
The Development of a CRISPR-FnCpf1 System for Large-Fragment Deletion and Multiplex Gene Editing in Acinetobacter baumannii


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, Primers, Culture Conditions, and Antibiotics
2.2. Plasmid Construction
2.3. Preparation of Electrocompetent Cells
2.4. Electrocompetent
2.5. Spacer Insertion
2.6. Genome Editing with CRISPR-FnCpf1
2.7. Plasmid Curing
2.8. Twitching Motility Assay
2.9. Statistical Analyses
3. Results
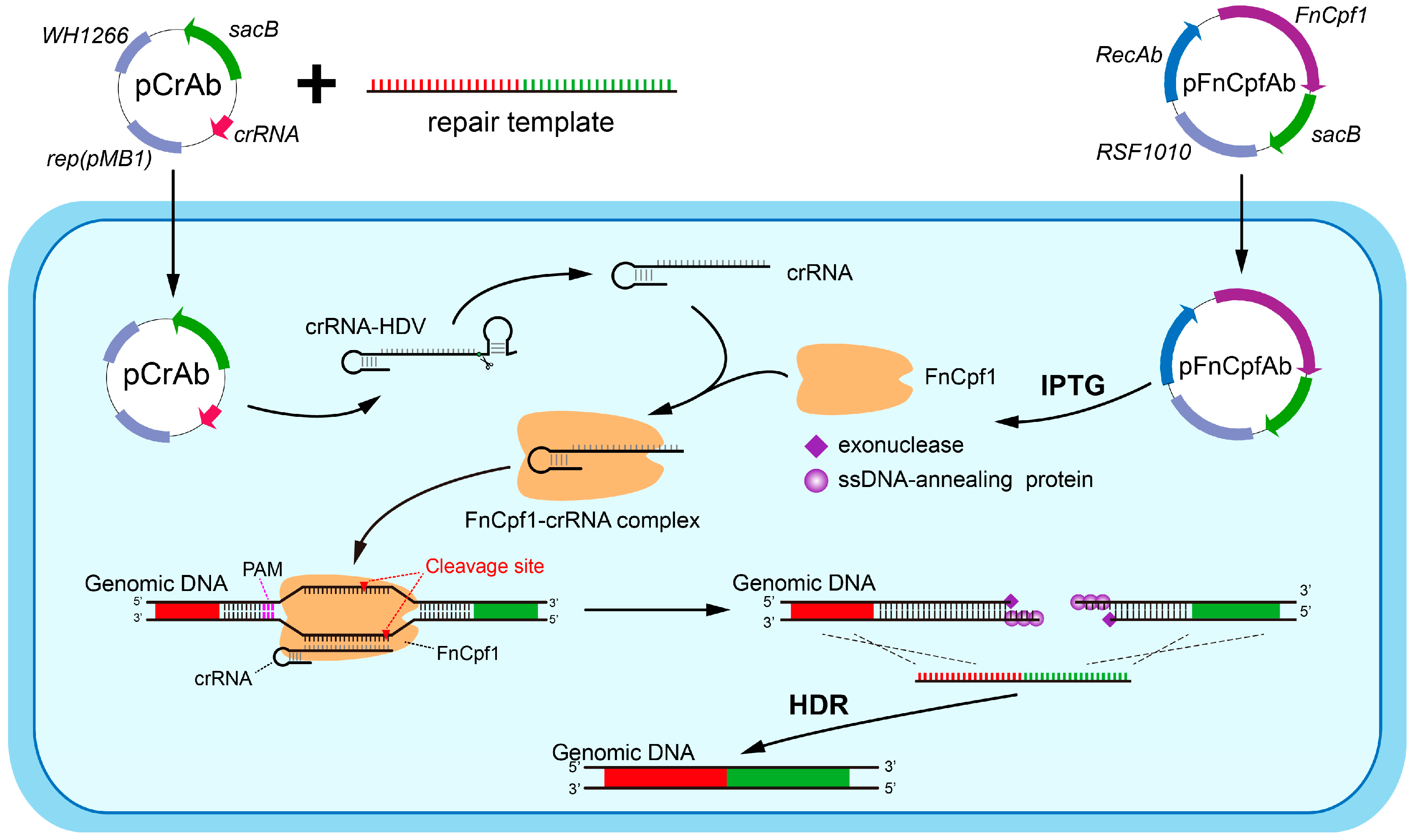
3.1. Construction of a CRISPR-FnCpf1-Based Two-Plasmid Genome Editing System in A. baumannii
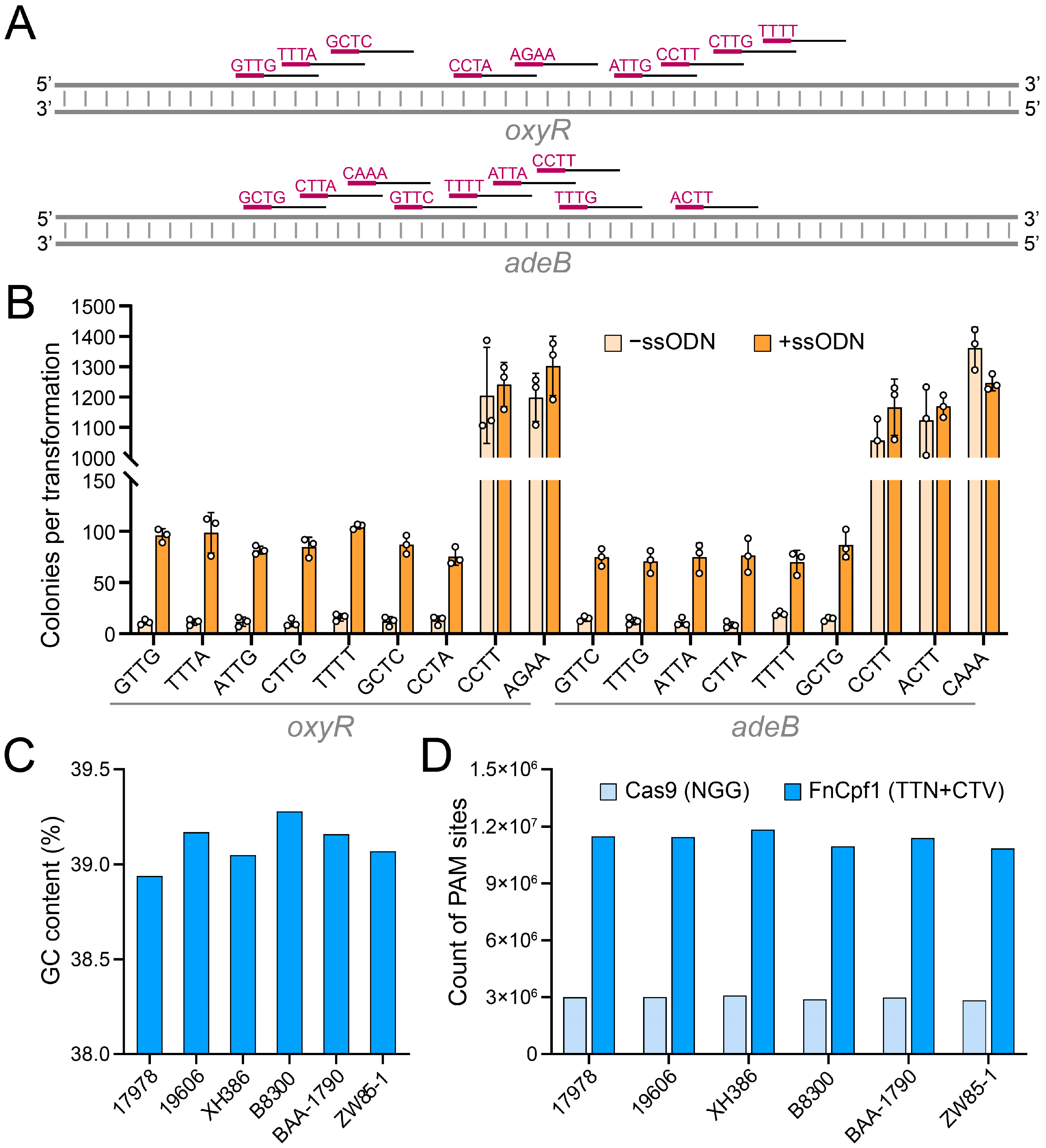
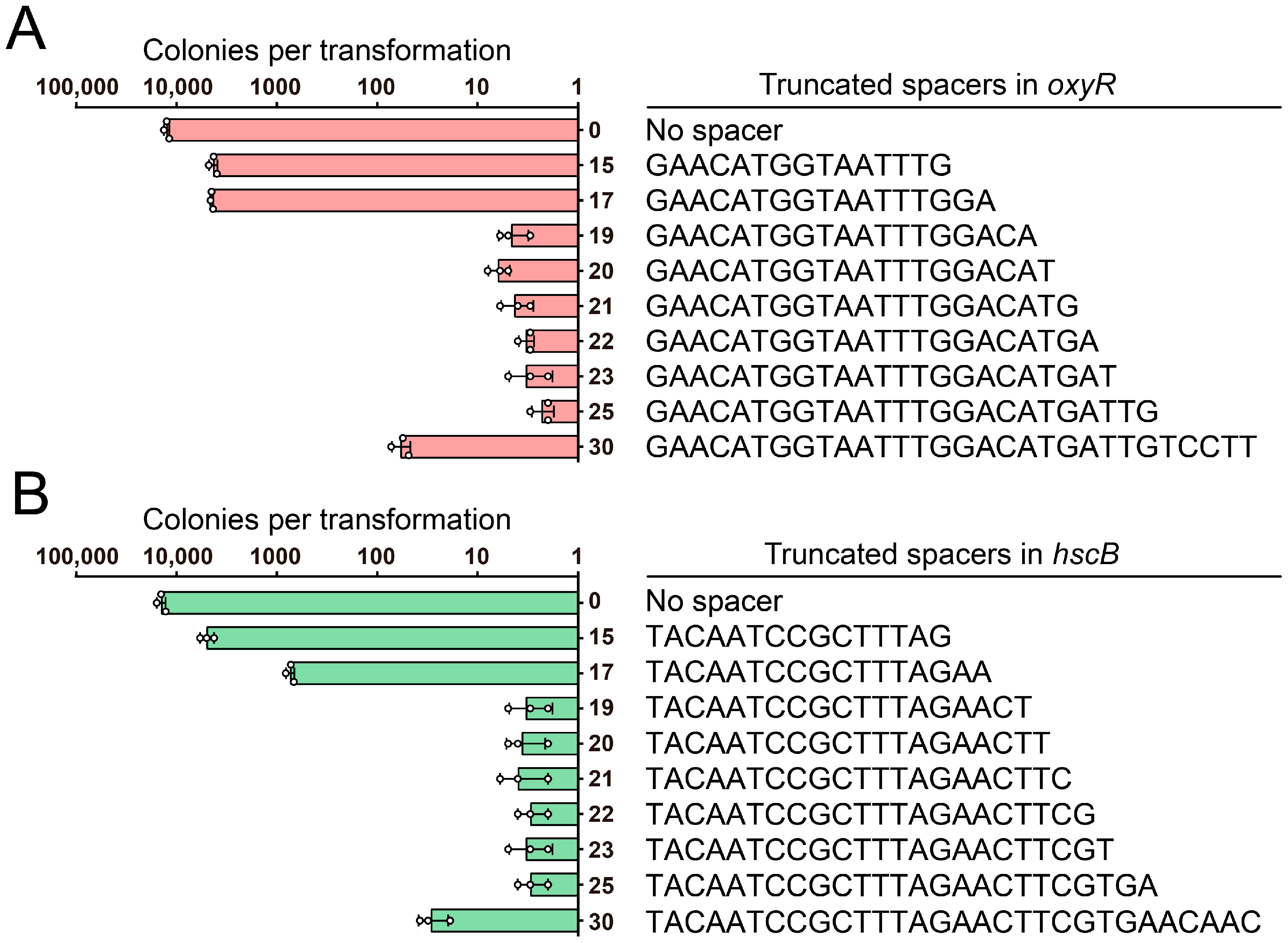
3.2. Determination of the FnCpf1 PAM Sequence and Spacer Length in A. baumannii
3.3. Comparison of Cas9 and FnCpf1 Editing Performance in A. baumannii
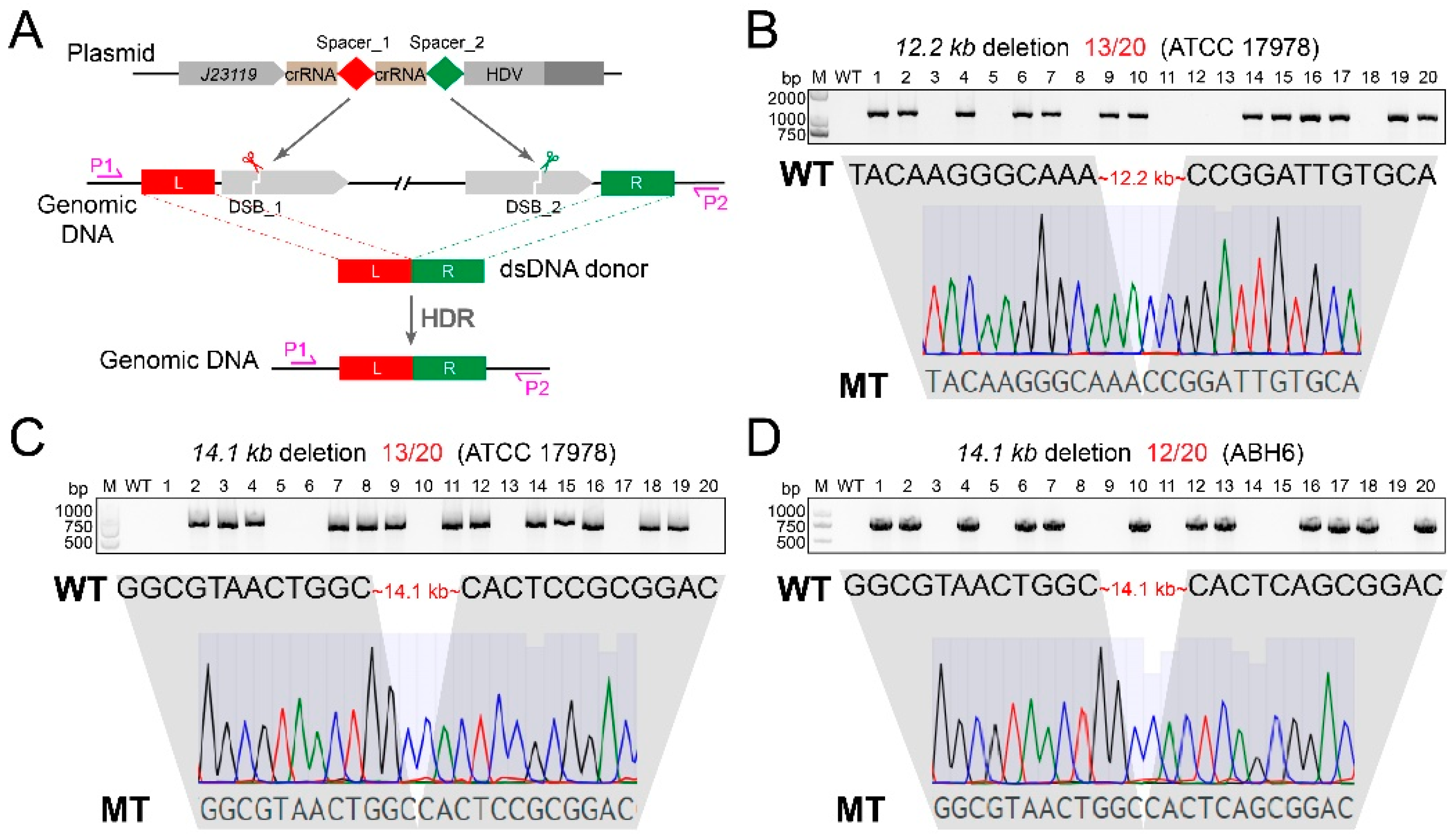
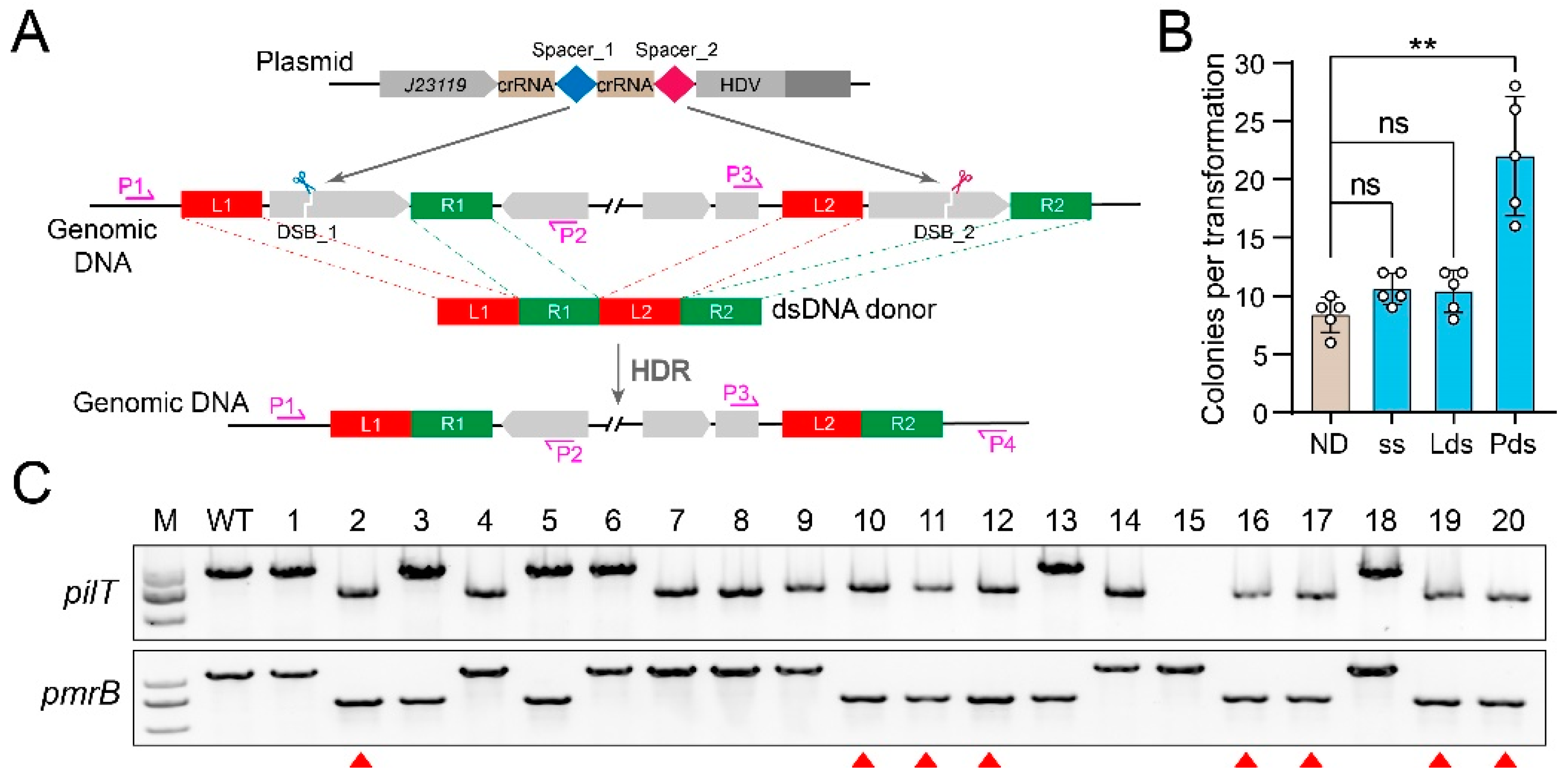
3.4. Large-Fragment Deletion by CRISPR-FnCpf1 in A. baumannii
3.5. Multiplex Gene Editing by CRISPR-FnCpf1 in A. baumannii
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harding, C.M.; Hennon, S.W.; Feldman, M.F. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat. Rev. Microbiol. 