
Genetic Predisposition for White Matter Hyperintensities and Risk of Mild Cognitive Impairment and Alzheimer’s Disease: Results from the HELIAD Study

 , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Participants and Procedures
2.2. Diagnostic Procedures
2.3. Genotyping and Imputation
2.4. Polygenic Risk Score Calculation
2.5. Statistical Analysis
3. Results
3.1. Baseline Participants’ Characteristics and Missing Data Analysis
3.2. PRS WMH and aMCI/AD Incidence
3.3. PRS WMH: Stratification Analysis
4. Discussion
4.1. Our Findings in Regard to Existing Literature
4.2. Strengths and Limitations of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Leeuw, F.E.; de Groot, J.C.; Achten, E.; Oudkerk, M.; Ramos, L.M.; Heijboer, R.; Hofman, A.; Jolles, J.; van Gijn, J.; Breteleret, M.M. Prevalence of cerebral white matter lesions in elderly people: A population based magnetic resonance imaging study. The Rotterdam Scan Study. J. Neurol. Neurosurg. Psychiatry 2001, 70, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Enzinger, C.; Ropele, S.; Schmidt, H.; Fazekas, F.; Austrian Stroke Prevention Study. Progression of cerebral white matter lesions: 6-year results of the Austrian Stroke Prevention Study. Lancet 2003, 361, 2046–2048. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.A.; Kant, I.M.J.; Slooter, A.J.C.; van Montfort, S.J.T.; van Buchem, M.A.; van Osch, M.J.P.; Hendrikse, J.; de Bresser, J. Different cardiovascular risk factors are related to distinct white matter hyperintensity MRI phenotypes in older adults. Neuroimage Clin. 2022, 35, 103131. [Google Scholar] [CrossRef] [PubMed]
- Garnier-Crussard, A.; Cotton, F.; Krolak-Salmon, P.; Chételat, G. White matter hyperintensities in Alzheimer’s disease: Beyond vascular contribution. Alzheimer’s Dement. 2023, 19, 3738–3748. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Chappell, F.M.; Valdés Hernández, M.D.C.; Makin, S.D.J.; Staals, J.; Shuler, K.; Thrippleton, M.J.; Armitage, P.A.; Muñoz-Maniega, S.; Heye, A.K.; et al. White matter hyperintensity reduction and outcomes after minor stroke. Neurology 2017, 89, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Atwood, L.D.; Wolf, P.A.; Heard-Costa, N.L.; Massaro, J.M.; Beiser, A.; D’Agostino, R.B.; DeCarli, C. Genetic variation in white matter hyperintensity volume in the Framingham Study. Stroke 2004, 35, 1609–1613. [Google Scholar] [CrossRef]
- DeStefano, A.L.; Atwood, L.D.; Massaro, J.M.; Heard-Costa, N.; Beiser, A.; Au, R.; Wolf, P.A.; DeCarli, C. Genome-wide scan for white matter hyperintensity: The Framingham Heart Study. Stroke 2006, 37, 77–81. [Google Scholar] [CrossRef]
- Biesbroek, J.M.; Weaver, N.A.; Hilal, S.; Kuijf, H.J.; Ikram, M.K.; Xu, X.; Tan, B.Y.; Venketasubramanian, N.; Postma, A.; Biessels, G.J.; et al. Impact of strategically located white matter hyperintensities on cognition in memory clinic patients with small vessel disease. PLoS ONE 2016, 11, e0166261. [Google Scholar] [CrossRef]
- Ding, D.; Xiong, Y.; Zhao, Q.; Guo, Q.; Chu, S.; Chu, W.W.C.; Luo, J.; Liang, X.; Zheng, L.; Hong, Z.; et al. White matter hyperintensity predicts the risk of incident cognitive decline in community dwelling elderly. J. Alzheimer’s Dis. 2018, 61, 1333–1341. [Google Scholar] [CrossRef]
