
AG1® Induces a Favorable Impact on Gut Microbial Structure and Functionality in the Simulator of Human Intestinal Microbial Ecosystem® Model

 , ,
, ,
Abstract
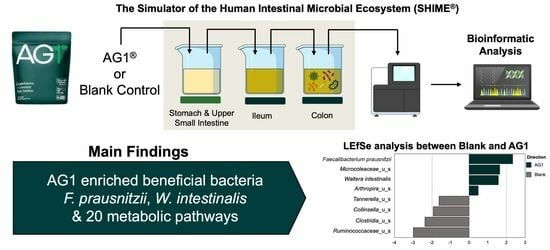
1. Introduction
2. Materials and Methods
2.1. Experimental Product and Model
2.2. DNA Extraction, Library Preparation, and Sequencing
2.3. Bioinformatics
2.4. Statistics
3. Results
3.1. Community Structure
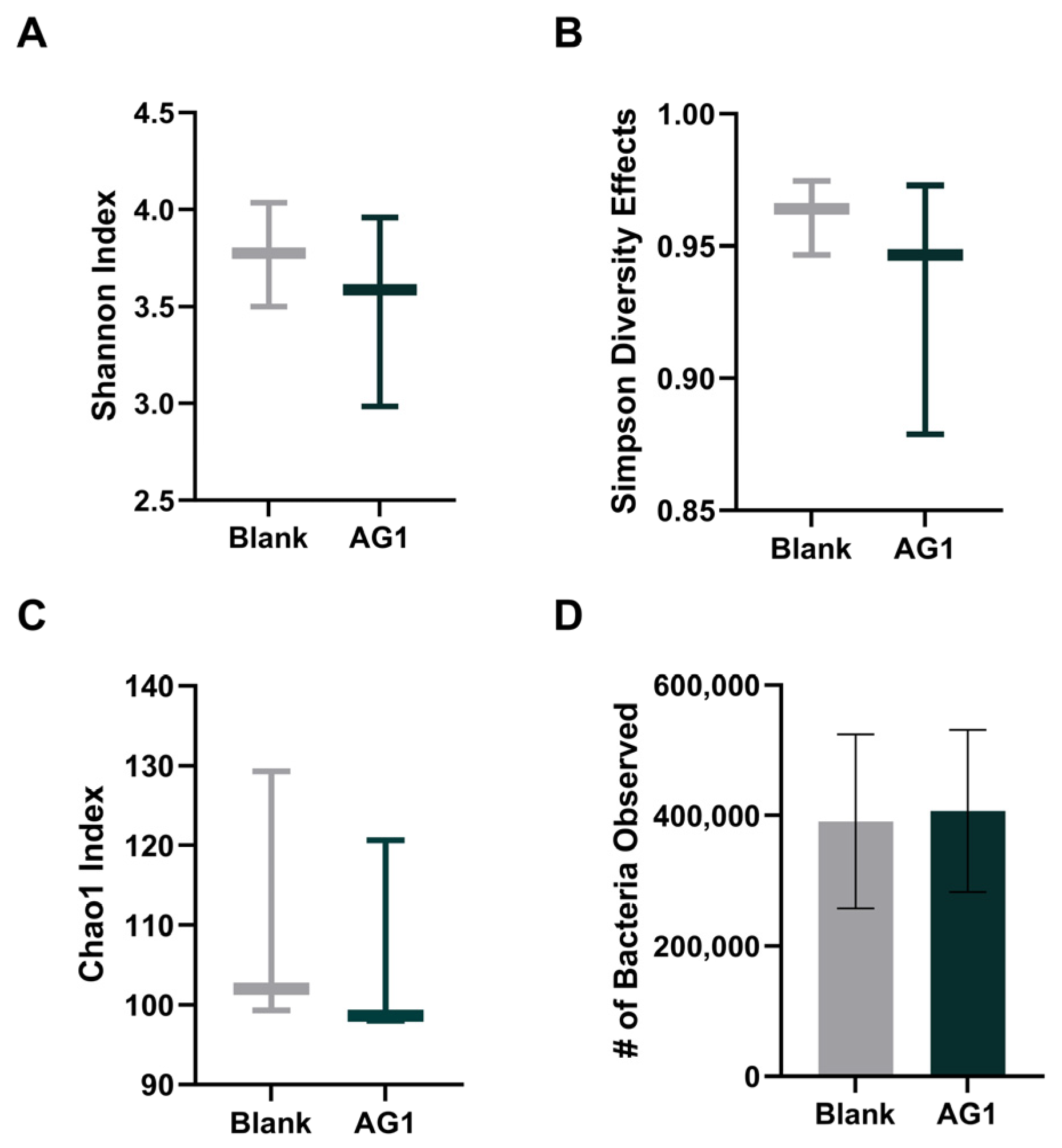
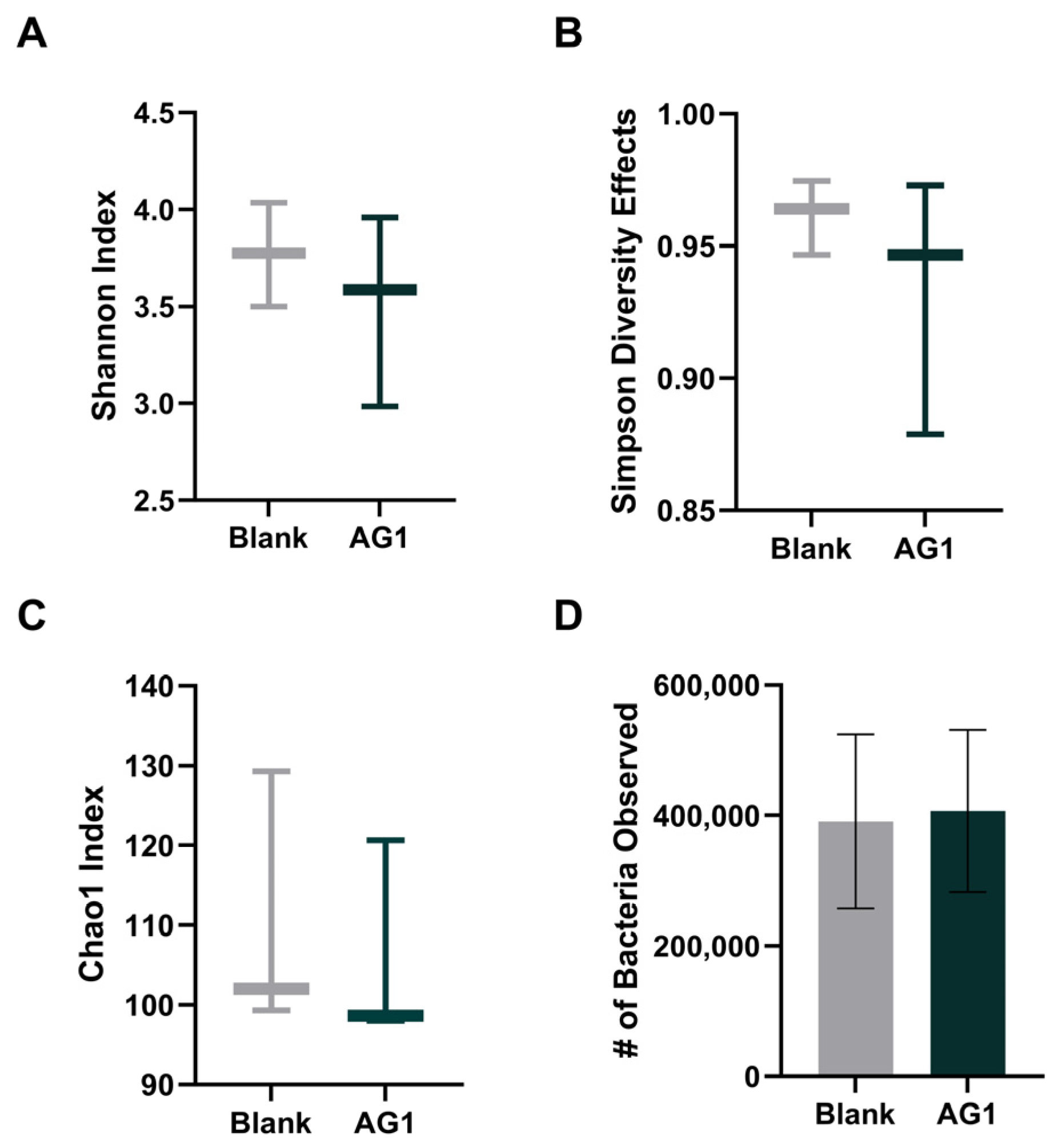
3.1.1. Alpha Diversity