2018, 16, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.A.; Ahmed, F.A.; Elkhateeb, A.F.; Mahmoud, E.E.; Ahmed, M.I.; Ahmed, R.I.; Hosni, A.; Alghamdi, S.; Kabrah, A.; Dablool, A.S. Virulence characteristics of biofilm-forming Acinetobacter baumannii in clinical isolates using a Galleria Mellonella Model. Microorganisms 2021, 9, 2365. [Google Scholar] [CrossRef] [PubMed]
- Levin-Reisman, I.; Ronin, I.; Gefen, O.; Braniss, I.; Shoresh, N.; Balaban, N.Q. Antibiotic tolerance facilitates the evolution of resistance. Science 2017, 355, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Kröger, C.; Kary, S.C.; Schauer, K.; Cameron, A.D. Genetic regulation of virulence and antibiotic resistance in Acinetobacter baumannii. Genes 2016, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Z.; Chen, Y.; Hua, X.; Yu, Y.; Ji, Q. A highly efficient CRISPR-Cas9-based genome engineering platform in Acinetobacter baumannii to understand the H2O2-sensing mechanism of OxyR. Cell Chem. Biol. 2019, 26, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P. Evolutionary classification of CRISPR–Cas systems: A burst of class 2 and derived variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.-Y.; Yan, H.-Q.; Ren, G.-X.; Zhao, J.-P.; Guo, X.-P.; Sun, Y.-C. CRISPR-Cas12a-assisted recombineering in bacteria. Appl. Environ. Microbiol. 2017, 83, e00947-17. [Google Scholar] [CrossRef]
