [Google Scholar] [CrossRef] [PubMed]
- Sozzani, S.; Luini, W.; Borsatti, A.; Polentarutti, N.; Zhou, D.; Piemonti, L.; D’Amico, G.; Power, C.A.; Wells, T.N.; Gobbi, M.; et al. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J. Immunol. 1997, 159, 1993–2000. [Google Scholar] [CrossRef]
- Weber, K.S.C.; Nelson, P.J.; Gröne, H.-J.; Weber, C. Expression of CCR2 by Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Charo, I.F.; Myers, S.J.; Herman, A.; Franci, C.; Connolly, A.J.; Coughlin, S.R. Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxyl-terminal tails. Proc. Natl. Acad. Sci. USA 1994, 91, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Yumimoto, K.; Sugiyama, S.; Mimori, K.; Nakayama, K.I. Potentials of C-C motif chemokine 2–C-C chemokine receptor type 2 blockers including propagermanium as anticancer agents. Cancer Sci. 2019, 110, 2090–2099. [Google Scholar] [CrossRef] [PubMed]
- Shahrara, S.; Amin, M.A.; Woods, J.M.; Haines, G.K.; Koch, A.E. Chemokine receptor expression and in vivo signaling pathways in the joints of rats with adjuvant-induced arthritis. Arthritis Rheum. 2003, 48, 3568–3583. [Google Scholar] [CrossRef]
- Zhu, S.; Liu, M.; Bennett, S.; Wang, Z.; Pfleger, K.D.G.; Xu, J. The molecular structure and role of CCL2 (MCP-1) and C-C chemokine receptor CCR2 in skeletal biology and diseases. J. Cell. Physiol. 2021, 236, 7211–7222. [Google Scholar] [CrossRef]
- Tian, Q.; Guo, Y.; Feng, S.; Liu, C.; He, P.; Wang, J.; Han, W.; Yang, C.; Zhang, Z.; Li, M. Inhibition of CCR2 attenuates neuroinflammation and neuronal apoptosis after subarachnoid hemorrhage through the PI3K/Akt pathway. J. Neuroinflamm. 2022, 19, 312. [Google Scholar] [CrossRef]
- Tao, L.-L.; Shi, S.-J.; Chen, L.-B.; Huang, G.-C. Expression of monocyte chemotactic protein-1/CCL2 in gastric cancer and its relationship with tumor hypoxia. World J. Gastroenterol. 2014, 20, 4421–4427. [Google Scholar] [CrossRef]
- Liu, Y.; Pan, J.; Pan, X.; Wu, L.; Bian, J.; Lin, Z.; Xue, M.; Su, T.; Lai, S.; Chen, F.; et al. Klotho-mediated targeting of CCL2 suppresses the induction of colorectal cancer progression by stromal cell senescent microenvironments. Mol. Oncol. 2019, 13, 2460–2475. [Google Scholar] [CrossRef]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yao, W.; Yuan, Y.; Chen, P.; Li, B.; Li, J.; Chu, R.; Song, H.; Xie, D.; Jiang, X.; et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut 2017, 66, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhou, Y.; Gao, Y.; Zhu, Z.; Zeng, X.; Liang, W.; Sun, S.; Chen, X.; Wang, H. Radiated glioblastoma cell-derived exosomal circ_0012381 induce M2 polarization of microglia to promote the growth of glioblastoma by CCL2/CCR2 axis. J. Transl. Med. 2022, 20, 388. [Google Scholar] [CrossRef] [PubMed]
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors in Physiology and Medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.D.; Dahlrot, R.H.; Boldt, H.B.; Hansen, S.; Kristensen, B.W. Tumour-associated microglia/macrophages predict poor prognosis in high-grade gliomas and correlate with an aggressive tumour subtype. Neuropathol. Appl. Neurobiol. 2018, 44, 185–206. [Google Scholar] [CrossRef]
- Zeiner, P.S.; Preusse, C.; Golebiewska, A.; Zinke, J.; Iriondo, A.; Muller, A.; Kaoma, T.; Filipski, K.; Müller-Eschner, M.; Bernatz, S.; et al. Distribution and prognostic impact of microglia/macrophage subpopulations in gliomas. Brain Pathol. 2019, 29, 513–529. [Google Scholar] [CrossRef]
- Caponegro, M.D.; Moffitt, R.A.; Tsirka, S.E. Expression of neuropilin-1 is linked to glioma associated microglia and macrophages and correlates with unfavorable prognosis in high grade gliomas. Oncotarget 2018, 9, 35655–35665. [Google Scholar] [CrossRef]
- Zhou, J.; Reddy, M.V.; Wilson, B.K.J.; Blair, D.A.; Taha, A.; Frampton, C.M.; Eiholzer, R.A.; Gan, P.Y.C.; Ziad, F.; Thotathil, Z.; et al. MR Imaging Characteristics Associate with Tumor-Associated Macrophages in Glioblastoma and Provide an Improved Signature for Survival Prognostication. Am. J. Neuroradiol. 2018, 39, 252–259. [Google Scholar] [CrossRef]
- Treps, L.; Edmond, S.; Harford-Wright, E.; Galan-Moya, E.M.; Schmitt, A.; Azzi, S.; Citerne, A.; Bidère, N.; Ricard, D.; Gavard, J. Extracellular vesicle-transported Semaphorin3A promotes vascular permeability in glioblastoma. Oncogene 2016, 35, 2615–2623. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Guan, X.; Hasan, M.N.; Maniar, S.; Jia, W.; Sun, D. Reactive Astrocytes in Glioblastoma Multiforme. Mol. Neurobiol. 2018, 55, 6927–6938. [Google Scholar] [CrossRef]
- Noronha, C.; Ribeiro, A.S.; Taipa, R.; Castro, D.S.; Reis, J.; Faria, C.; Paredes, J. Cadherin Expression and EMT: A Focus on Gliomas. Biomedicines 2021, 9, 1328. [Google Scholar] [CrossRef] [PubMed]
- Portela, M.; Mitchell, T.; Casas-Tintó, S. Cell to cell communication mediates glioblastoma progression in Drosophila. Biol. Open 2020, 9, bio.053405. [Google Scholar] [CrossRef]
- Yan, Q.; Yu, H.; Li, J. Study on the expression of BDNF in human gliomas. Sichuan Da Xue Xue Bao Yi Xue Ban 2009, 40, 415–417. [Google Scholar] [PubMed]
- Wang, X.; Prager, B.C.; Wu, Q.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Yang, K.; Morton, A.R.; Zhou, W.; Zhu, Z.; et al. Reciprocal Signaling between Glioblastoma Stem Cells and Differentiated Tumor Cells Promotes Malignant Progression. Cell Stem Cell 2018, 22, 514–528.e5. [Google Scholar] [CrossRef]
- Pan, Y.; Hysinger, J.D.; Barron, T.; Schindler, N.F.; Cobb, O.; Guo, X.; Yalçın, B.; Anastasaki, C.; Mulinyawe, S.B.; Ponnuswami, A.; et al. NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature 2021, 594, 277–282. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Chen, M.; Cheng, K.-W.; Qi, L.; Zhang, Z.; Peng, Y.; Li, K.; Liu, F.; Chen, G.; et al. Neuronal-driven glioma growth requires Gαi1 and Gαi3. Theranostics 2021, 11, 8535–8549. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.-J.; Kim, D.; Kim, S.; Hsieh, J.-T.; Baek, S.T. Targeting Wnt/β-catenin-mediated upregulation of oncogenic NLGN3 suppresses cancer stem cells in glioblastoma. Cell Death Dis. 2023, 14, 423. [Google Scholar] [CrossRef]
- Pei, Z.; Lee, K.-C.; Khan, A.; Erisnor, G.; Wang, H.-Y. Pathway analysis of glutamate-mediated, calcium-related signaling in glioma progression. Biochem. Pharmacol. 2020, 176, 113814. [Google Scholar] [CrossRef]
- Ramaswamy, P.; Dalavaikodihalli Nanjaiah, N.; Prasad, C.; Goswami, K. Transcriptional modulation of calcium-permeable AMPA receptor subunits in glioblastoma by MEK–ERK1/2 inhibitors and their role in invasion. Cell Biol. Int. 2020, 44, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S. Neuron-Cancer Synaptic and Other Electrical Signaling. In Cancer Neuroscience; Amit, M., Scheff, N.N., Eds.; Springer International Publishing: Cham, Germany, 2023; pp. 27–34. [Google Scholar] [CrossRef]