- Brickman, A.M.; Provenzano, F.A.; Muraskin, J.; Manly, J.J.; Blum, S.; Apa, Z.; Stern, Y.; Brown, T.R.; Luchsinger, J.A.; Mayeux, R. Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch. Neurol. 2012, 69, 1621–1627. [Google Scholar] [CrossRef]
- Chen, Y.F.; Wang, H.; Chu, Y.; Huang, Y.C.; Su, M.Y. Regional quantification of white matter hyperintensity in normal aging, mild cognitive impairment, and Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Lo, R.Y.; Jagust, W.J. Vascular burden and Alzheimer disease pathologic progression. Neurology 2012, 79, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Mortamais, M.; Artero, S.; Ritchie, K. White matter hyperintensities as early and independent predictors of Alzheimer’s disease risk. J. Alzheimer’s Dis. 2014, 42, S393–S400. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of 761 Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Silbert, L.C.; Dodge, H.H.; Perkins, L.G.; Sherbakov, L.; Lahna, D.; Erten-Lyons, D.; Woltjer, R.; Shinto, L.; Kaye, J.A. Trajectory of white matter hyperintensity burden preceding mild cognitive impairment. Neurology 2012, 79, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Opherk, C.; Gonik, M.; Duering, M.; Malik, R.; Jouvent, E.; Hervé, D.; Adib-Samii, P.; Bevan, S.; Pianese, L.; Silvestri, S.; et al. Genome-wide genotyping demonstrates a polygenic risk score associated with white matter hyperintensity volume in CADASIL. Stroke 2014, 45, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Verhaaren, B.F.; Debette, S.; Bis, J.C.; Smith, J.A.; Ikram, M.K.; Adams, H.H.; Beecham, A.H.; Rajan, K.B.; Lopez, L.M.; Barral, S.; et al. Multiethnic genome-wide association study of cerebral white matter hyperintensities on MRI. Circ. Cardiovasc. Genet. 2015, 8, 398–409. [Google Scholar] [CrossRef]
- Beecham, A.; Dong, C.; Wright, C.B.; Dueker, N.; Brickman, A.M.; Wang, L.; DeCarli, C.; Blanton, S.H.; Rundek, T.; Mayeux, R.; et al. Genome-wide scan in Hispanics highlights candidate loci for brain white matter hyperintensities. Neurol. Genet. 2017, 3, e185. [Google Scholar] [CrossRef]
- Guo, Y.; Shen, X.N.; Hou, X.H.; Ou, Y.N.; Huang, Y.Y.; Dong, Q.; Tan, L.; Yu, J.T.; Alzheimer’s Disease Neuroimaging Initiative. Genome-wide association study of white matter hyperintensity volume in elderly persons without dementia. Neuroimage Clin. 2020, 26, 102209. [Google Scholar] [CrossRef]
- Sargurupremraj, M.; Suzuki, H.; Jian, X.; Sarnowski, C.; Evans, T.E.; Bis, J.C.; Eiriksdottir, G.; Sakaue, S.; Terzikhan, N.; Habes, M.; et al. Cerebral small vessel disease genomics and its implications across the lifespan. Nat. Commun. 2020, 11, 6285. [Google Scholar] [CrossRef]
- Rutten-Jacobs, L.C.A.; Tozer, D.J.; Duering, M.; Malik, R.; Dichgans, M.; Markus, H.S.; Traylor, M. Genetic Study of White Matter Integrity in UK Biobank (N = 8448) and the Overlap With Stroke, Depression, and Dementia. Stroke 2018, 49, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Dardiotis, E.; Kosmidis, M.H.; Yannakoulia, M.; Hadjigeorgiou, G.M.; Scarmeas, N. The Hellenic Longitudinal Investigation of Aging and Diet (HELIAD): Rationale, study design, and cohort description. Neuroepidemiology 2014, 43, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Anastasiou, C.A.; Yannakoulia, M.; Kosmidis, M.H.; Dardiotis, E.; Hadjigeorgiou, G.M.; Sakka, P.; Arampatzi, X.; Bougea, A.; Labropoulos, I.; Scarmeas, N. Mediterranean diet and cognitive health: Initial results from the Hellenic longitudinal investigation of ageing and diet. PLoS ONE 2017, 12, e0182048. [Google Scholar] [CrossRef] [PubMed]