- Verwaal, R.; Buiting-Wiessenhaan, N.; Dalhuijsen, S.; Roubos, J.A. CRISPR/Cpf1 enables fast and simple genome editing of Saccharomyces cerevisiae. Yeast 2018, 35, 201–211. [Google Scholar] [CrossRef]
- Zhong, Z.; Zhang, Y.; You, Q.; Tang, X.; Ren, Q.; Liu, S.; Yang, L.; Wang, Y.; Liu, X.; Liu, B. Plant genome editing using FnCpf1 and LbCpf1 nucleases at redefined and altered PAM sites. Mol. Plant 2018, 11, 999–1002. [Google Scholar] [CrossRef]
- Kleinstiver, B.P.; Tsai, S.Q.; Prew, M.S.; Nguyen, N.T.; Welch, M.M.; Lopez, J.M.; McCaw, Z.R.; Aryee, M.J.; Joung, J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016, 34, 869–874. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Van Der Oost, J.; Regev, A. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Fagerlund, R.D.; Staals, R.H.; Fineran, P.C. The Cpf1 CRISPR-Cas protein expands genome-editing tools. Genome Biol. 2015, 16, 251. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Swarts, D.C.; van der Oost, J.; Jinek, M. Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol. Cell 2017, 66, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Fonfara, I.; Richter, H.; Bratovič, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef]
