

A Potential Role for the Receptor for Advanced Glycation End-Products (RAGE) in the Development of Secondhand Smoke-Induced Chronic Sinusitis

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Control and CS Samples
2.2. Mice and Animal Use
2.3. Histology
2.4. Tissue Isolation and Characterization
2.5. Statistical Analyses
3. Results
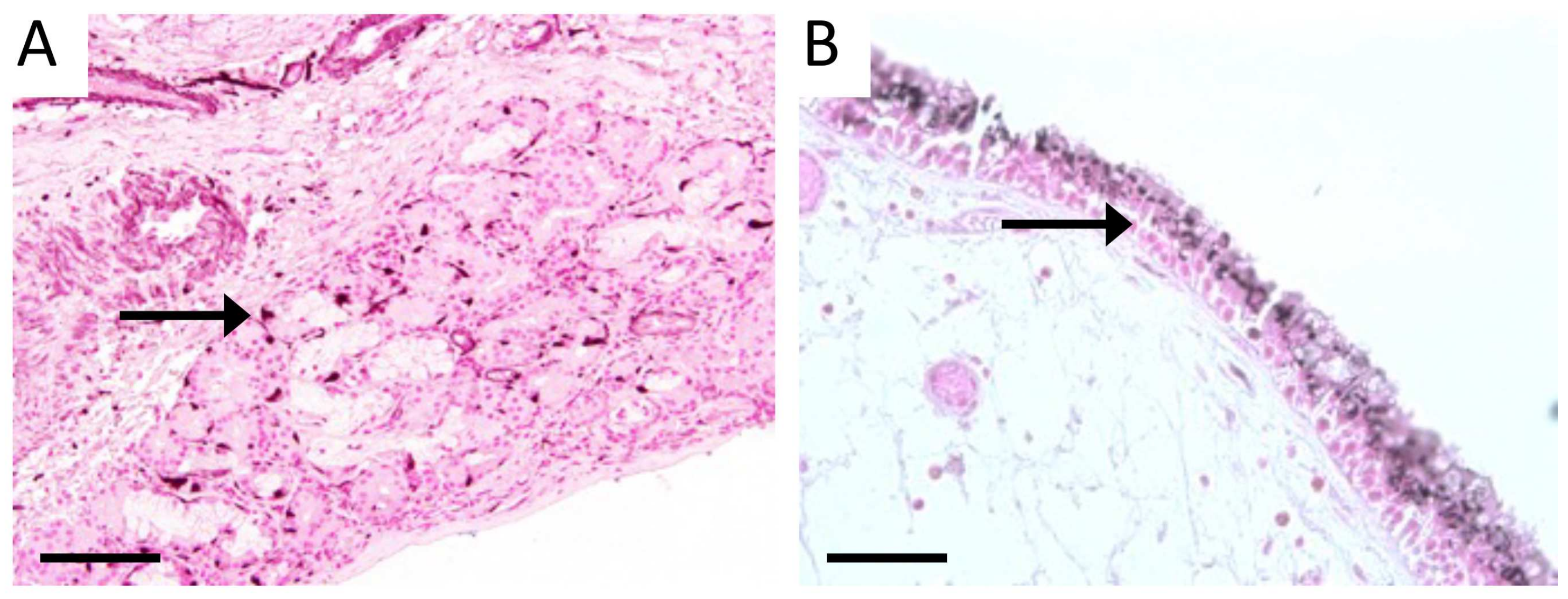
3.1. Characterization of Sinus Tissue from Human CS and Normal Patients
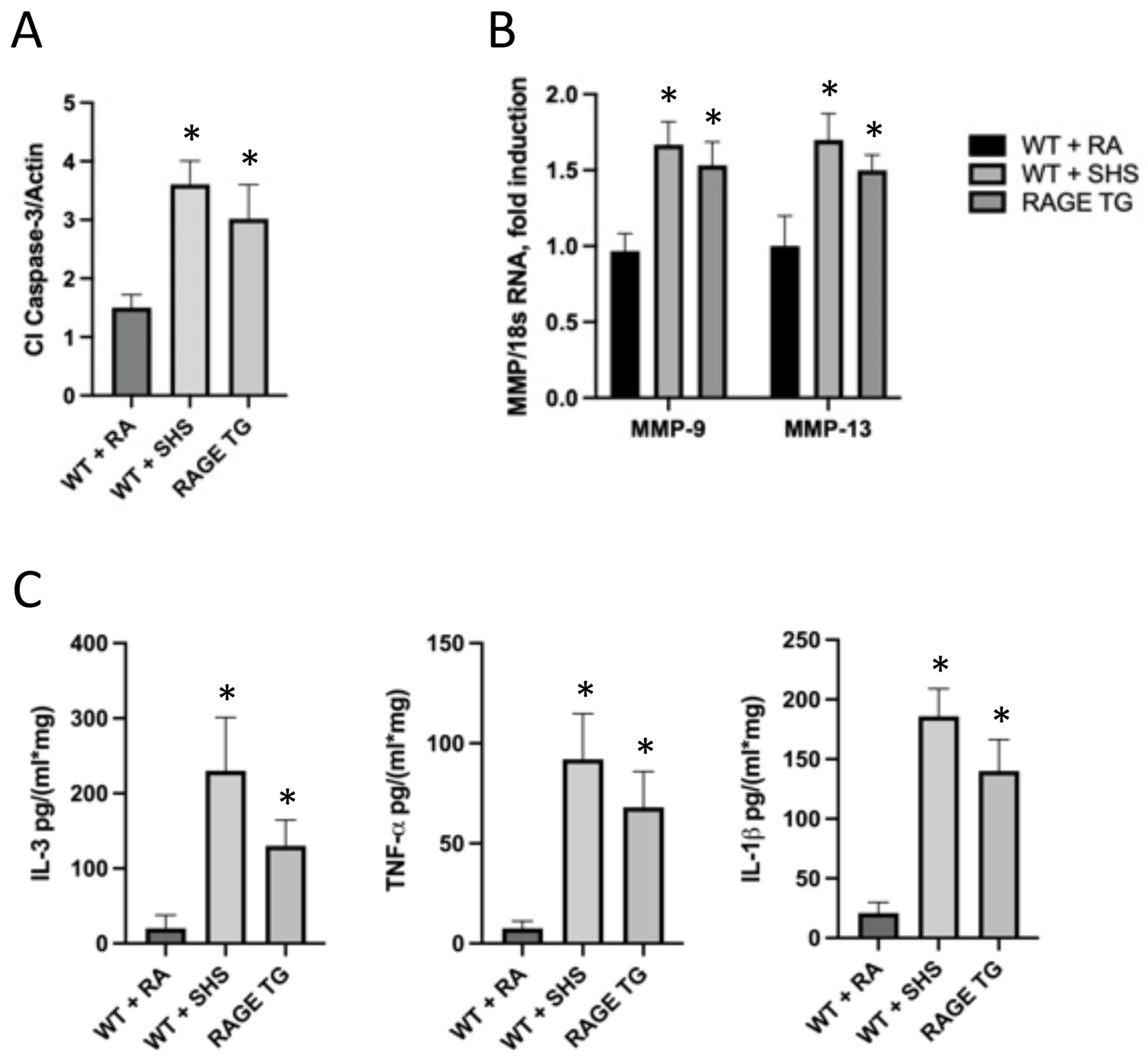
3.2. RAGE and Potential Mouse Models of CS
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adams, P.F.; Hendershot, G.E.; Marano, M.A. Current Estimates from the National Health Interview Survey; Centers for Disease Control and Prevention, National Center for Health Statistics: Bethesda, MD, USA, 1996; Volume 1999, pp. 1–203. [Google Scholar]
- Anand, V.K. Epidemiology and Economic Impact of Rhinosinusitis. Ann. Otol. Rhinol. Laryngol. 2004, 113, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, D.L.; Lucas, J.W.; Clarke, T.C. Summary health statistics for U.S. adults: National health interview survey, 2012. Vital Health Stat. 2014, 260, 1–161. [Google Scholar]
- Davis, K.S.; Casey, S.E.; Mulligan, J.K.; Mulligan, R.M.; Schlosser, R.J.; Atkinson, C. Murine complement deficiency ameliorates acute cigarette smoke-induced nasal damage. Otolaryngol. Neck Surg. 2010, 143, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Benninger, M.S.; Ferguson, B.J.; Hadley, J.A.; Hamilos, D.L.; Jacobs, M.; Kennedy, D.W.; Lanza, D.C.; Marple, B.F.; Osguthorpe, J.D.; Stankiewicz, J.A.; et al. Adult Chronic Rhinosinusitis: Definitions, Diagnosis, Epidemiology, and Pathophysiology. Otolaryngol. Head Neck Surg. 2003, 129, S1–S32. [Google Scholar] [CrossRef] [PubMed]
- Hamilos, D.L. Chronic sinusitis. J. Allergy Clin. Immunol. 2000, 106, 213–227. [Google Scholar] [CrossRef]
- Platts-Mills, T.A.; Rosenwasser, L.J. Chronic sinusitis consensus and the way forward. J. Allergy Clin. Immunol. 2004, 114, 1359–1361. [Google Scholar] [CrossRef]
- Bellanti, J.A.; Wallerstedt, D.B. Allergic Rhinitis Update: Epidemiology and Natural History. Allergy Asthma Proc. 2000, 21, 367–370. [Google Scholar] [CrossRef]
- Lindsay, R.; Slaughter, T.; Britton-Webb, J.; Mog, S.R.; Conran, R.; Tadros, M.; Earl, N.; Fox, D.; Roberts, J.; Bolger, W.E. Development of a Murine Model of Chronic Rhinosinusitis. Otolaryngol. Neck Surg. 2006, 134, 724–730. [Google Scholar] [CrossRef]
- Tammemagi, C.M.; Davis, R.M.; Benninger, M.S.; Holm, A.L.; Krajenta, R. Secondhand smoke as a potential cause of chronic rhinosinusitis: A case-control study. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 327–334. [Google Scholar] [CrossRef]