- Petersen, R.C.; Doody, R.; Kurz, A.; Mohs, R.C.; Morris, J.C.; Rabins, P.V.; Ritchie, K.; Rossor, M.; Thal, L.; Winblad, B. Current concepts in mild cognitive impairment. Arch. Neurol. 2001, 58, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Georgakis, M.K.; Ntanasi, E.; Ramirez, A.; Grenier-Boley, B.; Lambert, J.C.; Sakka, P.; Yannakoulia, M.; Kosmidis, M.H.; Dardiotis, E.; Hadjigeorgiou, G.M.; et al. Vascular burden and genetic risk in association with cognitive performance and dementia in a population-based study. Cereb. Circ. Cogn. Behav. 2022, 3, 100145. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; Maillard, P.; Harvey, D.; Fletcher, E.; Martinez, O.; Johnson, D.K.; Olichney, J.M.; Farias, S.T.; Villeneuve, S.; Jagust, W.; et al. Association of vascular brain injury, neurodegeneration, amyloid and cognitive trajectory. Neurology 2020, 95, e2622–e2634. [Google Scholar] [CrossRef]
- DeCarli, C.; Villeneuve, S.; Maillard, P.; Harvey, D.; Singh, B.; Carmichael, O.; Fletcher, E.; Olichney, J.; Farias, S.; Jagust, W.; et al. Vascular Burden Score Impacts Cognition Independent of Amyloid PET and MRI Measures of Alzheimer’s Disease and Vascular Brain Injury. J. Alzheimer’s Dis. 2019, 68, 187–196. [Google Scholar] [CrossRef]
- Bellenguez, C.; Küçükali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Mourtzi, N.; Charisis, S.; Tsapanou, A.; Ntanasi, E.; Hatzimanolis, A.; Ramirez, A.; Heilmann-Heimbach, S.; Grenier-Boley, B.; Lambert, J.C.; Yannakoulia, M.; et al. Genetic propensity for cerebral amyloidosis and risk of mild cognitive impairment and Alzheimer’s disease within a cognitive reserve framework. Alzheimer’s Dement. 2023, 19, 3794–3805. [Google Scholar] [CrossRef]
- Choi, S.W.; O’Reilly, P.F. PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience 2019, 8, giz082. [Google Scholar] [CrossRef] [PubMed]
- International Schizophrenia Consortium; Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Leonenko, G.; Baker, E.; Stevenson-Hoare, J.; Sierksma, A.; Fiers, M.; Williams, J.; de Strooper, B.; Escott-Price, V. Identifying individuals with high risk of Alzheimer’s disease using polygenic risk scores. Nat. Commun. 2021, 23, 4506. [Google Scholar] [CrossRef] [PubMed]
- Maraki, M.I.; Hatzimanolis, A.; Mourtzi, N.; Stefanis, L.; Yannakoulia, M.; Kosmidis, M.H.; Dardiotis, E.; Hadjigeorgiou, G.M.; Sakka, P.; Ramirez, A.; et al. Association of the Polygenic Risk Score with the Probability of Prodromal Parkinson’s Disease in Older Adults. Front. Mol. Neurosci. 2021, 14, 739571. [Google Scholar] [CrossRef] [PubMed]
- Tsapanou, A.; Mourtzi, N.; Charisism, S.; Hatzimanolis, A.; Ntanasi, E.; Kosmidis, M.H.; Yannakoulia, M.; Hadjigeorgiou, G.; Dardiotis, E.; Sakka, P.; et al. Sleep Polygenic Risk Score Is Associated with Cognitive Changes over Time. Genes 2021, 13, 63. [Google Scholar] [CrossRef] [PubMed]