- Taylor, K.R.; Barron, T.; Hui, A.; Spitzer, A.; Yalçin, B.; Ivec, A.E.; Geraghty, A.C.; Hartmann, G.G.; Arzt, M.; Gillespie, S.M.; et al. Glioma synapses recruit mechanisms of adaptive plasticity. Nature 2023, 623, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Almeida, C.E.; Santerre, A.; León-Moreno, L.C.; Aguilar-García, I.G.; Castañeda-Arellano, R.; Dueñas-Jiménez, S.H.; Dueñas-jiménez, J.M. Proliferation and apoptosis regulation by G protein-coupled estrogen receptor in glioblastoma C6 cells. Oncol. Lett. 2022, 24, 217. [Google Scholar] [CrossRef]
- Peña-Gutiérrez, K.M.; Hernández-Ortega, K.; Bello-Alvarez, C.; Camacho-Arroyo, I. Expression and estrogen regulation of G protein-coupled estrogen receptor in human glioblastoma cells. Oncol. Lett. 2022, 24, 397. [Google Scholar] [CrossRef] [PubMed]
- Dahlberg, D.; Rummel, J.; Distante, S.; De Souza, G.A.; Stensland, M.E.; Mariussen, E.; Rootwelt, H.; Voie, Ø.; Hassel, B. Glioblastoma microenvironment contains multiple hormonal and non-hormonal growth-stimulating factors. Fluids Barriers CNS 2022, 19, 45. [Google Scholar] [CrossRef] [PubMed]
- Del Moral-Morales, A.; González-Orozco, J.C.; Hernández-Vega, A.M.; Hernández-Ortega, K.; Peña-Gutiérrez, K.M.; Camacho-Arroyo, I. EZH2 Mediates Proliferation, Migration, and Invasion Promoted by Estradiol in Human Glioblastoma Cells. Front. Endocrinol. 2022, 13, 703733. Available online: https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2022.703733 (accessed on 27 February 2024). [CrossRef] [PubMed]
- Pratap, U.P.; Tidwell, M.; Balinda, H.U.; Clanton, N.A.; Yang, X.; Viswanadhapalli, S.; Sareddy, G.R.; Liang, D.; Xie, H.; Chen, Y.; et al. Preclinical Development of Brain Permeable ERβ Agonist for the Treatment of Glioblastoma. Mol. Cancer Ther. 2023, 22, 1248–1260. [Google Scholar] [CrossRef]
- Pagano, M.T.; Ortona, E.; Dupuis, M.L. A Role for Estrogen Receptor alpha36 in Cancer Progression. Front. Endocrinol. 2020, 11, 506. Available online: https://www.frontiersin.org/journals/endocrinology/articles/10.3389/fendo.2020.00506 (accessed on 27 February 2024). [CrossRef]
- Mahboobifard, F.; Dargahi, L.; Jorjani, M.; Ramezani Tehrani, F.; Pourgholami, M.H. The role of ERα36 in cell type-specific functions of estrogen and cancer development. Pharmacol. Res. 2021, 163, 105307. [Google Scholar] [CrossRef]
- Lan, Y.-L.; Zou, S.; Wang, X.; Lou, J.-C.; Xing, J.-S.; Yu, M.; Zhang, B. Update on the therapeutic significance of estrogen receptor beta in malignant gliomas. Oncotarget 2017, 8, 81686–81696. [Google Scholar] [CrossRef][Green Version]
- Liu, J.; Sareddy, G.R.; Zhou, M.; Viswanadhapalli, S.; Li, X.; Lai, Z.; Tekmal, R.R.; Brenner, A.; Vadlamudi, R.K. Differential Effects of Estrogen Receptor β Isoforms on Glioblastoma Progression. Cancer Res. 2018, 78, 3176–3189. [Google Scholar] [CrossRef] [PubMed]
- Pratap, U.P.; Sareddy, G.R.; Liu, Z.; Venkata, P.P.; Liu, J.; Tang, W.; Altwegg, K.A.; Ebrahimi, B.; Li, X.; Tekmal, R.R.; et al. Histone deacetylase inhibitors enhance estrogen receptor beta expression and augment agonist-mediated tumor suppression in glioblastoma. Neuro-Oncol. Adv. 2021, 3, vdab099. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Pratap, U.P.; Venkata, P.P.; Zhou, M.; Alejo, S.; Viswanadhapalli, S.; Tekmal, R.R.; Brenner, A.J.; Vadlamudi, R.K. Activation of estrogen receptor beta signaling reduces stemness of glioma stem cells. Stem Cells 2021, 39, 536–550. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Nair, B.C.; Gonugunta, V.K.; Zhang, Q.; Brenner, A.; Brann, D.W.; Tekmal, R.R.; Vadlamudi, R.K. Therapeutic Significance of Estrogen Receptor β Agonists in Gliomas. Mol. Cancer Ther. 2012, 11, 1174–1182. [Google Scholar] [CrossRef]
- Matejka, N.; Reindl, J. Perspectives of cellular communication through tunneling nanotubes in cancer cells and the connection to radiation effects. Radiat. Oncol. 2019, 14, 218. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, A.-L.; Heiland, D.H.; Evert, B.O.; Almeida, F.R.; Behringer, S.P.; Dolf, A.; Güresir, Á.; Güresir, E.; Joseph, K.; Pietsch, T.; et al. Inhibition of Gap Junctions Sensitizes Primary Glioblastoma Cells for Temozolomide. Cancers 2019, 11, 858. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.-M.; Kim, S.-H.; Jin, X.; Park, J.B.; Kim, S.H.; Joshi, K.; Nakano, I.; Kim, H. Crosstalk between Glioma-Initiating Cells and Endothelial Cells Drives Tumor Progression. Cancer Res. 2014, 74, 4482–4492. [Google Scholar] [CrossRef]
- Wang, G.-G.; Wang, Y.; Wang, S.-L.; Zhu, L.-C. Down-regulation of CX43 expression by miR-1 inhibits the proliferation and invasion of glioma cells. Transl. Cancer Res. 2022, 11, 4126–4136. [Google Scholar] [CrossRef]
- Moon, C.S.; Moon, D.; Kang, S.K. Aquaporins in Cancer Biology. Front. Oncol. 2022, 12, 782829. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2022.782829 (accessed on 6 December 2023). [CrossRef]
- Varricchio, A. Pharmacologically Impeding Glioblastoma Tumour Motility through Simultaneous Inhibition of Aquaporin-1 and Ion Channels. Ph.D. Thesis, Adelaide Medical School, Adelaide, Australia, 2023. Available online: https://digital.library.adelaide.edu.au/dspace/handle/2440/138780 (accessed on 6 December 2023).
- McCoy, E.; Sontheimer, H. Expression and function of water channels (aquaporins) in migrating malignant astrocytes. Glia 2007, 55, 1034–1043. [Google Scholar] [CrossRef]
- Lan, Y.-L.; Nie, T.; Zou, S. Identification of the prognostic and immunological roles of aquaporin 4: A potential target for survival and immunotherapy in glioma patients. Front. Cell. Neurosci. 2022, 16, 1061428. Available online: https://www.frontiersin.org/articles/10.3389/fncel.2022.1061428 (accessed on 6 December 2023). [CrossRef] [PubMed]
- Tan, Y.; Zhang, H.; Zhao, R.-F.; Wang, X.-C.; Qin, J.-B.; Wu, X.-F. Comparison of the values of MRI diffusion kurtosis imaging and diffusion tensor imaging in cerebral astrocytoma grading and their association with aquaporin-4. Neurol. India 2016, 64, 265. [Google Scholar] [CrossRef]
- Zhao, Z.; Meng, F.; Wang, W.; Wang, Z.; Zhang, C.; Jiang, T. Comprehensive RNA-seq transcriptomic profiling in the malignant progression of gliomas. Sci. Data 2017, 4, 170024. [Google Scholar] [CrossRef] [PubMed]
- Siddaway, R.; Milos, S.; Vadivel, A.K.A.; Dobson, T.H.W.; Swaminathan, J.; Ryall, S.; Pajovic, S.; Patel, P.G.; Nazarian, J.; Becher, O.; et al. Splicing is an alternate oncogenic pathway activation mechanism in glioma. Nat. Commun. 2022, 13, 588. [Google Scholar] [CrossRef] [PubMed]
- Vasileva, N.S.; Kuligina, E.V.; Dymova, M.A.; Savinovskaya, Y.I.; Zinchenko, N.D.; Ageenko, A.B.; Mishinov, S.V.; Dome, A.S.; Stepanov, G.A.; Richter, V.A.; et al. Transcriptome Changes in Glioma Cells Cultivated under Conditions of Neurosphere Formation. Cells 2022, 11, 3106. [Google Scholar] [CrossRef] [PubMed]
- Jing, S.-Y.; Lu, Y.-Y.; Yang, J.-K.; Deng, W.-Y.; Zhou, Q.; Jiao, B.-H. Expression of long non-coding RNA CRNDE in glioma and its correlation with tumor progression and patient survival. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3992–3996. [Google Scholar]
