
Modulating Golgi Stress Signaling Ameliorates Cell Morphological Phenotypes Induced by CHMP2B with Frontotemporal Dementia-Associated p.Asp148Tyr

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies, siRNAs, and Plasmids
2.2. Isolation and Culture of Primary Cortical Neuronal Cells
2.3. Cell Culture and Differentiation
2.4. Plasmid and siRNA Transfection
2.5. Denatured Polyacrylamide Electrophoresis and Immunoblotting
2.6. Fluorescence Images
2.7. Statistical Analyses
2.8. Ethics Statement
3. Results
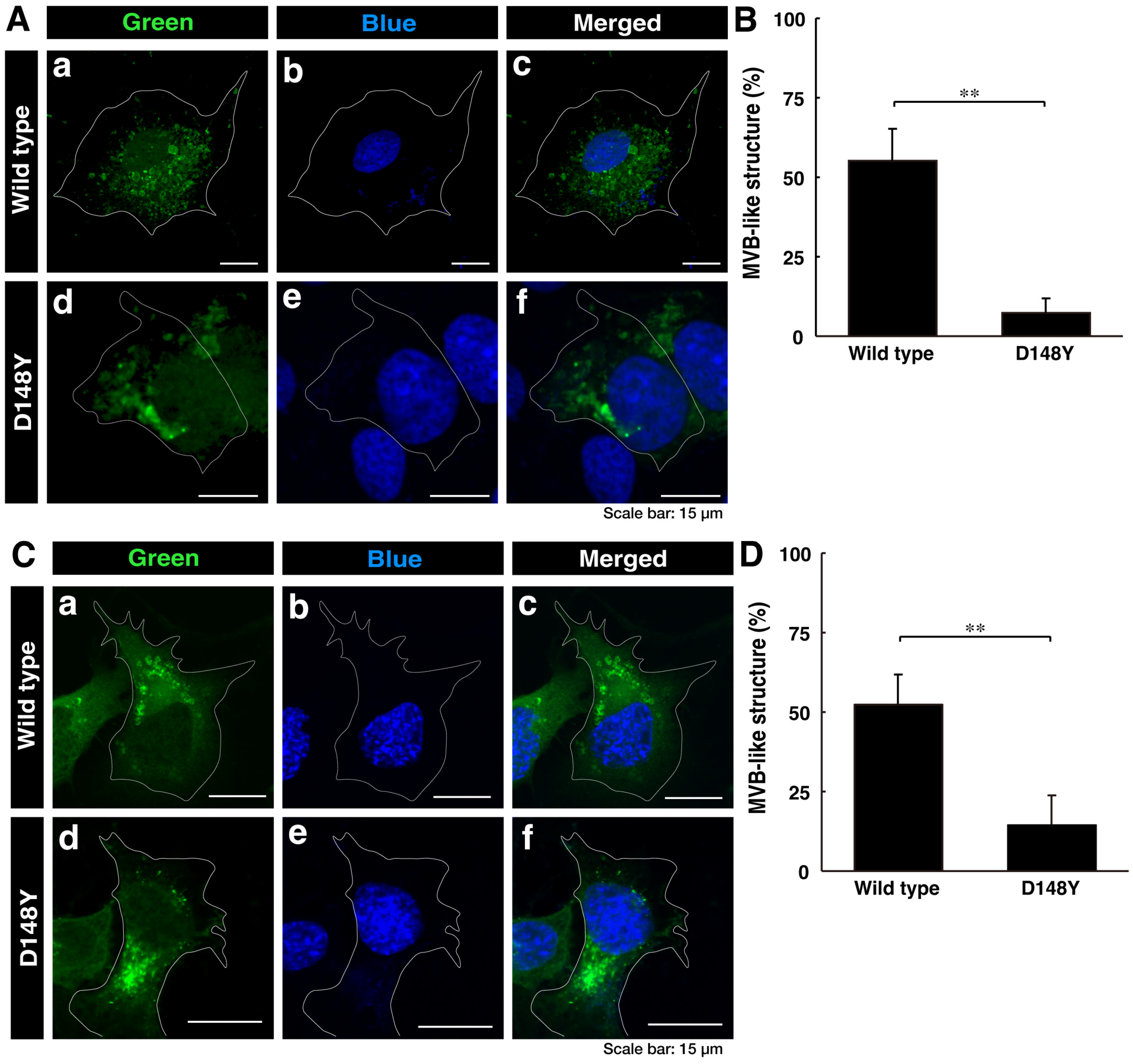
3.1. Cells Harboring CHMP2B with the D148Y Mutation form Aggregate-like Structures in the Golgi Body, Whereas Wild-Type CHMP2B Is Contained in MVB-like Structures
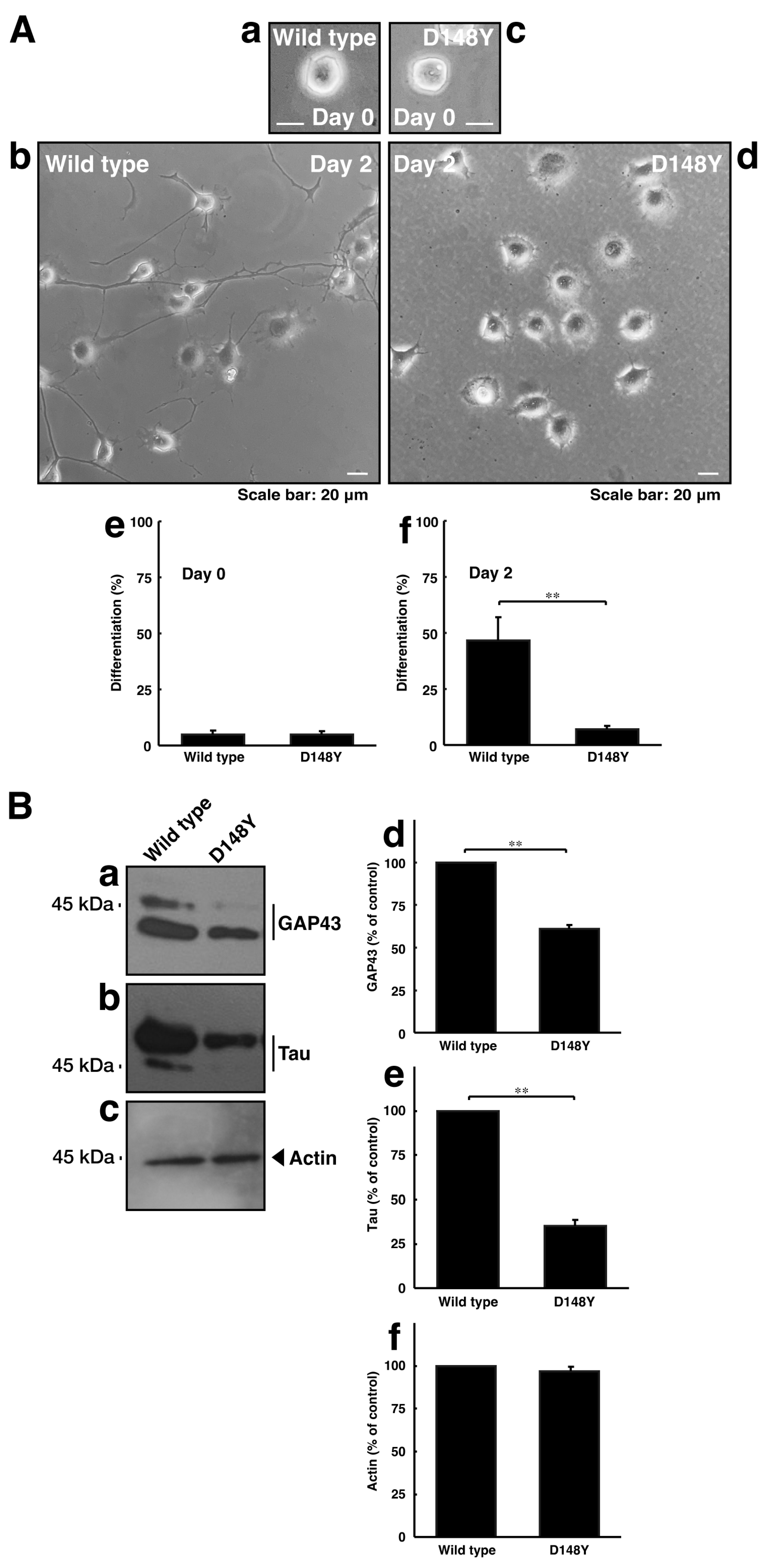
3.2. Cells Harboring CHMP2B with the D148Y Mutation Display Blunted Neuronal Morphological Changes
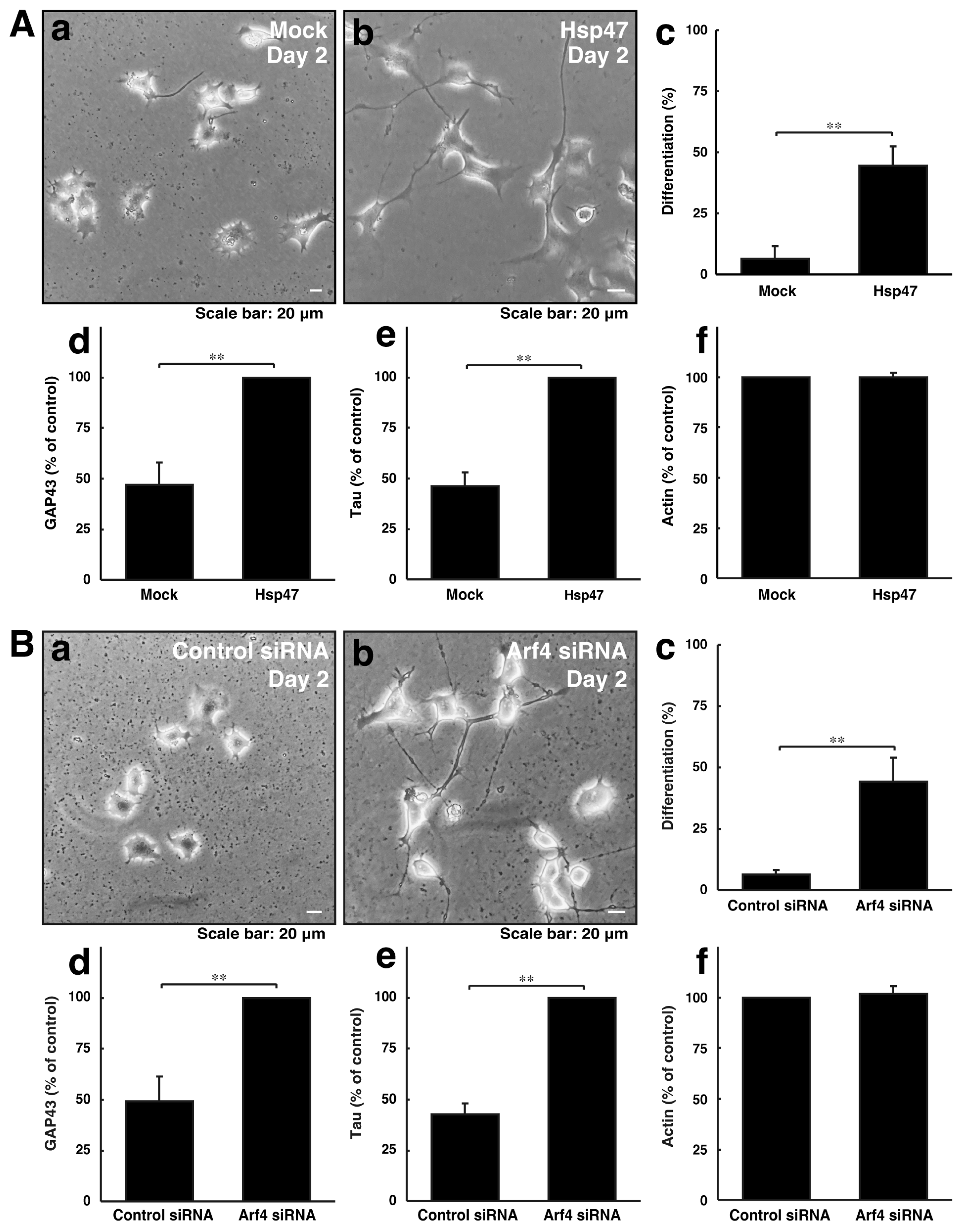
3.3. Neuronal Morphological Changes Can Be Recovered by Compensating Golgi Stress Signaling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Smeyers, J.; Banchi, E.G.; Latouche, M. FC9ERF72: What it is, what it does, and why it matters. Front. Cell Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.; Hu, F. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2021, 157, 334–350. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.A.; Lally, C.; Kupelian, V.; Flanders, W.D. Estimated prevalence and incidence of amyotrophic lateral sclerosis and SOD1 and C9orf72 genetic variants. Neuroepidemiology 2021, 5, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Olney, N.T.; Spina, S.; Miller, B.L. Frontotemporal dementia. Neurol. Clin. 2017, 35, 339–374. [Google Scholar] [CrossRef]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef]
- Greaves, C.V.; Rohrer, J.D. An update on genetic frontotemporal dementia. J. Neurol. 2019, 266, 2075–2086. [Google Scholar] [CrossRef]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef]
- Bang, J.; Spina, S.; Miller, B.L. Frontotemporal dementia. Lancet 2015, 386, 1672–1682. [Google Scholar] [CrossRef]