- Fürste, J.P.; Pansegrau, W.; Frank, R.; Blöcker, H.; Scholz, P.; Bagdasarian, M.; Lanka, E. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene 1986, 48, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Tucker, A.T.; Nowicki, E.M.; Boll, J.M.; Knauf, G.A.; Burdis, N.C.; MStephen, T.; Davies, B.W. Defining gene-phenotype relationships in Acinetobacter baumannii through one-step chromosomal gene inactivation. mBio 2014, 5, 01313–01314. [Google Scholar] [CrossRef]
- Hunger, M.; Schmucker, R.; Kishan, V.; Hillen, W. Analysis and nucleotide sequence of an origin of an origin of DNA replication in Acinetobacter calcoaceticus and its use for Escherichia coli shuttle plasmids. Gene 1990, 87, 45–51. [Google Scholar] [CrossRef]
- Been, M.D.; Wickham, G.S. Self-cleaving ribozymes of hepatitis delta virus RNA. Eur. J. Biochem. 1997, 247, 741–753. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Liu, P.; Nielsen, A.A.; Brophy, J.A.; Clancy, K.; Peterson, T.; Voigt, C.A. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 2013, 10, 659–664. [Google Scholar] [CrossRef]
- Quandt, J.; Hynes, M.F. Versatile suicide vectors which allow direct selection for gene replacement in gram-negative bacteria. Gene 1993, 127, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Clemmer, K.M.; Bonomo, R.A.; Rather, P.N. Genetic analysis of surface motility in Acinetobacter baumannii. Microbiology 2011, 157, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.; Lin, L.; Cheng, Y.; He, X.; Sun, H.; Xie, H.; Fu, J.; Liu, C.; Li, J.; Chen, D. A ‘new lease of life’: FnCpf1 possesses DNA cleavage activity for genome editing in human cells. Nucleic Acids Res. 2017, 45, 11295–11304. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Wang, Y.; Chen, W.; Ji, Q. CRISPR/Cpf1-mediated multiplex and large-fragment gene editing in Staphylococcus aureus. ACS Synth. Biol. 2022, 11, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Rostain, W.; Grebert, T.; Vyhovskyi, D.; Pizarro, P.T.; Tshinsele-Van Bellingen, G.; Cui, L.; Bikard, D. Cas9 off-target binding to the promoter of bacterial genes leads to silencing and toxicity. Nucleic Acids Res. 2023, 51, 3485–3496. [Google Scholar] [CrossRef] [PubMed]
- Meliawati, M.; Schilling, C.; Schmid, J. Recent advances of Cas12a applications in bacteria. Appl. Microbiol. Biotechnol. 2021, 105, 2981–2990. [Google Scholar] [CrossRef]