- Wang, S.; Hui, Y.; Li, X.; Jia, Q. Silencing of lncRNA CCDC26 Restrains the Growth and Migration of Glioma Cells In Vitro and In Vivo via Targeting miR-203. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 1143–1154. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.; He, J.; Zhang, C.; Chen, J.; Shi, D. Long Noncoding RNA H19 Promotes Proliferation and Invasion in Human Glioma Cells by Downregulating miR-152. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2018, 26, 1419–1428. [Google Scholar] [CrossRef]
- Ma, C.-C.; Xiong, Z.; Zhu, G.-N.; Wang, C.; Zong, G.; Wang, H.-L.; Bian, E.-B.; Zhao, B. Long non-coding RNA ATB promotes glioma malignancy by negatively regulating miR-200a. J. Exp. Clin. Cancer Res. 2016, 35, 90. [Google Scholar] [CrossRef]
- Yao, J.; Zhou, B.; Zhang, J.; Geng, P.; Liu, K.; Zhu, Y.; Zhu, W. A new tumor suppressor LncRNA ADAMTS9-AS2 is regulated by DNMT1 and inhibits migration of glioma cells. Tumor Biol. 2014, 35, 7935–7944. [Google Scholar] [CrossRef]
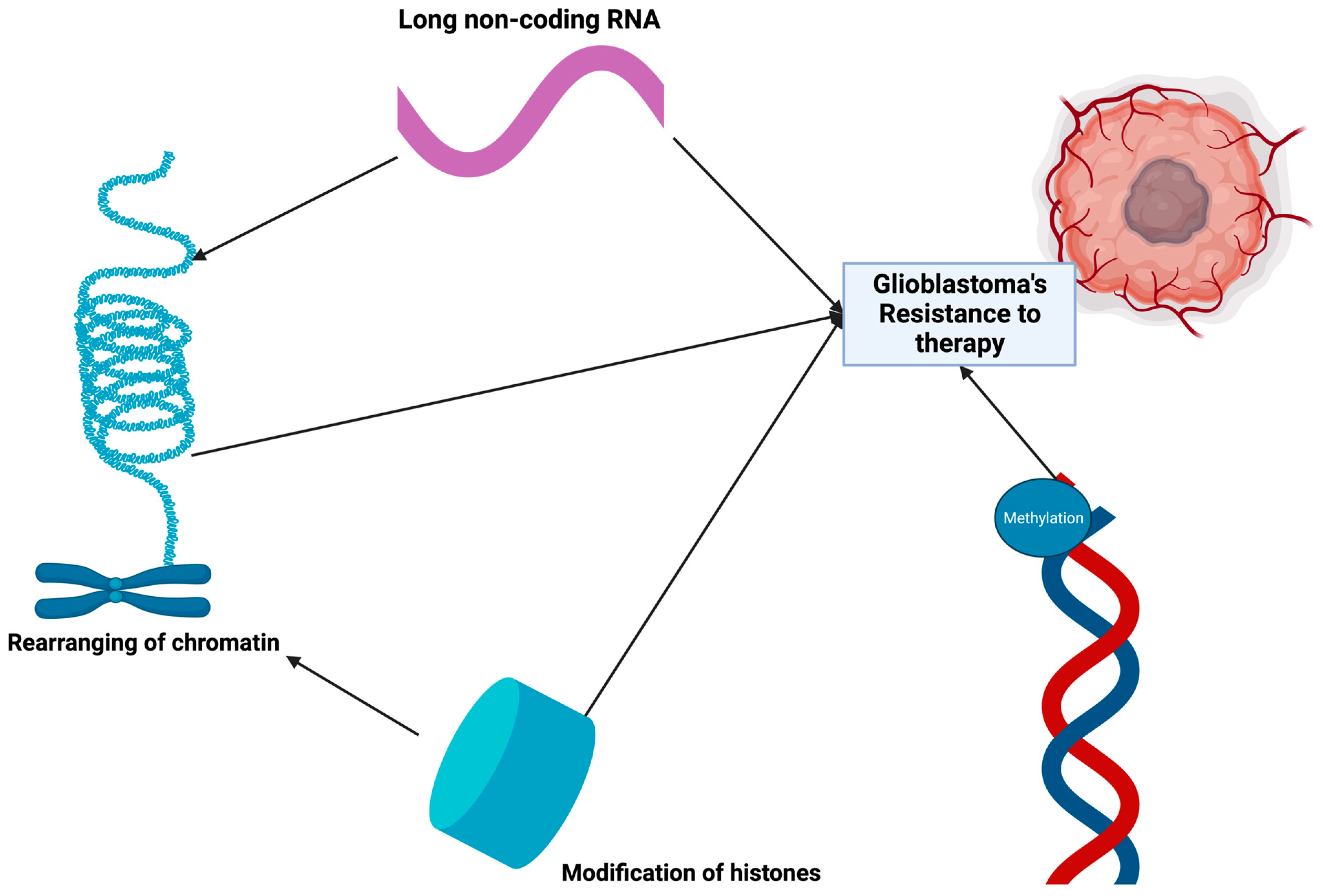
- Court, F.; Le Boiteux, E.; Fogli, A.; Müller-Barthélémy, M.; Vaurs-Barrière, C.; Chautard, E.; Pereira, B.; Biau, J.; Kemeny, J.-L.; Khalil, T.; et al. Transcriptional alterations in glioma result primarily from DNA methylation–independent mechanisms. Genome Res. 2019, 29, 1605–1621. [Google Scholar] [CrossRef]
- Binder, H.; Willscher, E.; Loeffler-Wirth, H.; Hopp, L.; Jones, D.T.W.; Pfister, S.M.; Kreuz, M.; Gramatzki, D.; Fortenbacher, E.; Hentschel, B.; et al. DNA methylation, transcriptome and genetic copy number signatures of diffuse cerebral WHO grade II/III gliomas resolve cancer heterogeneity and development. Acta Neuropathol. Commun. 2019, 7, 59. [Google Scholar] [CrossRef]
- Reifenberger, G.; Wirsching, H.-G.; Knobbe-Thomsen, C.B.; Weller, M. Advances in the molecular genetics of gliomas—Implications for classification and therapy. Nat. Rev. Clin. Oncol. 2017, 14, 434–452. [Google Scholar] [CrossRef]
- Fan, F.; Zhang, H.; Dai, Z.; Zhang, Y.; Xia, Z.; Cao, H.; Yang, K.; Hu, S.; Guo, Y.; Ding, F.; et al. A comprehensive prognostic signature for glioblastoma patients based on transcriptomics and single cell sequencing. Cell. Oncol. 2021, 44, 917–935. [Google Scholar] [CrossRef]
- Sorokin, M.; Raevskiy, M.; Zottel, A.; Šamec, N.; Skoblar Vidmar, M.; Matjašič, A.; Zupan, A.; Mlakar, J.; Suntsova, M.; Kuzmin, D.V.; et al. Large-Scale Transcriptomics-Driven Approach Revealed Overexpression of CRNDE as a Poor Survival Prognosis Biomarker in Glioblastoma. Cancers 2021, 13, 3419. [Google Scholar] [CrossRef]
- Bahar Halpern, K.; Vana, T.; Walker, M.D. Paradoxical Role of DNA Methylation in Activation of FoxA2 Gene Expression during Endoderm Development. J. Biol. Chem. 2014, 289, 23882–23892. [Google Scholar] [CrossRef] [PubMed]
- Piccioni, D.E.; Achrol, A.S.; Kiedrowski, L.A.; Banks, K.C.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 2019, 8, CNS34. [Google Scholar] [CrossRef] [PubMed]
- Tough, D.F.; Lewis, H.D.; Rioja, I.; Lindon, M.J.; Prinjha, R.K. Epigenetic pathway targets for the treatment of disease: Accelerating progress in the development of pharmacological tools: IUPHAR Review 11. Br. J. Pharmacol. 2014, 171, 4981–5010. [Google Scholar] [CrossRef] [PubMed]
- Gangoso, E.; Southgate, B.; Bradley, L.; Rus, S.; Galvez-Cancino, F.; McGivern, N.; Güç, E.; Kapourani, C.-A.; Byron, A.; Ferguson, K.M.; et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell 2021, 184, 2454–2470.e26. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Al-Zahrani, A.; Beylerli, O.; Sufianov, R.; Talybov, R.; Meshcheryakova, S.; Sufianova, G.; Gareev, I.; Sufianov, A. Circulating miRNAs as Diagnostic and Prognostic Biomarkers in High-Grade Gliomas. Front. Oncol. 2022, 12, 898537. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2022.898537 (accessed on 6 December 2023). [CrossRef] [PubMed]
- Sun, J.; Sun, Z.; Gareev, I.; Yan, T.; Chen, X.; Ahmad, A.; Zhang, D.; Zhao, B.; Beylerli, O.; Yang, G.; et al. Exosomal miR-2276-5p in Plasma Is a Potential Diagnostic and Prognostic Biomarker in Glioma. Front. Cell Dev. Biol. 2021, 9, 671202. Available online: https://www.frontiersin.org/articles/10.3389/fcell.2021.671202 (accessed on 6 December 2023). [CrossRef] [PubMed]
- Beylerli, O.; Khasanov, D.; Gareev, I.; Valitov, E.; Sokhatskii, A.; Wang, C.; Pavlov, V.; Khasanova, G.; Ahmad, A. Differential non-coding RNAs expression profiles of invasive and non-invasive pituitary adenomas. Non-Coding RNA Res. 2021, 6, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Kadiyala, P.; Carney, S.V.; Gauss, J.C.; Garcia-Fabiani, M.B.; Haase, S.; Alghamri, M.S.; Núñez, F.J.; Liu, Y.; Yu, M.; Taher, A.; et al. Inhibition of 2-hydroxyglutarate elicits metabolic reprogramming and mutant IDH1 glioma immunity in mice. J. Clin. Investig. 2021, 131, e139542. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Long, Y.; Yang, C.; Jin, L.; Tao, H.; Ge, H.; Chang, Y.E.; Karachi, A.; Kubilis, P.S.; De Leon, G.; et al. The IDH1 Mutation-Induced Oncometabolite, 2-Hydroxyglutarate, May Affect DNA Methylation and Expression of PD-L1 in Gliomas. Front. Mol. Neurosci. 2018, 11, 82. Available online: https://www.frontiersin.org/articles/10.3389/fnmol.2018.00082 (accessed on 6 December 2023). [CrossRef]
- Hu, C.; Wang, K.; Damon, C.; Fu, Y.; Ma, T.; Kratz, L.; Lal, B.; Ying, M.; Xia, S.; Cahill, D.P.; et al. ATRX loss promotes immunosuppressive mechanisms in IDH1 mutant glioma. Neuro-Oncol. 2022, 24, 888–900. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Kanamori, M.; Kawaguchi, T.; Nigro, J.M.; Feuerstein, B.G.; Berger, M.S.; Miele, L.; Pieper, R.O. Contribution of Notch signaling activation to human glioblastoma multiforme. J. Neurosurg. 2007, 106, 417–427. [Google Scholar] [CrossRef]
- Xing, Z.; Sun, L.; Guo, W. Elevated expression of Notch-1 and EGFR induced apoptosis in glioblastoma multiforme patients. Clin. Neurol. Neurosurg. 2015, 131, 54–58. [Google Scholar] [CrossRef]
- Han, N.; Hu, G.; Shi, L.; Long, G.; Yang, L.; Xi, Q.; Guo, Q.; Wang, J.; Dong, Z.; Zhang, M. Notch1 ablation radiosensitizes glioblastoma cells. Oncotarget 2017, 8, 88059–88068. [Google Scholar] [CrossRef]
- Dell’Albani, P.; Rodolico, M.; Pellitteri, R.; Tricarichi, E.; Torrisi, S.A.; D’Antoni, S.; Zappia, M.; Albanese, V.; Caltabiano, R.; Platania, N.; et al. Differential patterns of NOTCH1–4 receptor expression are markers of glioma cell differentiation. Neuro-Oncol. 2014, 16, 204–216. [Google Scholar] [CrossRef]
- Hori, K.; Sen, A.; Artavanis-Tsakonas, S. Notch signaling at a glance. J. Cell Sci. 2013, 126, 2135–2140. [Google Scholar] [CrossRef]
- Carlén, M.; Meletis, K.; Göritz, C.; Darsalia, V.; Evergren, E.; Tanigaki, K.; Amendola, M.; Barnabé-Heider, F.; Yeung, M.S.Y.; Naldini, L.; et al. Forebrain ependymal cells are Notch-dependent and generate neuroblasts and astrocytes after stroke. Nat. Neurosci. 2009, 12, 259–267. [Google Scholar] [CrossRef]
- Lasky, J.L.; Wu, H. Notch Signaling, Brain Development, and Human Disease. Pediatr. Res. 2005, 57, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.-J.; Koo, B.-K.; Im, S.-K.; Jeong, H.-W.; Ghim, J.; Kwon, M.; Moon, J.-S.; Miyata, T.; Kong, Y.-Y. Mind Bomb 1-Expressing Intermediate Progenitors Generate Notch Signaling to Maintain Radial Glial Cells. Neuron 2008, 58, 519–531. [Google Scholar] [CrossRef]