- Magrath Guimet, N.; Zapata-Restrepo, L.M.; Miller, B.L. Advances in Treatment of Frontotemporal Dementia. J. Neuropsychiatry Clin. Neurosci. 2022, 34, 316–327. [Google Scholar] [CrossRef]
- Boeve, B.F.; Boxer, A.L.; Kumfor, F.; Pijnenburg, Y.; Rohrer, J.D. Advances and controversies in frontotemporal dementia: Diagnosis, biomarkers, and therapeutic considerations. Lancet Neurol. 2022, 21, 258–272. [Google Scholar] [CrossRef]
- Antonioni, A.; Raho, E.M.; Lopriore, P.; Pace, A.P.; Latino, R.R.; Assogna, M.; Mancuso, M.; Gragnaniello, D.; Granieri, E.; Pugliatti, M.; et al. Frontotemporal dementia, where do we stand? A narrative review. Int. J. Mol. Sci. 2023, 24, 11732. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A.; Beigneux, A.; Ahmad, S.T.; Young, S.G.; Gao, F.B. ESCRT-III dysfunction causes autophagosome accumulation and neurodegeneration. Curr. Biol. 2007, 17, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Tolnay, M.; Mackenzie, I.R. The molecular basis of frontotemporal dementia. Expert Rev. Mol. Med. 2009, 11, e23. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Sadoul, R.; Laporte, M.H.; Chassefeyre, R.; Chi, K.I.; Goldberg, Y.; Chatellard, C.; Hemming, F.J.; Fraboulet, S. The role of ESCRT during development and functioning of the nervous system. Semin. Cell Dev. Biol. 2018, 74, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef] [PubMed]
- Ugbode, C.; West, R.J.H. Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 147, 105144. [Google Scholar] [CrossRef]
- García-Roldán, E.; Rivas-Infante, E.; Medina-Rodríguez, M.; Arriola-Infante, J.E.; Rodrigo-Herrero, S.; Paradas, C.; Rábano-Gutiérrez, A.; Franco-Macías, E. Lessons learned from a sporadic FUSopathy in a young man: A case report. BMC Neurol. 2023, 23, 55. [Google Scholar] [CrossRef]
- Craig, A.M.; Banker, G. Neuronal polarity. Annu. Rev. Neurosci. 1994, 17, 267–310. [Google Scholar] [CrossRef] [PubMed]