- Litvack, J.R.; Fong, K.; Mace, J.; James, K.E.; Smith, T.L. Predictors of olfactory dysfunction in patients with chronic rhinosinusitis. Laryngoscope 2008, 118, 2225–2230. [Google Scholar] [CrossRef]
- Houser, S.M.; Keen, K.J. The Role of Allergy and Smoking in Chronic Rhinosinusitis and Polyposis. Laryngoscope 2008, 118, 1521–1527. [Google Scholar] [CrossRef]
- Thornalley, P.J. Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell. Mol. Biol. 1998, 44, 1013–1023. [Google Scholar] [PubMed]
- Dahlin, K.; Mager, E.M.; Allen, L.; Tigue, Z.; Goodglick, L.; Wadehra, M.; Dobbs, L. Identification of Genes Differentially Expressed in Rat Alveolar Type I Cells. Am. J. Respir. Cell Mol. Biol. 2004, 31, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Shirasawa, M.; Fujiwara, N.; Hirabayashi, S.; Ohno, H.; Iida, J.; Makita, K.; Hata, Y. Receptor for advanced glycation end-products is a marker of type I lung alveolar cells. Genes Cells 2004, 9, 165–174. [Google Scholar] [CrossRef]
- Tanaka, N.; Yonekura, H.; Yamagishi, S.; Fujimori, H.; Yamamoto, Y.; Yamamoto, H. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-alpha through nuclear factor-kappa B, and by 17beta-estradiol through Sp-1 in human vascular endothelial cells. J. Biol. Chem. 2000, 275, 25781–25790. [Google Scholar] [CrossRef] [PubMed]
- Abel, M.; Ritthaler, U.; Zhang, Y.; Deng, Y.; Schmidt, A.M.; Greten, J.; Sernau, T.; Wahl, P.; Andrassy, K.; Ritz, E.; et al. Expression of receptors for advanced glycosylated end-products in renal disease. Nephrol. Dial. Transplant. 1995, 10, 1662–1667. [Google Scholar] [CrossRef]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humbert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Kloting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef]
- Drinda, S.; Franke, S.; Rüster, M.; Petrow, P.; Pullig, O.; Stein, G.; Hein, G. Identification of the receptor for advanced glycation end products in synovial tissue of patients with rheumatoid arthritis. Rheumatol. Int. 2004, 25, 411–413. [Google Scholar] [CrossRef]
- Greten, J.; Kreis, I.; Wiesel, K.; Stier, E.; Schmidt, A.M.; Stern, D.M.; Ritz, E.; Waldherr, R.; Nawroth, P.P. Receptors for advanced glycation end-products (AGE)—Expression by endothelial cells in non-diabetic uraemic patients. Nephrol. Dial. Transplant. 1996, 11, 786–790. [Google Scholar] [CrossRef][Green Version]