- Irvin, D.K.; Nakano, I.; Paucar, A.; Kornblum, H.I. Patterns of Jagged1, Jagged2, Delta-like 1 and Delta-like 3 expression during late embryonic and postnatal brain development suggest multiple functional roles in progenitors and differentiated cells. J. Neurosci. Res. 2004, 75, 330–343. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Sanchez, J.-C.; Gooley, A.A.; Appel, R.D.; Humphery-Smith, I.; Hochstrasser, D.F.; Williams, K.L. Progress with Proteome Projects: Why all Proteins Expressed by a Genome Should be Identified and How To Do It. Biotechnol. Genet. Eng. Rev. 1996, 13, 19–50. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Varshney, R.; Sethy, N.K. Human adaptation to high altitude: A review of convergence between genomic and proteomic signatures. Hum. Genom. 2022, 16, 21. [Google Scholar] [CrossRef] [PubMed]
- Thorarinsdottir, H.K.; Santi, M.; McCarter, R.; Rushing, E.J.; Cornelison, R.; Jales, A.; MacDonald, T.J. Protein Expression of Platelet-Derived Growth Factor Receptor Correlates with Malignant Histology and PTEN with Survival in Childhood Gliomas. Clin. Cancer Res. 2008, 14, 3386–3394. [Google Scholar] [CrossRef][Green Version]
- Sharifzad, F.; Ghavami, S.; Verdi, J.; Mardpour, S.; Mollapour Sisakht, M.; Azizi, Z.; Taghikhani, A.; Łos, M.J.; Fakharian, E.; Ebrahimi, M.; et al. Glioblastoma cancer stem cell biology: Potential theranostic targets. Drug Resist. Updates 2019, 42, 35–45. [Google Scholar] [CrossRef]
- Arora, A.; Patil, V.; Kundu, P.; Kondaiah, P.; Hegde, A.S.; Arivazhagan, A.; Santosh, V.; Pal, D.; Somasundaram, K. Serum biomarkers identification by iTRAQ and verification by MRM: S100A8/S100A9 levels predict tumor-stroma involvement and prognosis in Glioblastoma. Sci. Rep. 2019, 9, 2749. [Google Scholar] [CrossRef]
- Gaca-Tabaszewska, M.; Bogusiewicz, J.; Bojko, B. Metabolomic and Lipidomic Profiling of Gliomas—A New Direction in Personalized Therapies. Cancers 2022, 14, 5041. [Google Scholar] [CrossRef]
- Pandey, R.; Caflisch, L.; Lodi, A.; Brenner, A.J.; Tiziani, S. Metabolomic signature of brain cancer. Mol. Carcinog. 2017, 56, 2355–2371. [Google Scholar] [CrossRef]
- Uribe, D.; Niechi, I.; Rackov, G.; Erices, J.I.; San Martín, R.; Quezada, C. Adapt to Persist: Glioblastoma Microenvironment and Epigenetic Regulation on Cell Plasticity. Biology 2022, 11, 313. [Google Scholar] [CrossRef]
- Poff, A.; Koutnik, A.P.; Egan, K.M.; Sahebjam, S.; D’Agostino, D.; Kumar, N.B. Targeting the Warburg effect for cancer treatment: Ketogenic diets for management of glioma. Semin. Cancer Biol. 2019, 56, 135–148. [Google Scholar] [CrossRef]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Guido, C.; Whitaker-Menezes, D.; Bonuccelli, G.; Balliet, R.; Pestell, T.G.; Goldberg, A.F.; Pestell, R.G.; Howell, A.; Sneddon, S.; et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis, via glycolysis and ketone production. Cell Cycle 2012, 11, 2285–2302. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.E.; Lin, Z.; Whitaker-Menezes, D.; Howell, A.; Lisanti, M.P.; Sotgia, F. Ketone bodies and two-compartment tumor metabolism: Stromal ketone production fuels mitochondrial biogenesis in epithelial cancer cells. Cell Cycle 2012, 11, 3956–3963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, C.; Zheng, X.; Li, S.; Zhang, W.; Kang, Z.; Yin, S.; Chen, J.; Chen, F.; Li, W. Warburg effect-related risk scoring model to assess clinical significance and immunity characteristics of glioblastoma. Cancer Med. 2023, 12, 20639–20654. [Google Scholar] [CrossRef] [PubMed]
- Brito, C.; Tomás, A.; Azevedo, A.; Esteves, S.; Mafra, M.; Roque, L.; Pojo, M. PIK3CA Mutations in Diffuse Gliomas: An Update on Molecular Stratification, Prognosis, Recurrence, and Aggressiveness. Clin. Med. Insights Oncol. 2022, 16, 11795549211068804. Available online: https://journals.sagepub.com/doi/10.1177/11795549211068804 (accessed on 6 December 2023). [CrossRef] [PubMed]
- Fruman, D.A.; Mauvais-Jarvis, F.; Pollard, D.A.; Yballe, C.M.; Brazil, D.; Bronson, R.T.; Kahn, C.R.; Cantley, L.C. Hypoglycaemia, liver necrosis and perinatal death in mice lacking all isoforms of phosphoinositide 3-kinase p85α. Nat. Genet. 2000, 26, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Geering, B.; Cutillas, P.R.; Nock, G.; Gharbi, S.I.; Vanhaesebroeck, B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 7809–7814. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.Y.; Chute, D.J.; et al. Molecular Determinants of the Response of Glioblastomas to EGFR Kinase Inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Majewska, E.; Márquez, J.; Albrecht, J.; Szeliga, M. Transfection with GLS2 Glutaminase (GAB) Sensitizes Human Glioblastoma Cell Lines to Oxidative Stress by a Common Mechanism Involving Suppression of the PI3K/AKT Pathway. Cancers 2019, 11, 115. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, K.; Wu, Q.; Kim, L.J.Y.; Morton, A.R.; Gimple, R.C.; Prager, B.C.; Shi, Y.; Zhou, W.; Bhargava, S.; et al. Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells. Sci. Transl. Med. 2019, 11, eaau4972. [Google Scholar] [CrossRef]
- Akkulak, A.; Dağdelen, D.N.; Yalçın, A.; Oktay, E.; Diniz, G.; Kahraman, D.S.; Şenoğlu, M.; Yalcin, G.D. The expression of glutamate metabolism modulators in the intracranial tumors and glioblastoma cell line. Mol. Biol. Rep. 2022, 49, 1077–1083. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Martell, E.; Kuzmychova, H.; Senthil, H.; Kaul, E.; Chokshi, C.R.; Venugopal, C.; Anderson, C.M.; Singh, S.K.; Sharif, T. Compensatory cross-talk between autophagy and glycolysis regulates senescence and stemness in heterogeneous glioblastoma tumor subpopulations. Acta Neuropathol. Commun. 2023, 11, 110. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Wang, Z.; Dai, Z.; Zhang, H.; Zhang, J.; Luo, P.; Liu, Z.; Liu, Z.; Yang, K.; Cheng, Q.; et al. Glioblastoma glycolytic signature predicts unfavorable prognosis, immunological heterogeneity, and ENO1 promotes microglia M2 polarization and cancer cell malignancy. Cancer Gene Ther. 2023, 30, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, D.; Zhang, Z.; Wang, Y.; Zhang, Z.; Liang, Z.; Liu, F.; Chen, L. Photodynamic therapy enhances the cytotoxicity of temozolomide against glioblastoma via reprogramming anaerobic glycolysis. Photodiagnosis Photodyn. Ther. 2023, 42, 103342. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Bao, H.; Long, J.; Zhao, P.; Hu, X.; Wang, H.; Zhang, Y.; Yang, J.; Zhuge, Q.; Xia, L. GBE1 Promotes Glioma Progression by Enhancing Aerobic Glycolysis through Inhibition of FBP1. Cancers 2023, 15, 1594. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, W.; Liu, Y.; Ma, Z.; Xiang, W.; Wen, Y.; Zhang, D.; Li, Y.; Li, Y.; Li, T.; et al. PDIA4 promotes glioblastoma progression via the PI3K/AKT/m-TOR pathway. Biochem. Biophys. Res. Commun. 2022, 597, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Poteet, E.; Choudhury, G.R.; Winters, A.; Li, W.; Ryou, M.-G.; Liu, R.; Tang, L.; Ghorpade, A.; Wen, Y.; Yuan, F.; et al. Reversing the Warburg Effect as a Treatment for Glioblastoma *. J. Biol. Chem. 2013, 288, 9153–9164. [Google Scholar] [CrossRef]