- da Silva, J.S.; Dotti, C.G. Breaking the neuronal sphere: Regulation of the actin cytoskeleton in neuritogenesis. Nat. Rev. Neurosci. 2002, 3, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Arimura, N.; Kaibuchi, K. Neuronal polarity: From extracellular signals to intracellular mechanisms. Nat. Rev. Neurosci. 2007, 8, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Bray, D. Surface movements during the growth of single explanted neurons. Proc. Natl. Acad. Sci. USA 1970, 65, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Bray, D. Actin and myosin in neurons: A first review. Biochimie 1977, 59, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef]
- Hirose, M.; Ishizaki, T.; Watanabe, N.; Uehata, M.; Kranenburg, O.; Moolenaar, W.H.; Matsumura, F.; Maekawa, M.; Bito, H.; Narumiya, S. Molecular dissection of the Rho-associated protein kinase (p160ROCK)-regulated neurite remodeling in neuroblastoma N1E-115 cells. J. Cell Biol. 1998, 141, 1625–1636. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Torii, T.; Yamamori, N.; Ogata, T.; Tanoue, A.; Yamauchi, J. Akt and PP2A reciprocally regulate the guanine nucleotide exchange factor Dock6 to control axon growth of sensory neurons. Sci. Signal. 2013, 6, ra15. [Google Scholar] [CrossRef]
- Kato, Y.; Shirai, R.; Ohbuchi, K.; Oizumi, H.; Yamamoto, M.; Miyata, W.; Iguchi, T.; Mimaki, Y.; Miyamoto, Y.; Yamauchi, J. Hesperetin ameliorates inhibition of neuronal and oligodendroglial cell differentiation phenotypes induced by knockdown of Rab2b, an autism spectrum disorder-associated gene product. Neurol. Int. 2023, 15, 371–391. [Google Scholar] [CrossRef]
- Shirai, R.; Cho, M.; Isogai, M.; Fukatsu, S.; Okabe, M.; Okawa, M.; Miyamoto, Y.; Torii, T.; Yamauchi, J. FTD/ALS Type 7-Associated Thr104Asn Mutation of CHMP2B Blunts Neuronal Process Elongation, and Is Recovered by Knockdown of Arf4, the Golgi Stress Regulator. Neurol. Int. 2023, 15, 980–993. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Yokomaku, D.; Kiyosue, K.; Adachi, N.; Matsumoto, T.; Numakawa, Y.; Taguchi, T.; Hatanaka, H.; Yamada, M. Basic fibroblast growth factor evokes a rapid glutamate release through activation of the MAPK pathway in cultured cortical neurons. J. Biol. Chem. 2002, 277, 28861–28869. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Lee, S.E.; Ahn, S.G.; Lee, G.H. Psoralidin stimulates expression of immediate-early genes and synapse development in primary cortical neurons. Neurochem. Res. 2018, 43, 2460–2472. [Google Scholar] [CrossRef]