- Jungwirth, S.; Zehetmayer, S.; Hinterberger, M.; Tragl, K.H.; Fischer, P. The validity of amnestic MCI and non-amnestic MCI at age 75 in the prediction of Alzheimer’s dementia and vascular dementia. Int. Psychogeriatr. 2012, 24, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Reese, S.E.; Archer, K.J.; Therneau, T.M.; Atkinson, E.J.; Vachon, C.M.; de Andrade, M.; Kocher, J.P.; Eckel-Passow, J.E. A new statistic for identifying batch effects in high-throughput genomic data that uses guided principal component analysis. Bioinformatics 2013, 29, 2877–2883. [Google Scholar] [CrossRef]
- Van der Lee, S.J.; Wolters, F.J.; Ikram, M.K.; Hofman, A.; Ikram, M.A.; Amin, N.; van Duijn, C.M. The effect of APOE and other common genetic variants on the onset of Alzheimer’s disease and dementia: A community-based cohort study. Lancet Neurol. 2018, 17, 434–444. [Google Scholar] [CrossRef]
- Brownson, R.C.; Petitti, D.B. Applied Epidemiology: Theory to Practice, 2nd ed.; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Penke, B.; Szűcs, M.; Bogár, F. New Pathways Identify Novel Drug Targets for the Prevention and Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 5383. [Google Scholar] [CrossRef]
- Georgakis, M.K.; Duering, M.; Wardlaw, J.M.; Dichgans, M. WMH and long-term outcomes in ischemic stroke: A systematic review and meta-analysis. Neurology 2019, 92, e1298–e1308. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Viqar, F.; Zimmerman, M.E.; Tosto, G.; Benzinger, T.L.; Marcus, D.S.; Fagan, A.M.; Goate, A.; Fox, N.C.; Cairns, N.J.; et al. White matter hyperintensities are a core feature of Alzheimer’s disease: Evidence from the Dominantly Inherited Alzheimer Network: Whitematter hyperintensities in familial AD. Ann. Neurol. 2016, 79, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Garnier-Crussard, A.; Bougacha, S.; Wirth, M.; Dautricourt, S.; Sherif, S.; Landeau, B.; Gonneaud, J.; De Flores, R.; de la Sayette, V.; Vivien, D.; et al. White matter hyperintensity topography in Alzheimer’s disease and links to cognition. Alzheimer’s Dement. 2022, 18, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Walsh, P.; Sudre, C.H.; Fiford, C.M.; Ryan, N.S.; Lashley, T.; Frost, C.; Barnes, J.; ADNI Investigators. CSF amyloid is a consistent predictor of white matter hyperintensities across the disease course from aging to Alzheimer’s disease. Neurobiol. Aging 2020, 91, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Choi, S.H.; Lee, Y.M.; Kim, M.J.; Kim, Y.D.; Kim, J.Y.; Park, J.H.; Myung, W.; Na, H.R.; Han, H.J.; et al. Periventricular white matter hyperintensities and the risk of dementia: A CREDOS study. Int. Psychogeriatr. 2015, 27, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- Pålhaugen, L.; Sudre, C.H.; Tecelao, S.; Nakling, A.; Almdahl, I.S.; Kalheim, L.F.; Cardoso, M.J.; Johnsen, S.H.; Rongve, A.; Aarsland, D.; et al. Brain amyloid and vascular risk are related to distinct white matter hyperintensity patterns. J. Cereb. Blood Flow Metab. 2021, 41, 1162–1174. [Google Scholar] [CrossRef]
- De Groot, J.C.; de Leeuw, F.E.; Oudkerk, M.; Hofman, A.; Jolles, J.; Breteler, M.M. Cerebral white matter lesions and subjective cognitive dysfunction: The Rotterdam Scan Study. Neurology 2001, 56, 1539–1545. [Google Scholar] [CrossRef]