- Jones, J.E.C.; Esler, W.P.; Patel, R.; Lanba, A.; Vera, N.B.; Pfefferkorn, J.A.; Vernochet, C. Inhibition of Acetyl-CoA Carboxylase 1 (ACC1) and 2 (ACC2) Reduces Proliferation and De Novo Lipogenesis of EGFRvIII Human Glioblastoma Cells. PLoS ONE 2017, 12, e0169566. [Google Scholar] [CrossRef]
- Martins, F.; van der Kellen, D.; Gonçalves, L.G.; Serpa, J. Metabolic Profiles Point Out Metabolic Pathways Pivotal in Two Glioblastoma (GBM) Cell Lines, U251 and U-87MG. Biomedicines 2023, 11, 2041. Available online: https://www.mdpi.com/2227-9059/11/7/2041 (accessed on 27 February 2024). [CrossRef]
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa, K.J.; Singh, D.K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo, S.G.; Kovacs, Z.; Foong, C.; et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614. [Google Scholar] [CrossRef]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef]
- Jatyan, R.; Sahel, D.K.; Singh, P.; Sakhuja, R.; Mittal, A.; Chitkara, D. Temozolomide-fatty acid conjugates for glioblastoma multiforme: In vitro and in vivo evaluation. J. Control. Release 2023, 359, 161–174. [Google Scholar] [CrossRef]
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J.; Czernin, J.; Shyy, J.Y.-J.; Watson, A.D.; Phelps, M.; et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932–12937. [Google Scholar] [CrossRef]
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR Agonist Promotes Glioblastoma Cell Death through Inhibition of an EGFR/AKT/SREBP-1/LDLR–Dependent Pathway. Cancer Discov. 2011, 1, 442–456. [Google Scholar] [CrossRef]
- Xu, D.S.; Awad, A.-W.; Mehalechko, C.; Wilson, J.R.; Ashby, L.S.; Coons, S.W.; Sanai, N. An extent of resection threshold for seizure freedom in patients with low-grade gliomas. J. Neurosurg. 2018, 128, 1084–1090. [Google Scholar] [CrossRef]
- Jakola, A.S.; Myrmel, K.S.; Kloster, R.; Torp, S.H.; Lindal, S.; Unsgård, G.; Solheim, O. Comparison of a Strategy Favoring Early Surgical Resection vs a Strategy Favoring Watchful Waiting in Low-Grade Gliomas. JAMA 2012, 308, 1881–1888. [Google Scholar] [CrossRef]
- Brown, T.J.; Brennan, M.C.; Li, M.; Church, E.W.; Brandmeir, N.J.; Rakszawski, K.L.; Patel, A.S.; Rizk, E.B.; Suki, D.; Sawaya, R.; et al. Association of the Extent of Resection with Survival in Glioblastoma: A Systematic Review and Meta-analysis. JAMA Oncol. 2016, 2, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Sattari, S.A.; Rincon-Torroella, J.; Sattari, A.R.; Feghali, J.; Yang, W.; Kim, J.E.; Xu, R.; Jackson, C.M.; Mukherjee, D.; Lin, S.-C.; et al. Awake Versus Asleep Craniotomy for Patients with Eloquent Glioma: A Systematic Review and Meta-Analysis. Neurosurgery 2022, 94, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, K.; Takahashi, S.; Mori, T.; Uchinami, Y.; Yamaguchi, S.; Kinoshita, M.; Yamashina, M.; Higaki, H.; Maebayashi, K.; Aoyama, H. The need of radiotherapy optimization for glioblastomas considering immune responses. Jpn. J. Radiol. 2023, 41, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhai, X.; Yin, H.; Zhou, F.; Hu, Y.; Wang, K.; Yan, R.; Han, D. Multimodality MRI Radiomics Based on Machine Learning for Identifying True Tumor Recurrence and Treatment-Related Effects in Patients with Postoperative Glioma. Neurol. Ther. 2023, 12, 1729–1743. [Google Scholar] [CrossRef]
- Wei, R.; Chen, H.; Cai, Y.; Chen, J. Application of intraoperative ultrasound in the resection of high-grade gliomas. Front. Neurol. 2023, 14, 1240150. Available online: https://www.frontiersin.org/articles/10.3389/fneur.2023.1240150 (accessed on 7 December 2023). [CrossRef] [PubMed]
- Kyrgias, G.; Hajiioannou, J.; Tolia, M.; Kouloulias, V.; Lachanas, V.; Skoulakis, C.; Skarlatos, I.; Rapidis, A.; Bizakis, I. Intraoperative radiation therapy (IORT) in head and neck cancer: A systematic review. Medicine 2016, 95, e5035. [Google Scholar] [CrossRef]
- Leroy, H.-A.; Delmaire, C.; Le Rhun, E.; Drumez, E.; Lejeune, J.-P.; Reyns, N. High-field intraoperative MRI and glioma surgery: Results after the first 100 consecutive patients. Acta Neurochir. 2019, 161, 1467–1474. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, X.; Mei, S.; Sun, Y.; Li, J. Acquisition of temozolomide resistance by the rat C6 glioma cell line increases cell migration and side population phenotype. Oncol. Rep. 2019, 42, 2355–2362. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Stenken, J.A.; Poschenrieder, A.J. Bioanalytical chemistry of cytokines—A review. Anal. Chim. Acta 2015, 853, 95–115. [Google Scholar] [CrossRef] [PubMed]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Fang, S.; Dai, Y.; Mei, Y.; Yang, M.; Hu, L.; Yang, H.; Guan, X.; Li, J. Clinical significance and biological role of cancer-derived Type I collagen in lung and esophageal cancers. Thorac. Cancer 2019, 10, 277–288. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Yu, M.; Chang, Y.; Zhai, Y.; Pang, B.; Wang, P.; Li, G.; Jiang, T.; Zeng, F. TREM2 is associated with tumor immunity and implies poor prognosis in glioma. Front. Immunol. 2023, 13, 1089266. Available online: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1089266 (accessed on 6 December 2023). [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Giannoni, E.; Bianchini, F.; Masieri, L.; Serni, S.; Torre, E.; Calorini, L.; Chiarugi, P. Reciprocal Activation of Prostate Cancer Cells and Cancer-Associated Fibroblasts Stimulates Epithelial-Mesenchymal Transition and Cancer Stemness. Cancer Res. 2010, 70, 6945–6956. [Google Scholar] [CrossRef] [PubMed]
- Hawinkels, L.J.a.C.; Paauwe, M.; Verspaget, H.W.; Wiercinska, E.; van der Zon, J.M.; van der Ploeg, K.; Koelink, P.J.; Lindeman, J.H.N.; Mesker, W.; ten Dijke, P.; et al. Interaction with colon cancer cells hyperactivates TGF-β signaling in cancer-associated fibroblasts. Oncogene 2014, 33, 97–107. [Google Scholar] [CrossRef]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Rick, J.W.; Joshi, R.; Beniwal, A.; Spatz, J.; Chang, A.C.-C.; Nguyen, A.T.; Sudhir, S.; Chandra, A.; Haddad, A.; et al. Identification of Cancer-Associated Fibroblasts in Glioblastoma and Defining Their Pro-tumoral Effects. Cancer Biol. 2021. [Google Scholar] [CrossRef]
- 258. Jain, S.; Rick, J.W.; Joshi, R.S.; Beniwal, A.; Spatz, J.; Gill, S.; Chang, A.C.; Choudhary, N.; Nguyen, A.T.; Sudhir, S.; et al. Single-cell RNA sequencing and spatial transcriptomics reveal cancer-associated fibroblasts in glioblastoma with protumoral effects. J. Clin. Investig. 2023, 133, e147087. [Google Scholar] [CrossRef] [PubMed]
- Galbo, P.M.; Liu, Y.; Peng, M.; Wei, Y.; Madsen, A.T.; Graff, S.; Montagna, C.; Segall, J.E.; Sidoli, S.; Zang, X.; et al. Functional Contribution of Cancer-Associated Fibroblasts in Glioblastoma. Cancer Biol. 2022. [Google Scholar] [CrossRef]