- Machamer, C.E. The Golgi complex in stress and death. Front. Neurosci. 2015, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Yoshida, H. TFE3, HSP47, and CREB3 pathways of the mammalian Golgi stress response. Cell Struct. Funct. 2017, 42, 27–36. [Google Scholar] [CrossRef]
- Sasaki, K.; Yoshida, H. Organelle zones. Cell Struct. Funct. 2019, 44, 85–94. [Google Scholar] [CrossRef]
- Sasaki, K.; Yoshida, H. Golgi stress response and organelle zones. FEBS Lett. 2019, 593, 2330–2340. [Google Scholar] [CrossRef]
- Mohan, A.G.; Calenic, B.; Ghiurau, N.A.; Duncea-Borca, R.M.; Constantinescu, A.E.; Constantinescu, I. The Golgi apparatus: A voyage through time, structure, function and implication in neurodegenerative disorders. Cells 2023, 12, 1972. [Google Scholar] [CrossRef]
- Kim, W.K.; Choi, W.; Deshar, B.; Kang, S.; Kim, J. Golgi stress response: New insights into the pathogenesis and therapeutic targets of human diseases. Mol. Cells 2023, 46, 191–199. [Google Scholar] [CrossRef]
- Miyata, S.; Mizuno, T.; Koyama, Y.; Katayama, T.; Tohyama, M. The endoplasmic reticulum-resident chaperone heat shock protein 47 protects the Golgi apparatus from the effects of O-glycosylation inhibition. PLoS ONE 2013, 8, e69732. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, N.; Okuno, D.; Tokito, T.; Yura, H.; Kido, T.; Ishimoto, H.; Tanaka, Y.; Mukae, H. Hsp47: A therapeutic target in pulmonary fibrosis. Biomedicines 2023, 11, 2387. [Google Scholar] [CrossRef] [PubMed]
- Reiling, J.H.; Olive, A.J.; Sanyal, S.; Carette, J.E.; Brummelkamp, T.R.; Ploegh, H.L.; Starnbach, M.N.; Sabatini, D.M. A CREB3–ARF4 signalling pathway mediates the response to Golgi stress and susceptibility to pathogens. Nat. Cell Biol. 2013, 15, 1473–1485. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Forsberg, J.; Zhivotovsky, B. Caspase-2: The reinvented enzyme. Oncogene 2015, 34, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Nadanaka, S.; Tanakura, S.; Sawaguchi, S.; Midori, S.; Kawai, Y.; Yamaguchi, S.; Shimada, Y.; Nakamura, Y.; Matsumura, Y.; et al. TFE3 is a bHLH-ZIP-type transcription factor that regulates the mammalian Golgi stress response. Cell Struct. Funct. 2015, 40, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Jamaludin, M.I.; Wakabayashi, S.; Taniguchi, M.; Sasaki, K.; Komori, R.; Kawamura, H.; Takase, H.; Sakamoto, M.; Yoshida, H. MGSE regulates crosstalk from the mucin pathway to the TFE3 pathway of the Golgi stress response. Cell Struct. Funct. 2019, 44, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Wang, Y.; Wang, X.; Xu, J. CHCHD2 and CHCHD10: Future therapeutic targets in cognitive disorder and motor neuron disorder. Front. Neurosci. 2022, 16, 988265. [Google Scholar] [CrossRef]
- Shammas, M.K.; Huang, T.H.; Narendra, D.P. CHCHD2 and CHCHD10-related neurodegeneration: Molecular pathogenesis and the path to precision therapy. Biochem. Soc. Trans. 2023, 51, 797–809. [Google Scholar] [CrossRef]
- Nagata, K. Hsp47: A collagen-specific molecular chaperone. Trends. Biochem. Sci. 1996, 21, 22–26. [Google Scholar] [CrossRef]
- Ito, S.; Nagata, K. Quality control of procollagen in cells. Annu. Rev. Biochem. 2021, 90, 631–658. [Google Scholar] [CrossRef]
- Hwang, J.; Qi, L. Quality control in the endoplasmic reticulum: Crosstalk between ERAD and UPR pathways. Trends. Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Cela, I.; Dufrusine, B.; Rossi, C.; Luini, A.; De Laurenzi, V.; Federici, L.; Sallese, M. KDEL receptors: Pathophysiological functions, therapeutic options, and biotechnological opportunities. Biomedicines 2022, 10, 1234. [Google Scholar] [CrossRef] [PubMed]
- Cherfils, J. Arf GTPases and their effectors: Assembling multivalent membrane-binding platforms. Curr. Opin. Struct. Biol. 2014, 29, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Nawrotek, A.; Zeghouf, M.; Cherfils, J. Protein-membrane interactions in small GTPase signaling and pharmacology: Perspectives from Arf GTPases studies. Biochem. Soc. Trans. 2020, 48, 2721–2728. [Google Scholar] [CrossRef]