- A Hofmann, M.; Drury, S.; Hudson, B.I.; Gleason, M.R.; Qu, W.; Lu, Y.; Lalla, E.; Chitnis, S.; Monteiro, J.; Stickland, M.H.; et al. RAGE and arthritis: The G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002, 3, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Lalla, E.; Lu, Y.; Gleason, M.R.; Wolf, B.M.; Tanji, N.; Ferran, L.J.; Kohl, B.; Rao, V.; Kisiel, W.; et al. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J. Clin. Investig. 2001, 107, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J., Jr.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of Advanced Glycation End Products and Their Cellular Receptor RAGE in Diabetic Nephropathy and Nondiabetic Renal Disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.A.; Kafatos, F.C.; Janeway, C.A., Jr.; Ezekowitz, R.A.B. Phylogenetic Perspectives in Innate Immunity. Science 1999, 284, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Kislinger, T.; Tanji, N.; Wendt, T.; Qu, W.; Lu, Y.; Ferran, L.J., Jr.; Taguchi, A.; Olson, K.; Bucciarelli, L.; Goova, M.; et al. Receptor for Advanced Glycation End Products Mediates Inflammation and Enhanced Expression of Tissue Factor in Vasculature of Diabetic Apolipoprotein E–Null Mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 905–910. [Google Scholar] [CrossRef]
- Imler, J.L.; Hoffmann, J.A. Toll and Toll-like proteins: An ancient family of receptors signaling infection. Rev. Immunogenet. 2000, 2, 294–304. [Google Scholar]
- Kaisho, T.; Akira, S. Toll-like receptors and their signaling mechanism in innate immunity. Acta Odontol. Scand. 2001, 59, 124–130. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9. [Google Scholar] [CrossRef]
- Fu, M.X.; Requena, J.R.; Jenkins, A.J.; Lyons, T.J.; Baynes, J.W.; Thorpe, S.R. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J. Biol. Chem. 1996, 271, 9982–9986. [Google Scholar] [CrossRef]
- Degryse, B.; Bonaldi, T.; Scaffidi, P.; Müller, S.; Resnati, M.; Sanvito, F.; Arrigoni, G.; Bianchi, M.E. The High Mobility Group (Hmg) Boxes of the Nuclear Protein Hmg1 Induce Chemotaxis and Cytoskeleton Reorganization in Rat Smooth Muscle Cells. J. Cell Biol. 2001, 152, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, B.W.; Heizmann, C.W. The S100 family of EF-hand calcium-binding proteins: Functions and pathology. Trends Biochem. Sci. 1996, 21, 134–140. [Google Scholar] [CrossRef]
- Marenholz, I.; Heizmann, C.W.; Fritz, G. S100 proteins in mouse and man: From evolution to function and pathology (including an update of the nomenclature). Biochem. Biophys. Res. Commun. 2004, 322, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.S.; Papadimitriou, J.C.; Morales, R.E.; Califano, J.A.; Kline, N.L.; Bhatnagar, K.; Hebert, A.M.; Taylor, R.J. The association of active and passive tobacco smoke exposure with chronic rhinosinusitis symptom severity: A cross-sectional study. Int. Forum Allergy Rhinol. 2021, 12, 278–285. [Google Scholar] [CrossRef]