- Tavakoli, F.; Sartakhti, J.S.; Manshaei, M.H.; Basanta, D. Cancer immunoediting: A game theoretical approach. Silico Biol. 2021, 14, 1–12. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Voutouri, C.; Kirkpatrick, N.D.; Chung, E.; Mpekris, F.; Baish, J.W.; Munn, L.L.; Fukumura, D.; Stylianopoulos, T.; Jain, R.K. Experimental and computational analyses reveal dynamics of tumor vessel cooption and optimal treatment strategies. Proc. Natl. Acad. Sci. USA 2019, 116, 2662–2671. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Gerber, H.-P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Hovinga, K.E.; Shimizu, F.; Wang, R.; Panagiotakos, G.; Van Der Heijden, M.; Moayedpardazi, H.; Correia, A.S.; Soulet, D.; Major, T.; Menon, J.; et al. Inhibition of Notch Signaling in Glioblastoma Targets Cancer Stem Cells via an Endothelial Cell Intermediate. Stem Cells 2010, 28, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Wu, Q.; Yan, K.; MacSwords, J.M.; Chandler-Militello, D.; Misuraca, K.L.; Lathia, J.D.; Forrester, M.T.; Lee, J.; Stamler, J.S.; et al. Glioma Stem Cell Proliferation and Tumor Growth Are Promoted by Nitric Oxide Synthase-2. Cell 2011, 146, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.; Ozawa, T.; Squatrito, M.; Bleau, A.-M.; Brennan, C.W.; Hambardzumyan, D.; Holland, E.C. Perivascular Nitric Oxide Activates Notch Signaling and Promotes Stem-like Character in PDGF-Induced Glioma Cells. Cell Stem Cell 2010, 6, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.C.; Veeravagu, A.; Hsu, A.R.; Tse, V.C.K. Recurrent glioblastoma multiforme: A review of natural history and management options. Neurosurg. Focus 2006, 20, E3. [Google Scholar] [CrossRef] [PubMed]
- Baker, G.J.; Yadav, V.N.; Motsch, S.; Koschmann, C.; Calinescu, A.-A.; Mineharu, Y.; Camelo-Piragua, S.I.; Orringer, D.; Bannykh, S.; Nichols, W.S.; et al. Mechanisms of Glioma Formation: Iterative Perivascular Glioma Growth and Invasion Leads to Tumor Progression, VEGF-Independent Vascularization, and Resistance to Antiangiogenic Therapy. Neoplasia 2014, 16, 543–561. [Google Scholar] [CrossRef]
- Seifert, S.; Sontheimer, H. Bradykinin enhances invasion of malignant glioma into the brain parenchyma by inducing cells to undergo amoeboid migration. J. Physiol. 2014, 592, 5109–5127. [Google Scholar] [CrossRef]
- Alam, M.M.; Lee, S.-C.; Jung, Y.; Yun, H.J.; Min, H.-Y.; Lee, H.J.; Pham, P.C.; Moon, J.; Kwon, D.I.; Lim, B.; et al. Novel C6-substituted 1,3,4-oxadiazinones as potential anti-cancer agents. Oncotarget 2015, 6, 40598–40610. [Google Scholar] [CrossRef]
- Pham, K.; Luo, D.; Siemann, D.W.; Law, B.K.; Reynolds, B.A.; Hothi, P.; Foltz, G.; Harrison, J.K. VEGFR inhibitors upregulate CXCR4 in VEGF receptor-expressing glioblastoma in a TGFβR signaling-dependent manner. Cancer Lett. 2015, 360, 60–67. [Google Scholar] [CrossRef]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel Cooption, Regression, and Growth in Tumors Mediated by Angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef]
- McCoy, M.G.; Nyanyo, D.; Hung, C.K.; Goerger, J.P.; Zipfel, W.R.; Williams, R.M.; Nishimura, N.; Fischbach, C. Endothelial cells promote 3D invasion of GBM by IL-8-dependent induction of cancer stem cell properties. Sci. Rep. 2019, 9, 9069. [Google Scholar] [CrossRef]
- Infanger, D.W.; Cho, Y.; Lopez, B.S.; Mohanan, S.; Liu, S.C.; Gursel, D.; Boockvar, J.A.; Fischbach, C. Glioblastoma Stem Cells Are Regulated by Interleukin-8 Signaling in a Tumoral Perivascular Niche. Cancer Res. 2013, 73, 7079–7089. [Google Scholar] [CrossRef]
- Lindberg, O.R.; McKinney, A.; Engler, J.R.; Koshkakaryan, G.; Gong, H.; Robinson, A.E.; Ewald, A.J.; Huillard, E.; James, C.D.; Molinaro, A.M.; et al. GBM heterogeneity as a function of variable epidermal growth factor receptor variant III activity. Oncotarget 2016, 7, 79101–79116. [Google Scholar] [CrossRef][Green Version]
- Hyvönen, M.; Enbäck, J.; Huhtala, T.; Lammi, J.; Sihto, H.; Weisell, J.; Joensuu, H.; Rosenthal-Aizman, K.; El-Andaloussi, S.; Langel, U.; et al. Novel Target for Peptide-Based Imaging and Treatment of Brain Tumors. Mol. Cancer Ther. 2014, 13, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Jabouille, A.; Delugin, M.; Pineau, R.; Dubrac, A.; Soulet, F.; Lhomond, S.; Pallares-Lupon, N.; Prats, H.; Bikfalvi, A.; Chevet, E.; et al. Glioblastoma invasion and cooption depend on IRE1α; endoribonuclease activity. Oncotarget 2015, 6, 24922–24934. [Google Scholar] [CrossRef]
- Auf, G.; Jabouille, A.; Guérit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-requiring enzyme 1α is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef] [PubMed]
- Caspani, E.M.; Crossley, P.H.; Redondo-Garcia, C.; Martinez, S. Glioblastoma: A Pathogenic Crosstalk between Tumor Cells and Pericytes. PLoS ONE 2014, 9, e101402. [Google Scholar] [CrossRef] [PubMed]
- Griveau, A.; Seano, G.; Shelton, S.J.; Kupp, R.; Jahangiri, A.; Obernier, K.; Krishnan, S.; Lindberg, O.R.; Yuen, T.J.; Tien, A.-C.; et al. A Glial Signature and Wnt7 Signaling Regulate Glioma-Vascular Interactions and Tumor Microenvironment. Cancer Cell 2018, 33, 874–889.e7. [Google Scholar] [CrossRef] [PubMed]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.F.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef]
- Montana, V.; Sontheimer, H. Bradykinin Promotes the Chemotactic Invasion of Primary Brain Tumors. J. Neurosci. 2011, 31, 4858–4867. [Google Scholar] [CrossRef]
- Zagzag, D.; Esencay, M.; Mendez, O.; Yee, H.; Smirnova, I.; Huang, Y.; Chiriboga, L.; Lukyanov, E.; Liu, M.; Newcomb, E.W. Hypoxia- and Vascular Endothelial Growth Factor-Induced Stromal Cell-Derived Factor-1α/CXCR4 Expression in Glioblastomas. Am. J. Pathol. 2008, 173, 545–560. [Google Scholar] [CrossRef]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neuro-Oncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef] [PubMed]
- Labanieh, L.; Majzner, R.G.; Klysz, D.; Sotillo, E.; Fisher, C.J.; Vilches-Moure, J.G.; Pacheco, K.Z.B.; Malipatlolla, M.; Xu, P.; Hui, J.H.; et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell 2022, 185, 1745–1763.e22. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef] [PubMed]
- Nehama, D.; Di Ianni, N.; Musio, S.; Du, H.; Patané, M.; Pollo, B.; Finocchiaro, G.; Park, J.J.H.; Dunn, D.E.; Edwards, D.S.; et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine 2019, 47, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Meister, H.; Look, T.; Roth, P.; Pascolo, S.; Sahin, U.; Lee, S.; Hale, B.D.; Snijder, B.; Regli, L.; Ravi, V.M.; et al. Multifunctional mRNA-Based CAR T Cells Display Promising Antitumor Activity Against Glioblastoma. Clin. Cancer Res. 2022, 28, 4747–4756. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, Z.; Zhong, K.; Wang, Z.; Yang, N.; Tang, X.; Li, H.; Lu, Q.; Wu, Z.; Yuan, B.; et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cell therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol. Ther. 2023, 31, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Chalise, L.; Kato, A.; Ohno, M.; Maeda, S.; Yamamichi, A.; Kuramitsu, S.; Shiina, S.; Takahashi, H.; Ozone, S.; Yamaguchi, J.; et al. Efficacy of cancer-specific anti-podoplanin CAR-T cells and oncolytic herpes virus G47Δ combination therapy against glioblastoma. Mol. Ther.-Oncolytics 2022, 26, 265–274. [Google Scholar] [CrossRef]