- Song, Y.; Yuan, L.; Wang, Y.; Chen, M.; Deng, J.; Lv, Q.; Sui, T.; Li, Z.; Lai, L. Efficient dual sgRNA-directed large gene deletion in rabbit with CRISPR/Cas9 system. Cell. Mol. Life Sci. 2016, 73, 2959–2968. [Google Scholar] [CrossRef]
- Chen, X.; Xu, F.; Zhu, C.; Ji, J.; Zhou, X.; Feng, X.; Guang, S. Dual sgRNA-directed gene knockout using CRISPR/Cas9 technology in Caenorhabditis elegans. Sci. Rep. 2014, 4, 7581. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Chen, W.; Song, L.; Zhang, Y.; Shen, Z.; Yu, F.; Li, M.; Ji, Q. CRISPR-Cas9 and CRISPR-assisted cytidine deaminase enable precise and efficient genome editing in Klebsiella pneumoniae. Appl. Environ. Microbiol. 2018, 84, e01834-18. [Google Scholar] [CrossRef]
- Rumbo-Feal, S.; Perez, A.; Ramelot, T.A.; Alvarez-Fraga, L.; Vallejo, J.A.; Beceiro, A.; Ohneck, E.J.; Arivett, B.A.; Merino, M.; Fiester, S.E. Contribution of the A. baumannii A1S_0114 gene to the interaction with eukaryotic cells and virulence. Front. Cell. Infect. Microbiol. 2017, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.C. Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J. Bacteriol. 1998, 180, 2063–2071. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Buchholz, F.; Muyrers, J.P.; Stewart, A.F. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 1998, 20, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Ding, Y.; Rong, H.; Wang, Y. The Development of a CRISPR-FnCpf1 System for Large-Fragment Deletion and Multiplex Gene Editing in Acinetobacter baumannii. Curr. Issues Mol. Biol. 2024, 46, 570-584. https://doi.org/10.3390/cimb46010037
Wang S, Ding Y, Rong H, Wang Y. The Development of a CRISPR-FnCpf1 System for Large-Fragment Deletion and Multiplex Gene Editing in Acinetobacter baumannii. Current Issues in Molecular Biology. 2024; 46(1):570-584. https://doi.org/10.3390/cimb46010037
Chicago/Turabian StyleWang, Shuai, Yue Ding, Hua Rong, and Yu Wang. 2024. "The Development of a CRISPR-FnCpf1 System for Large-Fragment Deletion and Multiplex Gene Editing in Acinetobacter baumannii" Current Issues in Molecular Biology 46, no. 1: 570-584. https://doi.org/10.3390/cimb46010037
APA StyleWang, S., Ding, Y., Rong, H., & Wang, Y. (2024). The Development of a CRISPR-FnCpf1 System for Large-Fragment Deletion and Multiplex Gene Editing in Acinetobacter baumannii. Current Issues in Molecular Biology, 46(1), 570-584. https://doi.org/10.3390/cimb46010037