- Hirschi-Budge, K.M.; Tsai, K.Y.F.; Curtis, K.L.; Davis, G.S.; Theurer, B.K.; Kruyer, A.M.M.; Homer, K.W.; Chang, A.; Van Ry, P.M.; Arroyo, J.A.; et al. RAGE signaling during tobacco smoke-induced lung inflammation and potential therapeutic utility of SAGEs. BMC Pulm. Med. 2022, 22, 160. [Google Scholar] [CrossRef]
- Stogsdill, M.P.; Stogsdill, J.A.; Bodine, B.G.; Fredrickson, A.C.; Sefcik, T.L.; Wood, T.T.; Kasteler, S.D.; Reynolds, P.R. Conditional Overexpression of Receptors for Advanced Glycation End-Products in the Adult Murine Lung Causes Airspace Enlargement and Induces Inflammation. Am. J. Respir. Cell Mol. Biol. 2013, 49, 128–134. [Google Scholar] [CrossRef]
- Gunia, S.; Liebe, D.; Koch, S. Loss of basal cell keratin 14 reflects increased risk of recurrence in surgically resected sinonasal inverted papilloma. J. Clin. Pathol. 2007, 61, 707–712. [Google Scholar] [CrossRef]
- Davidson, D.J.; Gray, M.A.; Kilanowski, F.M.; Tarran, R.; Randell, S.H.; Sheppard, D.N.; Argent, B.E.; Dorin, J.R. Murine epithelial cells: Isolation and culture. J. Cyst. Fibros. 2004, 3, 59–62. [Google Scholar] [CrossRef]
- Agha-Hosseini, F.; Mirzaii-Dizgah, I. Serum and saliva collagenase-3 (MMP-13) in patients with oral lichen planus and oral squamous cell carcinoma. Med. J. Islam. Repub. Iran 2015, 29, 218. [Google Scholar]
- Caminha, G.P.; Pizzichini, E.; Neto, J.F.L.; Hopkins, C.; Moreira, J.D.S.; Pizzichini, M.M.M. Rhinosinusitis symptoms, smoking and COPD: Prevalence and associations. Clin. Otolaryngol. 2018, 43, 1560–1565. [Google Scholar] [CrossRef] [PubMed]
- Reh, D.D.; Lin, S.Y.; Clipp, S.L.; Irani, L.; Alberg, A.J.; Navas-Acien, A. Secondhand Tobacco Smoke Exposure and Chronic Rhinosinusitis: A Population-Based Case–Control Study. Am. J. Rhinol. Allergy 2009, 23, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Kuhar, H.N.; Ganti, A.; Brown, H.J.; Gattuso, P.; Ghai, R.; Mahdavinia, M.; Batra, P.S.; Tajudeen, B.A. Histopathologic Influences of Comorbid Smoking Status in Chronic Rhinosinusitis. Am. J. Rhinol. Allergy 2020, 34, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.B.; Stogsdill, J.A.; Lewis, J.B.; Wood, T.T.; Reynolds, P.R. RAGE and tobacco smoke: Insights into modeling chronic obstructive pulmonary disease. Front. Physiol. 2012, 3, 301. [Google Scholar] [CrossRef]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Ann. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Van Crombruggen, K.; Vogl, T.; Perez-Novo, C.; Holtappels, G.; Bachert, C. Differential release and deposition of S100A8/A9 proteins in inflamed upper airway tissue. Eur. Respir. J. 2016, 47, 264–274. [Google Scholar] [CrossRef]