- Cobb, D.A.; de Rossi, J.; Liu, L.; An, E.; Lee, D.W. Targeting of the alphav beta3 integrin complex by CAR-T cells leads to rapid regression of diffuse intrinsic pontine glioma and glioblastoma. J. Immunother. Cancer 2022, 10, e003816. [Google Scholar] [CrossRef]
- Tsoumakidou, M. The advent of immune stimulating CAFs in cancer. Nat. Rev. Cancer 2023, 23, 258–269. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast Heterogeneity and Immunosuppressive Environment in Human Breast Cancer. Cancer Cell 2018, 33, 463–479.e10. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein–α. Science 2010, 330, 827–830. Available online: https://www.science.org/doi/10.1126/science.1195300 (accessed on 7 December 2023). [CrossRef]
- Chauhan, V.P.; Chen, I.X.; Tong, R.; Ng, M.R.; Martin, J.D.; Naxerova, K.; Wu, M.W.; Huang, P.; Boucher, Y.; Kohane, D.S.; et al. Reprogramming the microenvironment with tumor-selective angiotensin blockers enhances cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2019, 116, 10674–10680. [Google Scholar] [CrossRef]
- Chen, Z.; Zhuo, S.; He, G.; Tang, J.; Hao, W.; Gao, W.-Q.; Yang, K.; Xu, H. Prognosis and Immunotherapy Significances of a Cancer-Associated Fibroblasts-Related Gene Signature in Gliomas. Front. Cell Dev. Biol. 2021, 9, 721897. Available online: https://www.frontiersin.org/articles/10.3389/fcell.2021.721897 (accessed on 7 December 2023). [CrossRef]
- Shin, S.Y.; Lee, K.S.; Choi, Y.-K.; Lim, H.J.; Lee, H.G.; Lim, Y.; Lee, Y.H. The antipsychotic agent chlorpromazine induces autophagic cell death by inhibiting the Akt/mTOR pathway in human U-87MG glioma cells. Carcinogenesis 2013, 34, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Matteoni, S.; Matarrese, P.; Ascione, B.; Buccarelli, M.; Ricci-Vitiani, L.; Pallini, R.; Villani, V.; Pace, A.; Paggi, M.G.; Abbruzzese, C. Anticancer Properties of the Antipsychotic Drug Chlorpromazine and Its Synergism with Temozolomide in Restraining Human Glioblastoma Proliferation In Vitro. Front. Oncol. 2021, 11, 635472. Available online: https://www.frontiersin.org/journals/oncology/articles/10.3389/fonc.2021.635472 (accessed on 27 February 2024). [CrossRef]
- Matteoni, S.; Matarrese, P.; Ascione, B.; Ricci-Vitiani, L.; Pallini, R.; Villani, V.; Pace, A.; Paggi, M.G.; Abbruzzese, C. Chlorpromazine induces cytotoxic autophagy in glioblastoma cells via endoplasmic reticulum stress and unfolded protein response. J. Exp. Clin. Cancer Res. 2021, 40, 347. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Xi, H.; Liao, M.; Zhang, Y.; Ma, H.; Wu, M.; Xue, Q.; Sun, H.; Zhang, Y.; Xia, Y. Repurposed antipsychotic chlorpromazine inhibits colorectal cancer and pulmonary metastasis by inducing G2/M cell cycle arrest, apoptosis, and autophagy. Cancer Chemother. Pharmacol. 2022, 89, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Jhou, A.-J.; Chang, H.-C.; Hung, C.-C.; Lin, H.-C.; Lee, Y.-C.; Liu, W.; Han, K.-F.; Lai, Y.-W.; Lin, M.-Y.; Lee, C.-H. Chlorpromazine, an antipsychotic agent, induces G2/M phase arrest and apoptosis via regulation of the PI3K/AKT/mTOR-mediated autophagy pathways in human oral cancer. Biochem. Pharmacol. 2021, 184, 114403. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Lee, J.M.; Jeon, B.; Elkamhawy, A.; Paik, S.; Hong, J.; Oh, S.-J.; Paek, S.H.; Lee, C.J.; Hassan, A.H.E.; et al. Repositioning of the antipsychotic trifluoperazine: Synthesis, biological evaluation and in silico study of trifluoperazine analogs as anti-glioblastoma agents. Eur. J. Med. Chem. 2018, 151, 186–198. [Google Scholar] [CrossRef]
- Kang, S.; Hong, J.; Lee, J.M.; Moon, H.E.; Jeon, B.; Choi, J.; Yoon, N.A.; Paek, S.H.; Roh, E.J.; Lee, C.J.; et al. Trifluoperazine, a Well-Known Antipsychotic, Inhibits Glioblastoma Invasion by Binding to Calmodulin and Disinhibiting Calcium Release Channel IP3R. Mol. Cancer Ther. 2017, 16, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kwon, H.N.; Kang, S.; Park, S. Interaction between IDH1 WT and calmodulin and its implications for glioblastoma cell growth and migration. Biochem. Biophys. Res. Commun. 2020, 524, 224–230. [Google Scholar] [CrossRef]
- Fishman, H.; Monin, R.; Dor-On, E.; Kinzel, A.; Haber, A.; Giladi, M.; Weinberg, U.; Palti, Y. Tumor Treating Fields (TTFields) increase the effectiveness of temozolomide and lomustine in glioblastoma cell lines. J. Neurooncol. 2023, 163, 83–94. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination with Extension of Survival Among Patients with Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Westberg, S.; Sadeghi, A.; Svensson, E.; Segall, T.; Dimopoulou, M.; Korsgren, O.; Hemminki, A.; Loskog, A.S.I.; Tötterman, T.H.; Von Euler, H. Treatment Efficacy and Immune Stimulation by AdCD40L Gene Therapy of Spontaneous Canine Malignant Melanoma. J. Immunother. 2013, 36, 350–358. [Google Scholar] [CrossRef]
- Gillard, M.; Cadieu, E.; De Brito, C.; Abadie, J.; Vergier, B.; Devauchelle, P.; Degorce, F.; Dréano, S.; Primot, A.; Dorso, L.; et al. Naturally occurring melanomas in dogs as models for non-UV pathways of human melanomas. Pigment Cell Melanoma Res. 2014, 27, 90–102. [Google Scholar] [CrossRef]
- Samadder, N.J.; Jasperson, K.; Burt, R.W. Hereditary and Common Familial Colorectal Cancer: Evidence for Colorectal Screening. Dig. Dis. Sci. 2015, 60, 734–747. [Google Scholar] [CrossRef]
- Ochocka, N.; Segit, P.; Walentynowicz, K.A.; Wojnicki, K.; Cyranowski, S.; Swatler, J.; Mieczkowski, J.; Kaminska, B. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021, 12, 1151. [Google Scholar] [CrossRef]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef]
- Roux, A.; Zanello, M.; Antonia Simboli, G.; Varlet, P.; Tauziede-Espariat, A.; Beccaria, K.; Blauwblomme, T.; Puget, S.; Oppenheim, C.; Dangouloff-Ros, V.; et al. Clinico-Radio-Histo-Molecular and Neurocognitive Characteristics of Diffuse Gliomas in Adolescent and Young Adults: A Comprehensive Review. Oncology 2023, 101, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yang, Q.; Xu, P.; Deng, M.; Jiang, T.; Cai, L.; Li, J.; Sai, K.; Xi, S.; Ouyang, H.; et al. Adjuvant Temozolomide Chemotherapy with or without Interferon Alfa Among Patients with Newly Diagnosed High-grade Gliomas: A Randomized Clinical Trial. JAMA Netw. Open 2023, 6, e2253285. [Google Scholar] [CrossRef]
- He, J.; Zhou, J.; Yang, W.; Zhou, Q.; Liang, X.; Pang, X.; Li, J.; Pan, F.; Liang, H. Dexamethasone affects cell growth/apoptosis/chemosensitivity of colon cancer via glucocorticoid receptor α/NF-κB. Oncotarget 2017, 8, 67670–67683. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, R.; Yao, Q.; Liu, S.; Tian, X.; Hao, C.; Lu, W.; Zhou, T. Time-dependent pharmacokinetics of dexamethasone and its efficacy in human breast cancer xenograft mice: A semi-mechanism-based pharmacokinetic/pharmacodynamic model. Acta Pharmacol. Sin. 2018, 39, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Jessurun, C.A.C.; Hulsbergen, A.F.C.; Lamba, N.; Nandoe Tewarie, R.D.S.; Smith, T.R.; Broekman, M.L.D. Practice Variation in Perioperative Steroid Dosing for Brain Tumor Patients: An International Survey. World Neurosurg. 2022, 159, e431–e441. [Google Scholar] [CrossRef]