- Chen, P.W.; Gasilina, A.; Yadav, M.P.; Randazzo, P.A. Control of cell signaling by Arf GTPases and their regulators: Focus on links to cancer and other GTPase families. Biochim. Biophys. Acta. Mol. Cell Res. 2022, 1869, 119171. [Google Scholar] [CrossRef]
- Ito, A.; Fukaya, M.; Okamoto, H.; Sakagami, H. Physiological and pathological roles of the cytohesin family in neurons. Int. J. Mol. Sci. 2022, 23, 5087. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent or Material | Company or Source | Cat. No. | Lot No. | Concentration Used |
|---|---|---|---|---|
| Antibody | ||||
| Anti-heat-shock protein 47 (Hsp47) | Santa Cruz Biotechnology (Santa Cruz, CA, USA) | sc-5293 | I2118 | Immunoblotting (IB), 1:200 |
| Anti-Arf4 | Proteintech Japan (Tokyo, Japan) | 11673-1-AP | 00048284 | IB, 1:1000 |
| Anti-Actin | MBL (Tokyo, Japan) | M177-3 | 007 | IB, 1:1000 |
| Anti-Growth associated protein 43 (GAP43) | Santa Cruz Biotechnology | sc-17790 | J0920 | IB, 1:8500 |
| Anti-Tau | Santa Cruz Biotechnology | sc-21796 | H2923 | IB, 1:500 |
| Anti-Lys-Asp-Glu-Leu (KDEL) | MBL | M181-3 | 004 | Immunofluorescence (IF), 1:200 |
| Anti-Golgi matrix protein of 130 kDa (GM130) | BD (Franklin Lakes, NJ, USA) | 610823 | 8352796 | IB, 1:500 and IF, 1:200 |
| Anti-Cathepsin D | Abcam (Cambridge, UK) | ab75852 | GR260148-33 | IF, 1:200 |
| Anti-IgG (H+L chain) (Rabbit) pAb-HRP | MBL | 458 | 353 | IB, 1:5000 |
| Anti-IgG (H+L chain) (Mouse) pAb-HRP | MBL | 330 | 365 | IB, 1:5000 |
| Alexa Fluor TM 594 goat anti-mouse IgG (H+L) | ThermoFisher Scientific (Waltham, MA, USA) | A11005 | 226-8383 | IF, 1:500 |
| Alexa Fluor TM 594 goat anti-rabbit IgG (H+L) | ThermoFisher Scientific | A11012 | 201-8240 | IF, 1:500 |
| siRNA and recombinant DNA | ||||
| 5′-GACGACAAUUCUGUAUAAA-dTdT-3′ and 5′-UUUAUACAGAAUUGUCGUC-dTdT-3′ | siRNA duplex for Arf4 (generated in this study) | Not applicable | 50 nM | |