- Ciprandi, G.; Bellussi, L.M.; Passali, G.C.; Damiani, V.; Passali, D. HMGB1 in nasal inflammatory diseases: A reappraisal 30 years after its discovery. Expert Rev. Clin. Immunol. 2020, 16, 457–463. [Google Scholar] [CrossRef]
- Dzaman, K.; Szczepanski, M.J.; Molinska-Glura, M.; Krzeski, A.; Zagor, M. Expression of the Receptor for Advanced Glycation End Products, a Target for High Mobility Group Box 1 Protein, and its Role in Chronic Recalcitrant Rhinosinusitis with Nasal Polyps. Arch. Immunol. Ther. Exp. 2014, 63, 223–230. [Google Scholar] [CrossRef]
- Lee, S.-H.; Cho, J.H.; Park, J.-H.; Cho, J.-S.; Lee, H.-M. High Mobility Group Box Chromosomal Protein-1 Induces Myofibroblast Differentiation and Extracellular Matrix Production via RAGE, p38, JNK and AP-1 Signaling Pathways in Nasal Fibroblasts. Am. J. Rhinol. Allergy 2021, 35, 774–780. [Google Scholar] [CrossRef]
- Cerami, C.; Founds, H.; Nicholl, I.; Mitsuhashi, T.; Giordano, D.; Vanpatten, S.; Lee, A.; Al-Abed, Y.; Vlassara, H.; Bucala, R.; et al. Tobacco smoke is a source of toxic reactive glycation products. Proc. Natl. Acad. Sci. USA 1997, 94, 13915–13920. [Google Scholar] [CrossRef]
- Kupper, D.S.; Valera, F.C.; Malinsky, R.; Milanezi, C.M.; Silva, J.S.; Tamashiro, E.; Anselmo-Lima, W.T. Expression of apoptosis mediators p53 and caspase 3, 7, and 9 in chronic rhinosinusitis with nasal polyposis. Am. J. Rhinol. Allergy 2014, 28, 187–191. [Google Scholar] [CrossRef]
- Lin, H.; Lin, D.; Xiong, X. Differential expression of Livin, caspase-3, and second mitochondria-derived activator of caspases in chronic rhinosinusitis with nasal polyps. Otolaryngol. Head Neck Surg. 2014, 51, 1067–1072. [Google Scholar] [CrossRef] [PubMed]
- Lucas, B.R.; Voegels, R.L.; do Amaral, J.B.; Bachi, A.L.L.; Pezato, R. BMP-7, MMP-9, and TGF-beta tissue remodeling proteins and their correlations with interleukins 6 and 10 in chronic rhinosinusitis. Eur. Arch. Otorhinolaryngol. 2021, 278, 4335–4343. [Google Scholar] [CrossRef] [PubMed]
- Lygeros, S.; Danielides, G.; Grafanaki, K.; Riga, M. Matrix metalloproteinases and chronic rhinosinusitis with nasal polyposis. Unravelling a puzzle through a systematic review. Rhinol. J. 2021, 59, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Wang, M.; Zhang, N.; Yu, P.; Wang, P.; Li, Y.; Wang, X.; Zhang, L.; Bachert, C. Involvement of the extracellular matrix proteins periostin and tenascin C in nasal polyp remodeling by regulating the expression of MMPs. Clin. Transl. Allergy 2021, 11, e12059. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Zhu, S.; Ma, R.; Wang, L.; Shen, X.; Li, Y.; Feng, N.; Wang, L.; Liu, M.; Xie, L.; et al. Correlation analysis of TGF-beta1, MMP-9, TIMP-1, IL-1, IL-4, IL-6, IL-17, and TNF-alpha in refractory chronic rhinosinusitis: A retrospective study. Allergol. Immunopathol. 2022, 50, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Lennard, C.M.; Mann, E.A.; Sun, L.L.; Chang, A.S.; Bolger, W.E. Interleukin-1 beta, interleukin-5, interleukin-6, interleukin-8, and tumor necrosis factor-alpha in chronic sinusitis: Response to systemic corticosteroids. Am. J. Rhinol. 2000, 14, 367–373. [Google Scholar] [CrossRef]
- Min, Y.G.; Lee, K.S. The role of cytokines in rhinosinusitis. J. Korean Med. Sci. 2000, 15, 255–259. [Google Scholar] [CrossRef]
- Mohamad, S.; Ab Hamid, S.S.; Azlina, A.; Shukri, N.M. Association of IL-1 gene polymorphisms with chronic rhinosinusitis with and without nasal polyp. Asia Pac. Allergy 2019, 9, e22. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robin, H.; Trudeau, C.; Robbins, A.; Chung, E.; Rahman, E.; Gangmark-Strickland, O.; Licari, F.W.; Winden, D.R.; Orr, D.L.; Arroyo, J.A.; et al. A Potential Role for the Receptor for Advanced Glycation End-Products (RAGE) in the Development of Secondhand Smoke-Induced Chronic Sinusitis. Curr. Issues Mol. Biol. 2024, 46, 729-740. https://doi.org/10.3390/cimb46010047
Robin H, Trudeau C, Robbins A, Chung E, Rahman E, Gangmark-Strickland O, Licari FW, Winden DR, Orr DL, Arroyo JA, et al. A Potential Role for the Receptor for Advanced Glycation End-Products (RAGE) in the Development of Secondhand Smoke-Induced Chronic Sinusitis. Current Issues in Molecular Biology. 2024; 46(1):729-740. https://doi.org/10.3390/cimb46010047
Chicago/Turabian StyleRobin, Hannah, Courtney Trudeau, Adam Robbins, Emily Chung, Erum Rahman, Olivia Gangmark-Strickland, Frank W. Licari, Duane R. Winden, Dan L. Orr, Juan A. Arroyo, and et al. 2024. "A Potential Role for the Receptor for Advanced Glycation End-Products (RAGE) in the Development of Secondhand Smoke-Induced Chronic Sinusitis" Current Issues in Molecular Biology 46, no. 1: 729-740. https://doi.org/10.3390/cimb46010047
APA StyleRobin, H., Trudeau, C., Robbins, A., Chung, E., Rahman, E., Gangmark-Strickland, O., Licari, F. W., Winden, D. R., Orr, D. L., Arroyo, J. A., & Reynolds, P. R. (2024). A Potential Role for the Receptor for Advanced Glycation End-Products (RAGE) in the Development of Secondhand Smoke-Induced Chronic Sinusitis. Current Issues in Molecular Biology, 46(1), 729-740. https://doi.org/10.3390/cimb46010047