- Kloppenborg, R.P.; Nederkoorn, P.J.; Grool, A.M.; Vincken, K.L.; Mali, W.P.; Vermeulen, M.; van der Graaf, Y.; Geerlings, M.I. Cerebral small-vessel disease and progression of brain atrophy: The SMART-MR study. Neurology 2012, 79, 2029–2036. [Google Scholar] [CrossRef]
- Gouw, A.A.; Seewann, A.; Vrenken, H.; van der Flier, W.M.; Rozemuller, J.M.; Barkhof, F.; Scheltens, P.; Geurts, J.J. Heterogeneity of white matter hyperintensities in Alzheimer’s disease: Post-mortem quantitative MRI and neuropathology. Brain 2008, 131, 3286–3298. [Google Scholar] [CrossRef]
- Tsai, H.H.; Chen, Y.F.; Yen, R.F.; Lo, Y.L.; Yang, K.C.; Jeng, J.S.; Tsai, L.K.; Chang, C.F. Plasma soluble TREM2 is associated with white matter lesions independent of amyloid and tau. Brain 2021, 144, 3371–3380. [Google Scholar] [CrossRef]
- Desmarais, P.; Gao, A.F.; Lanctôt, K.; Rogaeva, E.; Ramirez, J.; Herrmann, N.; Stuss, D.T.; Black, S.E.; Keith, J.; Masellis, M. White matter hyperintensities in autopsy-confirmed frontotemporal lobar degeneration and Alzheimer’s disease. Alzheimer’s Res. Ther. 2021, 13, 129. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lv, C.; Li, X.; Zhang, J.; Chen, K.; Liu, Z.; Li, H.; Fan, J.; Qin, T.; Luo, L.; et al. The positive impacts of early-life education on cognition, leisure activity, and brain structure in healthy aging. Aging 2019, 11, 4923–4942. [Google Scholar] [CrossRef] [PubMed]
- Then, F.S.; Luck, T.; Angermeyer, M.C.; Riedel-Heller, S.G. Education as protector against dementia, but what exactly do we mean by education? Age Ageing 2016, 45, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Seyedsalehi, A.; Warrier, V.; Bethlehem, R.A.I.; Perry, B.I.; Burgess, S.; Murray, G.K. Educational attainment, structural brain reserve and Alzheimer’s disease: A Mendelian randomization analysis. Brain 2023, 146, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Arola, A.; Laakso, H.M.; Pitkänen, J.; Koikkalainen, J.; Lötjönen, J.; Korvenoja, A.; Erkinjuntti, T.; Melkas, S.; Jokinen, H. Associations of cognitive reserve and psychological resilience with cognitive functioning in subjects with cerebral white matter hyperintensities. Eur. J. Neurol. 2021, 28, 2622–2630. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Zhu, H.; Chen, H.; Liu, R.; Huang, L.; Chen, H.; Cheng, Y.; Qin, R.; Shao, P.; Xu, H.; et al. Effects of cognitive reserve proxies on cognitive function and frontoparietal control network in subjects with white matter hyperintensities: A cross-sectional functional magnetic resonance imaging study. CNS Neurosci. Ther. 2022, 28, 932–941. [Google Scholar] [CrossRef]
- Zahodne, L.B.; Manly, J.J.; Brickman, A.M.; Siedlecki, K.L.; Decarli, C.; Stern, Y. Quantifying cognitive reserve in older adults by decomposing episodic memory variance: Replication and extension. J. Int. Neuropsychol. Soc. 2013, 19, 854–862. [Google Scholar] [CrossRef]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef]
- Chen, H.; Zhu, H.; Huang, L.; Chen, H.; Liu, R.; Qin, R.; Shao, P.; Xu, H.; Ma, J.; Cheng, Y.; et al. The flexibility of cognitive reserve in regulating the frontoparietal control network and cognitive function in subjects with white matter hyperintensities. Behav. Brain Res. 2022, 425, 113831. [Google Scholar] [CrossRef]