- Iorgulescu, J.B.; Gokhale, P.C.; Speranza, M.C.; Eschle, B.K.; Poitras, M.J.; Wilkens, M.K.; Soroko, K.M.; Chhoeu, C.; Knott, A.; Gao, Y.; et al. Concurrent Dexamethasone Limits the Clinical Benefit of Immune Checkpoint Blockade in Glioblastoma. Clin. Cancer Res. 2021, 27, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Bielecka-Wajdman, A.M.; Ludyga, T.; Smyk, D.; Smyk, W.; Mularska, M.; Świderek, P.; Majewski, W.; Mullins, C.S.; Linnebacher, M.; Obuchowicz, E. Glucose Influences the Response of Glioblastoma Cells to Temozolomide and Dexamethasone. Cancer Control 2022, 29, 10732748221075468. Available online: https://journals.sagepub.com/doi/10.1177/10732748221075468 (accessed on 7 December 2023). [CrossRef]
- Lin, Y.-M.; Jan, H.-J.; Lee, C.-C.; Tao, H.-Y.; Shih, Y.-L.; Wei, H.-W.; Lee, H.-M. Dexamethasone reduced invasiveness of human malignant glioblastoma cells through a MAPK phosphatase-1 (MKP-1) dependent mechanism. Eur. J. Pharmacol. 2008, 593, 1–9. [Google Scholar] [CrossRef]
- McPherson, R.C.; Konkel, J.E.; Prendergast, C.T.; Thomson, J.P.; Ottaviano, R.; Leech, M.D.; Kay, O.; Zandee, S.E.J.; Sweenie, C.H.; Wraith, D.C.; et al. Epigenetic modification of the PD-1 (Pdcd1) promoter in effector CD4+ T cells tolerized by peptide immunotherapy. eLife 2014, 3, e03416. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Zhang, T.; Deng, S.-C.; Wei, J.-C.; Yang, P.; Wang, Q.; Chen, Z.-P.; Li, W.-L.; Chen, H.-C.; Hu, H.; et al. PD-L1 promotes colorectal cancer stem cell expansion by activating HMGA1-dependent signaling pathways. Cancer Lett. 2019, 450, 1–13. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Morris, V.K.; Salem, M.E.; Nimeiri, H.; Iqbal, S.; Singh, P.; Ciombor, K.; Polite, B.; Deming, D.; Chan, E.; Wade, J.L.; et al. Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): A multicentre, single-arm, phase 2 study. Lancet Oncol. 2017, 18, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Nokihara, H.; Yamada, Y.; Shibata, T.; Tamura, Y.; Seki, Y.; Honda, K.; Tanabe, Y.; Wakui, H.; Tamura, T. Phase I study of Nivolumab, an anti-PD-1 antibody, in patients with malignant solid tumors. Investig. New Drugs 2017, 35, 207–216. [Google Scholar] [CrossRef]
- Stutvoet, T.S.; Kol, A.; de Vries, E.G.; de Bruyn, M.; Fehrmann, R.S.; Terwisscha van Scheltinga, A.G.; de Jong, S. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J. Pathol. 2019, 249, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Torasawa, M.; Yoshida, T.; Yagishita, S.; Shimoda, Y.; Shirasawa, M.; Matsumoto, Y.; Masuda, K.; Shinno, Y.; Okuma, Y.; Goto, Y.; et al. Nivolumab versus pembrolizumab in previously-treated advanced non-small cell lung cancer patients: A propensity-matched real-world analysis. Lung Cancer 2022, 167, 49–57. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Guo, G.; Cai, X.; Yu, H.; Cai, Y.; Zhang, B.; Hong, S.; Zhang, L. Nivolumab plus ipilimumab versus pembrolizumab as chemotherapy-free, first-line treatment for PD-L1-positive non-small cell lung cancer. Clin. Transl. Med. 2020, 10, 107–115. [Google Scholar] [CrossRef]
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Milani, M.; Volpari, T.; Chiodoni, C.; et al. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro-Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaih, A.; Steinbach, J.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.; Taylor, J.W.; Honnorat, J.; et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro-Oncol. 2022, 24, 1935–1949. [Google Scholar] [CrossRef]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaih, A.; Reardon, D.A.; Cloughesy, T.; Sumrall, A.; Baehring, J.; van den Bent, M.; Bähr, O.; et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro-Oncol. 2023, 25, 123–134. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chen, Y.; Guo, Y.; Xu, J.; Wang, X.; Ning, W.; Ma, L.; Qu, Y.; Zhang, M.; Zhang, H. Mutation-derived, genomic instability-associated lncRNAs are prognostic markers in gliomas. PeerJ 2023, 11, e15810. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; Van Der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Nassiri, F.; Patil, V.; Yefet, L.S.; Singh, O.; Liu, J.; Dang, R.M.A.; Yamaguchi, T.N.; Daras, M.; Cloughesy, T.F.; Colman, H.; et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: A phase 1/2 trial. Nat. Med. 2023, 29, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Y.; Gu, L.; Chen, R.; Zhu, H.; Zhang, X.; Zhang, Y.; Feng, S.; Qiu, S.; Jian, Z.; et al. Gene Targets of CAR-T Cell Therapy for Glioblastoma. Cancers 2023, 15, 2351. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Vicente, Y.; Castillejos-López, M.D.J.; Carmona-Aparicio, L.; Coballase-Urrutia, E.; Velasco-Hidalgo, L.; Niembro-Zúñiga, A.M.; Zapata-Tarrés, M.; Torres-Espíndola, L.M. Immunotherapy for Pediatric Gliomas: CAR-T Cells Against B7H3: AReview of the Literature. CNS Neurol. Disord.-Drug Targets 2024, 23, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Philip, A.; Samuel, B.; Bhatia, S.; Khalifa, S.; El-Seedi, H. Artificial Intelligence and Precision Medicine: A New Frontier for the Treatment of Brain Tumors. Life 2022, 13, 24. [Google Scholar] [CrossRef]
- Xu, J.; Meng, Y.; Qiu, K.; Topatana, W.; Li, S.; Wei, C.; Chen, T.; Chen, M.; Ding, Z.; Niu, G. Applications of Artificial Intelligence Based on Medical Imaging in Glioma: Current State and Future Challenges. Front. Oncol. 2022, 12, 892056. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onciul, R.; Brehar, F.-M.; Toader, C.; Covache-Busuioc, R.-A.; Glavan, L.-A.; Bratu, B.-G.; Costin, H.P.; Dumitrascu, D.-I.; Serban, M.; Ciurea, A.V. Deciphering Glioblastoma: Fundamental and Novel Insights into the Biology and Therapeutic Strategies of Gliomas. Curr. Issues Mol. Biol. 2024, 46, 2402-2443. https://doi.org/10.3390/cimb46030153
Onciul R, Brehar F-M, Toader C, Covache-Busuioc R-A, Glavan L-A, Bratu B-G, Costin HP, Dumitrascu D-I, Serban M, Ciurea AV. Deciphering Glioblastoma: Fundamental and Novel Insights into the Biology and Therapeutic Strategies of Gliomas. Current Issues in Molecular Biology. 2024; 46(3):2402-2443. https://doi.org/10.3390/cimb46030153
Chicago/Turabian StyleOnciul, Razvan, Felix-Mircea Brehar, Corneliu Toader, Razvan-Adrian Covache-Busuioc, Luca-Andrei Glavan, Bogdan-Gabriel Bratu, Horia Petre Costin, David-Ioan Dumitrascu, Matei Serban, and Alexandru Vlad Ciurea. 2024. "Deciphering Glioblastoma: Fundamental and Novel Insights into the Biology and Therapeutic Strategies of Gliomas" Current Issues in Molecular Biology 46, no. 3: 2402-2443. https://doi.org/10.3390/cimb46030153
APA StyleOnciul, R., Brehar, F.-M., Toader, C., Covache-Busuioc, R.-A., Glavan, L.-A., Bratu, B.-G., Costin, H. P., Dumitrascu, D.-I., Serban, M., & Ciurea, A. V. (2024). Deciphering Glioblastoma: Fundamental and Novel Insights into the Biology and Therapeutic Strategies of Gliomas. Current Issues in Molecular Biology, 46(3), 2402-2443. https://doi.org/10.3390/cimb46030153