| 5′-GCCAUUCUAUCCUCUAGAG-dTdT-3′ and 5′-CUCUAGAGGAUAGAAUGGC-dTdT-3′ | siRNA duplex for P. pyralis luciferase (generated in this study) | Not applicable | 50 nM | |
| pcDNA3.1(+)-human Hsp47 | GenScript (generated in this study) | Not applicable | 1.25 mg of DNA per 6 cm dish | |
| pEGFP-N3-human CHMP2B | Generated in this study | Not applicable | 1.25 mg of DNA per 6 cm dish | |
| pEGFP-N3-human CHMP2B with the D148Y mutation | Generated in this study | Not applicable | 1.25 mg of DNA per 6 cm dish |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fukatsu, S.; Okawa, M.; Okabe, M.; Cho, M.; Isogai, M.; Yokoi, T.; Shirai, R.; Oizumi, H.; Yamamoto, M.; Ohbuchi, K.; et al. Modulating Golgi Stress Signaling Ameliorates Cell Morphological Phenotypes Induced by CHMP2B with Frontotemporal Dementia-Associated p.Asp148Tyr. Curr. Issues Mol. Biol. 2024, 46, 1398-1412. https://doi.org/10.3390/cimb46020090
Fukatsu S, Okawa M, Okabe M, Cho M, Isogai M, Yokoi T, Shirai R, Oizumi H, Yamamoto M, Ohbuchi K, et al. Modulating Golgi Stress Signaling Ameliorates Cell Morphological Phenotypes Induced by CHMP2B with Frontotemporal Dementia-Associated p.Asp148Tyr. Current Issues in Molecular Biology. 2024; 46(2):1398-1412. https://doi.org/10.3390/cimb46020090
Chicago/Turabian StyleFukatsu, Shoya, Maho Okawa, Miyu Okabe, Mizuka Cho, Mikinori Isogai, Takanori Yokoi, Remina Shirai, Hiroaki Oizumi, Masahiro Yamamoto, Katsuya Ohbuchi, and et al. 2024. "Modulating Golgi Stress Signaling Ameliorates Cell Morphological Phenotypes Induced by CHMP2B with Frontotemporal Dementia-Associated p.Asp148Tyr" Current Issues in Molecular Biology 46, no. 2: 1398-1412. https://doi.org/10.3390/cimb46020090
APA StyleFukatsu, S., Okawa, M., Okabe, M., Cho, M., Isogai, M., Yokoi, T., Shirai, R., Oizumi, H., Yamamoto, M., Ohbuchi, K., Miyamoto, Y., & Yamauchi, J. (2024). Modulating Golgi Stress Signaling Ameliorates Cell Morphological Phenotypes Induced by CHMP2B with Frontotemporal Dementia-Associated p.Asp148Tyr. Current Issues in Molecular Biology, 46(2), 1398-1412. https://doi.org/10.3390/cimb46020090