- Jian, X.; Satizabal, C.L.; Smith, A.V.; Wittfeld, K.; Bis, J.C.; Smith, J.A.; Hsu, F.C.; Nho, K.; Hofer, E.; Hagenaars, S.P.; et al. Exome Chip Analysis Identifies Low-Frequency and Rare Variants in MRPL38 for White Matter Hyperintensities on Brain Magnetic Resonance Imaging. Stroke 2018, 49, 1812–1819. [Google Scholar] [CrossRef]
- Freudenberger, P.; Schmidt, R.; Schmidt, H. Genetics of age-related white matter lesions from linkage to genome wide association studies. J. Neurol. Sci. 2012, 322, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Knol, M.J.; Wang, R.; Mishra, A.; Liu, D.; Luciano, M.; Teumer, A.; Armstrong, N.; Bis, J.C.; Jhun, M.A.; et al. Epigenetic and integrative cross-omics analyses of cerebral white matter hyperintensities on MRI. Brain 2023, 146, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Balado, J.; Pizarro, J.; Riba-Llena, I.; Penalba, A.; Faura, J.; Palà, E.; Montaner, J.; Hernández-Guillamon, M.; Delgado, P. New candidate blood biomarkers potentially associated with white matter hyperintensities progression. Sci. Rep. 2021, 11, 14324. [Google Scholar] [CrossRef] [PubMed]
- Bejanin, A.; Schonhaut, D.R.; La Joie, R.; Kramer, J.H.; Baker, S.L.; Sosa, N.; Ayakta, N.; Cantwell, A.; Janabi, M.; Lauriola, M.; et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 2017, 140, 3286–3300. [Google Scholar] [CrossRef] [PubMed]
- Daly, E.; Zaitchik, D.; Copeland, M.; Schmahmann, J.; Gunther, J.; Albert, M. Predicting conversion to Alzheimer disease using standardized clinical information. Arch. Neurol. 2000, 57, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Ye, B.S.; Woo, S.; Kim, S.W.; Chin, J.; Choi, S.H.; Jeong, J.H.; Yoon, S.J.; Yoon, B.; Park, K.W.; et al. Prediction Model of Conversion to Dementia Risk in Subjects with Amnestic Mild Cognitive Impairment: A Longitudinal, Multi-Center Clinic-Based Study. J. Alzheimer’s Dis. 2017, 60, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Kosmidis, M.H.; Vlachos, G.S.; Anastasiou, C.A.; Yannakoulia, M.; Dardiotis, E.; Hadjigeorgiou, G.; Sakka, P.; Ntanasi, E.; Scarmeas, N. Dementia Prevalence in Greece: The Hellenic Longitudinal Investigation of Aging and Diet (HELIAD). Alzheimer Dis. Assoc. Disord. 2018, 32, 232–239. [Google Scholar] [CrossRef]
- Vlachos, G.S.; Kosmidis, M.H.; Yannakoulia, M.; Dardiotis, E.; Hadjigeorgiou, G.; Tzoulaki, I.; Georgiou, A.N.; Sakka, P.; Anastasiou, C.A.; Stefanis, L.; et al. Dementia Incidence in the Elderly Population of Greece: Results from the HELIAD Study. Alzheimer Dis. Assoc. Disord. 2021, 35, 48–54. [Google Scholar] [CrossRef]
- Vlachos, G.S.; Kosmidis, M.H.; Yannakoulia, M.; Dardiotis, E.; Hadjigeorgiou, G.; Tzoulaki, I.; Georgiou, A.N.; Sakka, P.; Anastasiou, C.A.; Stefanis, L.; et al. Incidence of mild cognitive impairment in the elderly population in Greece: Results from the HELIAD study. Aging Clin. Exp. Res. 2021, 33, 2679–2688. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. Mini-mental state. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Vlahou, C.H.; Kosmidis, M.H.; Dardagani, A.; Tsotsi, S.; Giannakou, M.; Giazkoulidou, A.; Zervoudakis, E.; Pontikakis, N. Development of the Greek verbal learning test: Reliability, construct validity, and normative standards. Arch Clin. Neuropsychol. 2013, 28, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Ingram, F.; Soukup, V.M.; Ingram, P.T. The Medical College of Georgia Complex Figures: Reliability and preliminary normative data using an intentional learning paradigm in older adults. Neuropsychiatry Neuropsychol. Behav. Neurol. 1997, 10, 144–146. [Google Scholar] [PubMed]
- Kosmidis, M.H.; Vlahou, C.H.; Panagiotaki, P.; Kiosseoglou, G. The verbal fluency task in the Greek population: Normative data, and clustering and switching strategies. J. Int. Neuropsychol. Soc. 2004, 10, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Tsapkini, K.; Vlahou, C.H.; Potagas, C. Adaptation and validation of standardized aphasia tests in different languages: Lessons from the Boston diagnostic aphasia examination—short form in Greek. Behav. Neurol. 2010, 22, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Vlahou, C.H.; Kosmidis, M.H. The Greek trail making test: Preliminary norms for clinical and research use. Psychol. J. Hell Psychol. Soc. 2002, 9, 336–352. (In Greek) [Google Scholar]
- Kosmidis, M.H.; Tsotsi, S.; Karambela, O.; Takou, E.; Vlahou, C.H. Cultural factors influencing performance on visuo- perceptual neuropsychological tasks. Behav. Neurol. 2010, 23, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Bozikas, V.P.; Giazkoulidou, A.; Hatzigeorgiadou, M.; Karavatos, A.; Kosmidis, M.H. Do age and education contribute to performance on the clock drawing test? Normative data for the Greek population. J. Clin. Exp. Neuropsychol. 2008, 30, 199–203. [Google Scholar] [CrossRef]
- Lezak, M.D.; Howieson, D.B.; Loring, D.W. Neuropsychological Assessment; Oxford University Press: New York, NY, USA, 2004. [Google Scholar]
- Grove, M.L.; Yu, B.; Cochran, B.J.; Haritunians, T.; Bis, J.C.; Taylor, K.D.; Hansen, M.; Borecki, I.B.; Cupples, L.A.; Fornage, M.; et al. Best practices and joint calling of the HumanExome BeadChip: The CHARGE Consortium. PLoS ONE 2013, 8, e68095. [Google Scholar] [CrossRef]
- Abraham, G.; Qiu, Y.; Inouye, M. FlashPCA2: Principal component analysis of Biobank-scale genotype datasets. Bioinformatics 2017, 33, 2776–2778. [Google Scholar] [CrossRef]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Marchini, J.; Howie, B.; Myers, S.; McVean, G.; Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007, 39, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Hu, X.; Peng, Y. An analytical comparison of the principal component method and the mixed effects model for association studies in the presence of cryptic relatedness and population stratification. Hum. Hered. 2013, 76, 1–9. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| All Participants | Non-aMCI 1/AD 2 at Follow Up | aMCI/AD at Follow Up | ||
|---|---|---|---|---|
| N = 537 | N = 475 | N = 62 | p-Value | |
| Age, y, mean ± SD 3 | 73.07 ± 4.84 | 72.84 ± 4.74 | 74.83 ± 5.24 | 0.002 |
| Sex, female (%) | 318 (59.2) | 286 (60.2) | 32 (51.6) | 0.217 |
| Education, y 4, mean ± SD | 7.53 ± 4.42 | 7.80 ± 4.38 | 5.44 ± 4.22 | <0.001 |
| Duration of follow-up, y, mean ± SD | 2.91 ± 0.80 | 2.89 ± 0.79 | 3.06 ± 0.89 | 0.132 |
| Global cognition score | −0.351 ± 0.779 | −0.242 ± 0.709 | −1.189 ± 0.792 | <0.001 |
| APOE ε4 carrier, positive (n, %) | 383/459 (83.4) | 336/404 (83.2) | 47/55 (85.4) | 0.573 |
| PRS WMH 5, mean ± SD | −0.003 ± 1.010 | −0.278 ± 1.017 | 0.189 ± 0.943 | 0.095 |
| VBS 6, mean ± SD | 1.322 ± 1.056 | 1.295 ± 1.054 | 1.539 ± 1.059 | 0.091 |
| Model 1 (N = 537) | Model 2 (N = 537) | Model 3 (N = 458) | ||||
|---|---|---|---|---|---|---|
| HR 1 (95% CI 2) | p-Value | HR (95% CI) | p-Value | HR (95% CI) | p-Value | |
| PRS WMH 3 | 1.452 (1.104, 1.910) | 0.008 | 1.364 (1.028, 1.810) | 0.031 | 1.472 (1.079, 2.007) | 0.015 |
| CR 4 | - | 0.935 (0.865, 1.010) | 0.090 | 0.925 (0.866, 0.984) | 0.044 | |
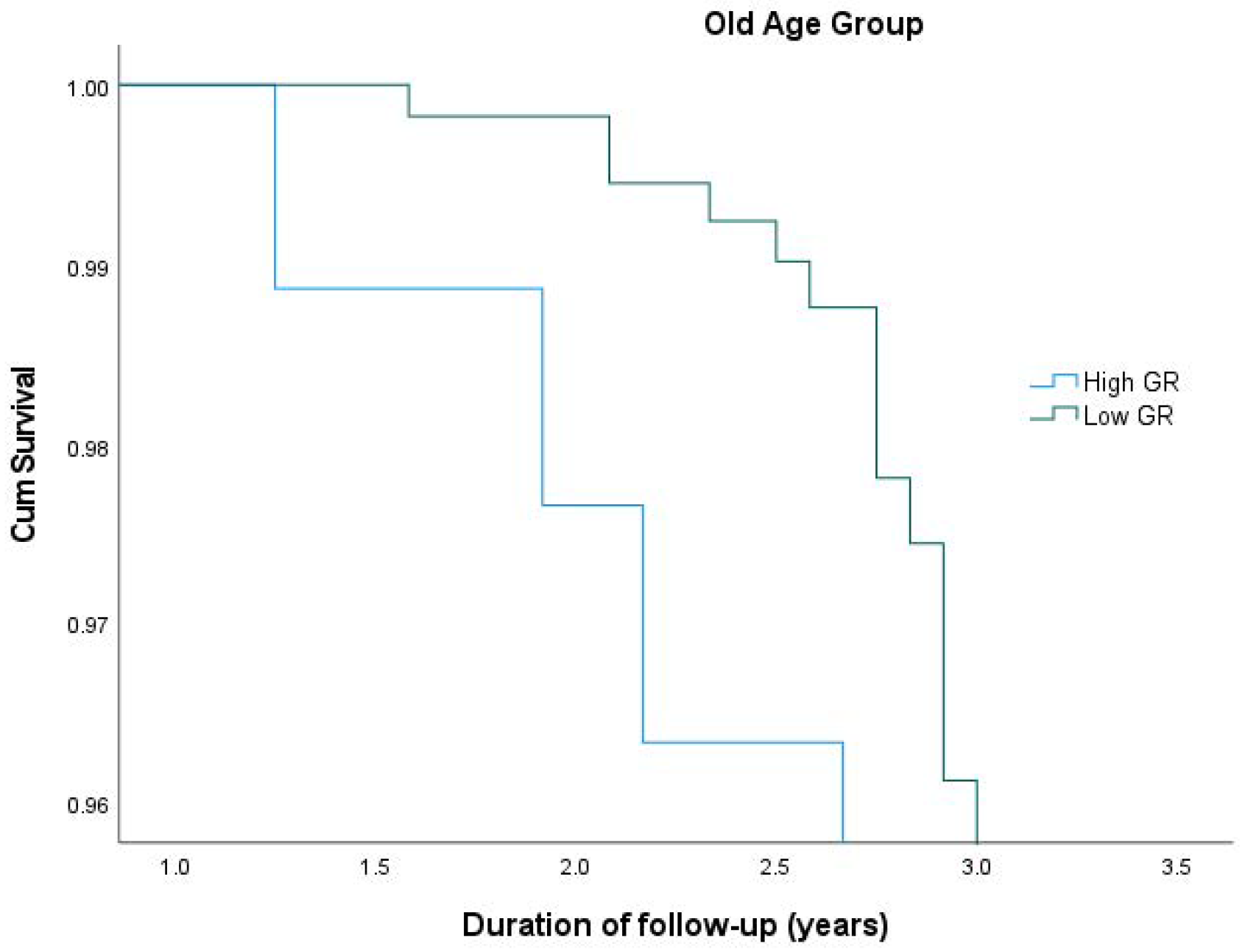
| Young Age Group(N = 223) | Old Age Group (N = 223) | |||||
|---|---|---|---|---|---|---|
| N | HR 1 (95% CI 2) | p-Value | N | HR (95% CI) | p-Value | |
| Low Genetic Risk | 111 | 1 (reference) | 111 | 1 (reference) | ||
| High Genetic Risk | 112 | 0.649 (0.278, 1.514) | 0.317 | 112 | 3.423 (1.713, 6.840) | <0.001 |
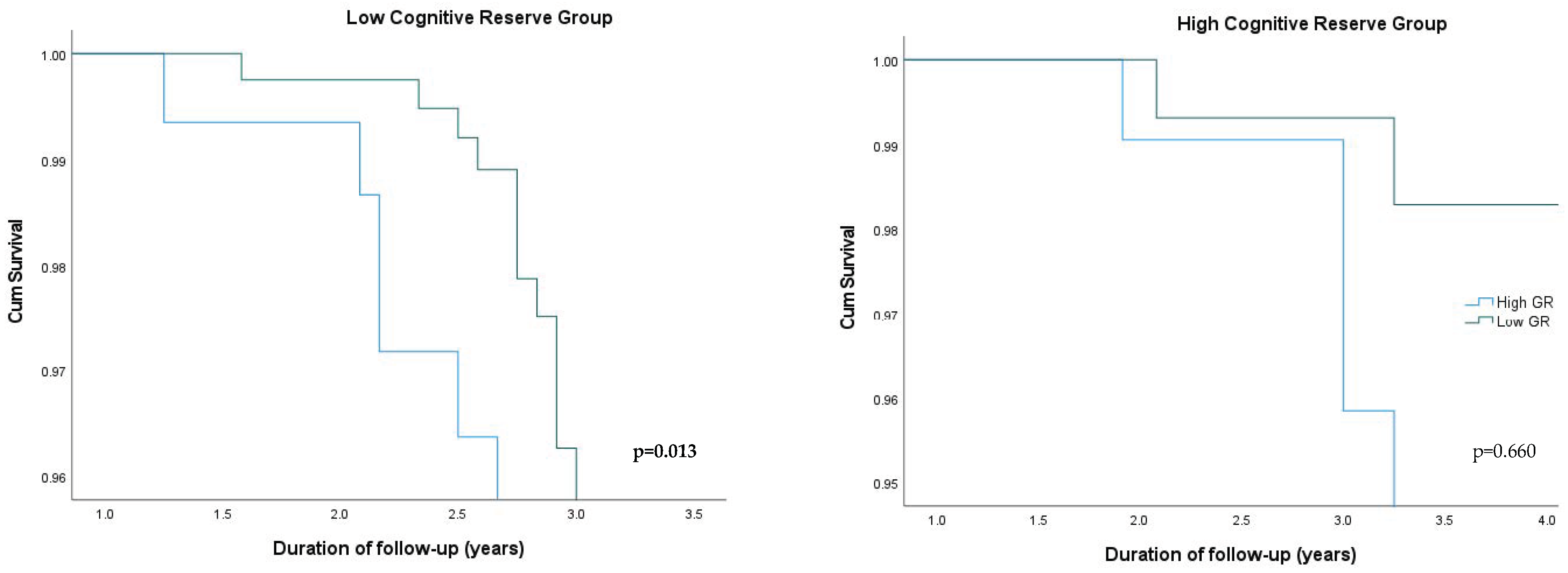
| Low CR 1 Group (N = 320) | High CR Group (N = 138) | |||||
|---|---|---|---|---|---|---|
| N | HR 2 (95% CI 3) | p-Value | N | HR (95% CI) | p-Value | |
| Low Genetic Risk | 160 | 1 (reference) | 69 | 1 (reference) | ||
| High Genetic Risk | 160 | 2.059 (1.086, 3.279) | 0.013 | 69 | 1.228 (0.744, 1.702) | 0.660 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sampatakakis, S.N.; Mourtzi, N.; Charisis, S.; Mamalaki, E.; Ntanasi, E.; Hatzimanolis, A.; Ramirez, A.; Lambert, J.-C.; Yannakoulia, M.; Kosmidis, M.H.; et al. Genetic Predisposition for White Matter Hyperintensities and Risk of Mild Cognitive Impairment and Alzheimer’s Disease: Results from the HELIAD Study. Curr. Issues Mol. Biol. 2024, 46, 934-947. https://doi.org/10.3390/cimb46010060
Sampatakakis SN, Mourtzi N, Charisis S, Mamalaki E, Ntanasi E, Hatzimanolis A, Ramirez A, Lambert J-C, Yannakoulia M, Kosmidis MH, et al. Genetic Predisposition for White Matter Hyperintensities and Risk of Mild Cognitive Impairment and Alzheimer’s Disease: Results from the HELIAD Study. Current Issues in Molecular Biology. 2024; 46(1):934-947. https://doi.org/10.3390/cimb46010060
Chicago/Turabian StyleSampatakakis, Stefanos N., Niki Mourtzi, Sokratis Charisis, Eirini Mamalaki, Eva Ntanasi, Alexandros Hatzimanolis, Alfredo Ramirez, Jean-Charles Lambert, Mary Yannakoulia, Mary H. Kosmidis, and et al. 2024. "Genetic Predisposition for White Matter Hyperintensities and Risk of Mild Cognitive Impairment and Alzheimer’s Disease: Results from the HELIAD Study" Current Issues in Molecular Biology 46, no. 1: 934-947. https://doi.org/10.3390/cimb46010060
APA StyleSampatakakis, S. N., Mourtzi, N., Charisis, S., Mamalaki, E., Ntanasi, E., Hatzimanolis, A., Ramirez, A., Lambert, J.-C., Yannakoulia, M., Kosmidis, M. H., Dardiotis, E., Hadjigeorgiou, G., Sakka, P., & Scarmeas, N. (2024). Genetic Predisposition for White Matter Hyperintensities and Risk of Mild Cognitive Impairment and Alzheimer’s Disease: Results from the HELIAD Study. Current Issues in Molecular Biology, 46(1), 934-947. https://doi.org/10.3390/cimb46010060