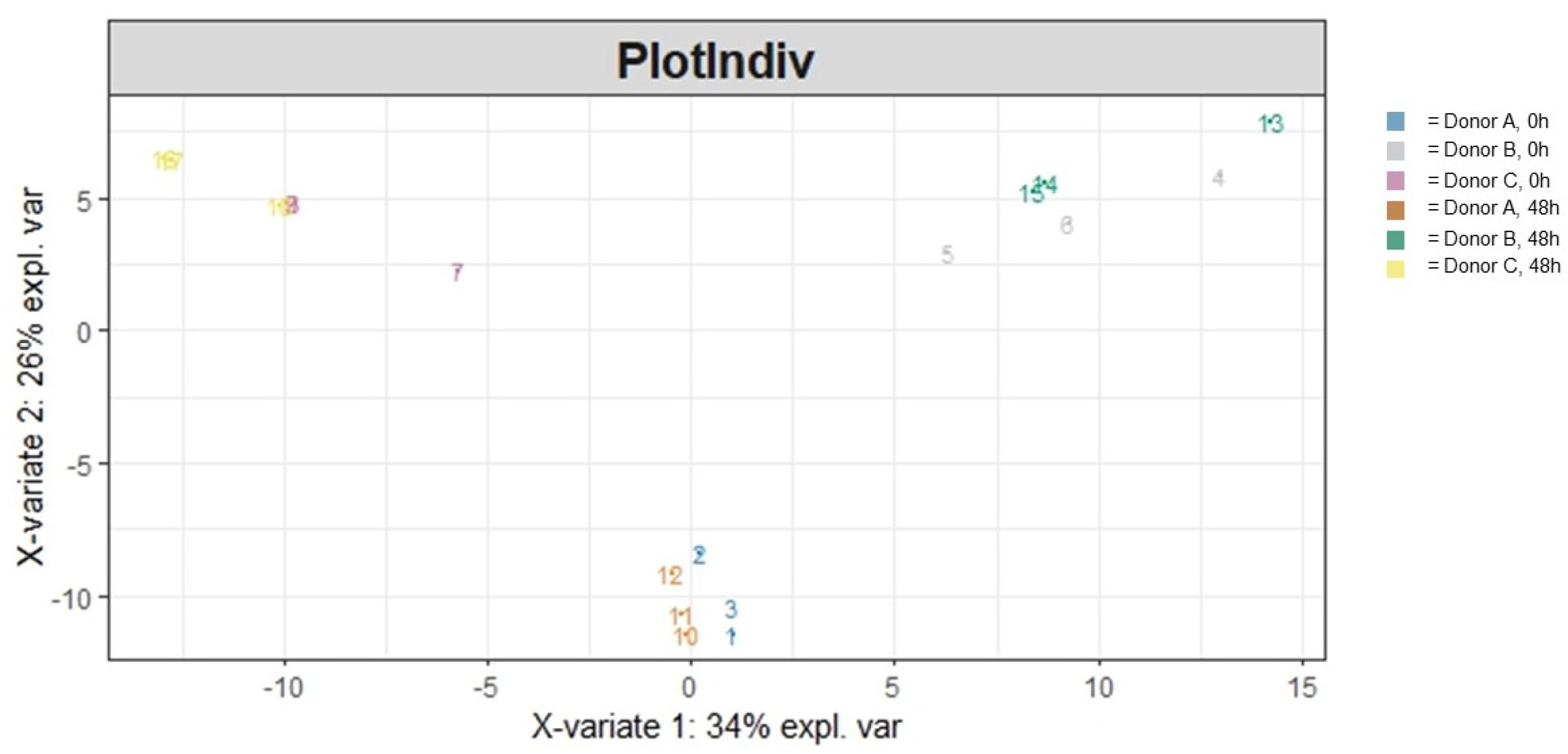
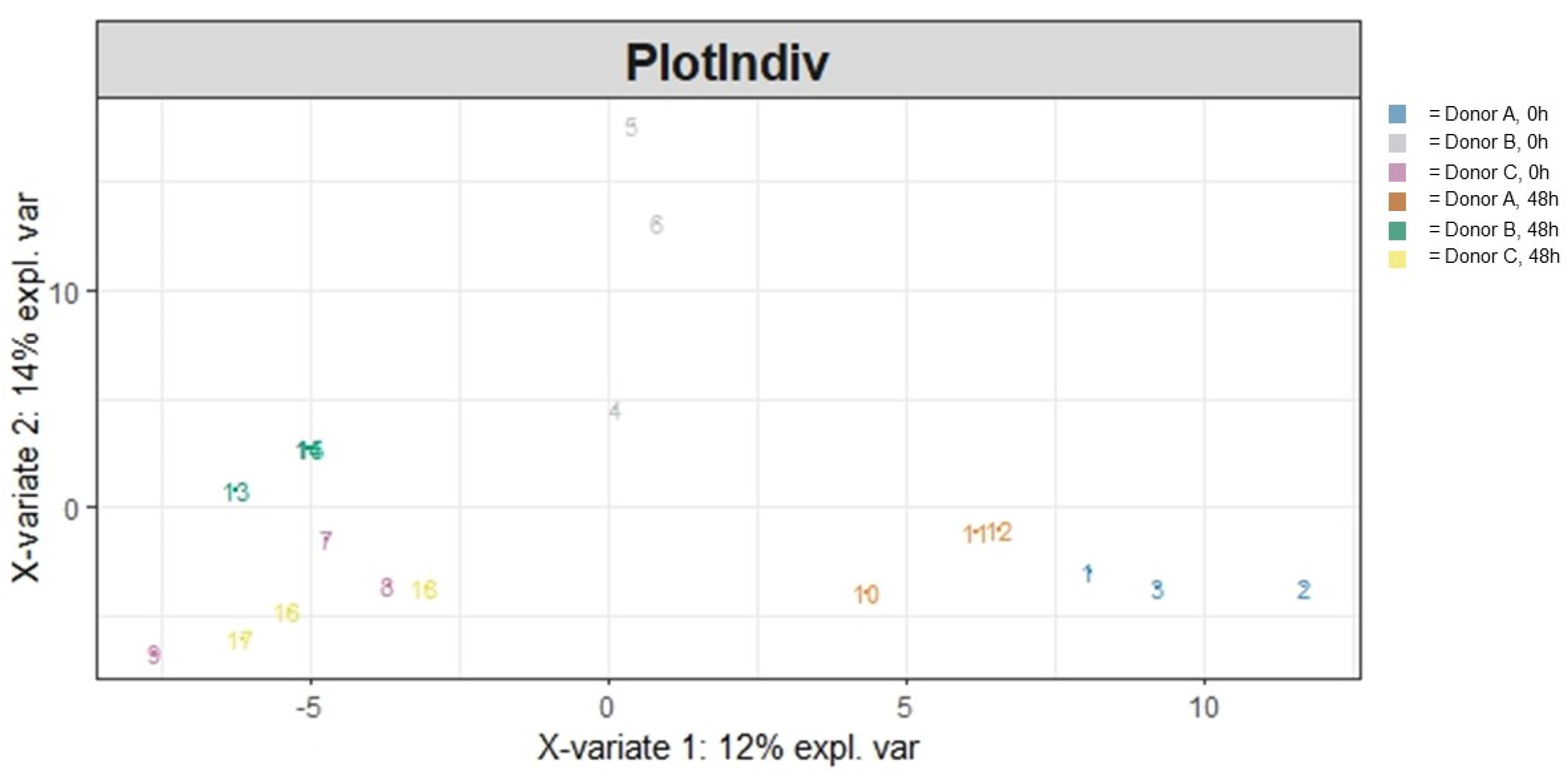
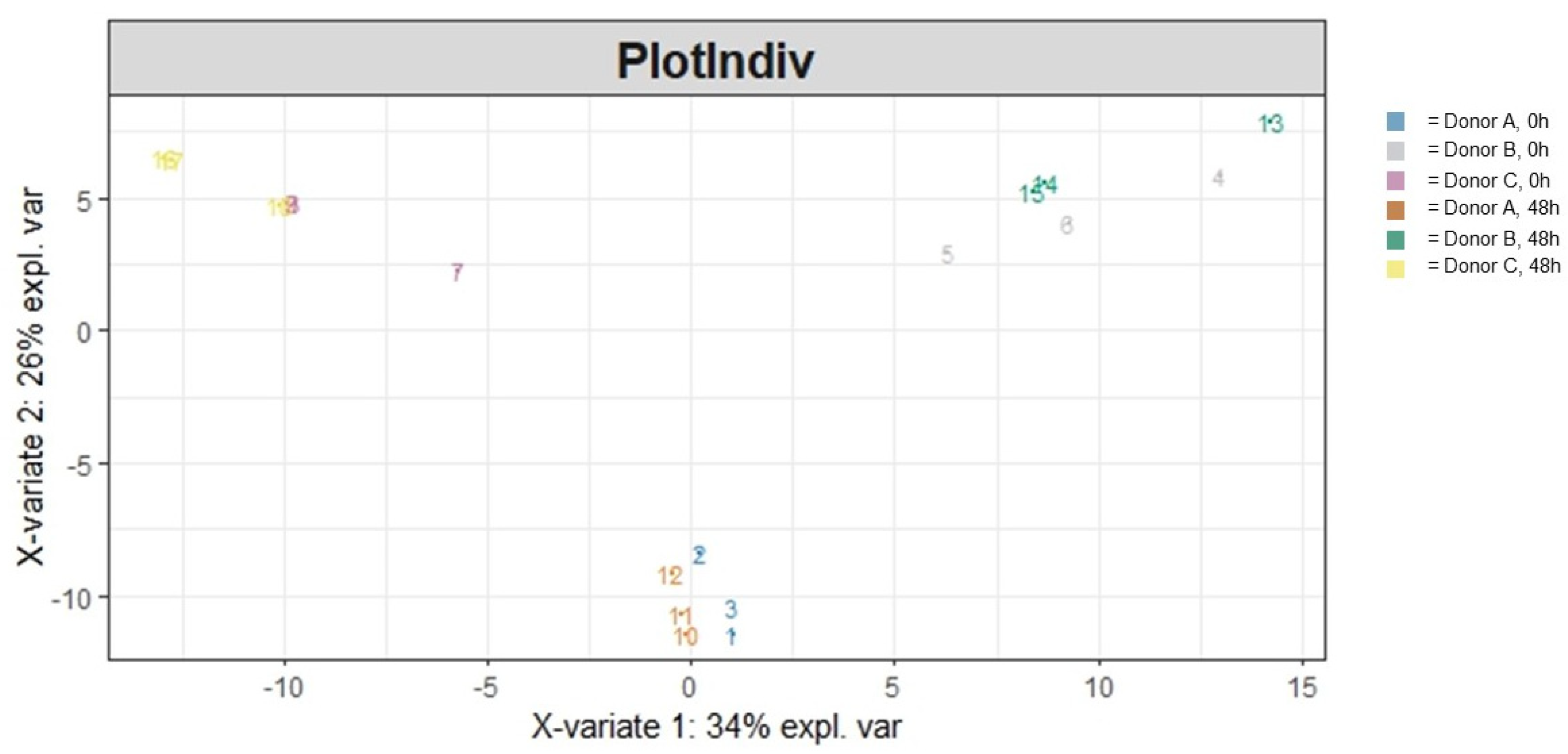
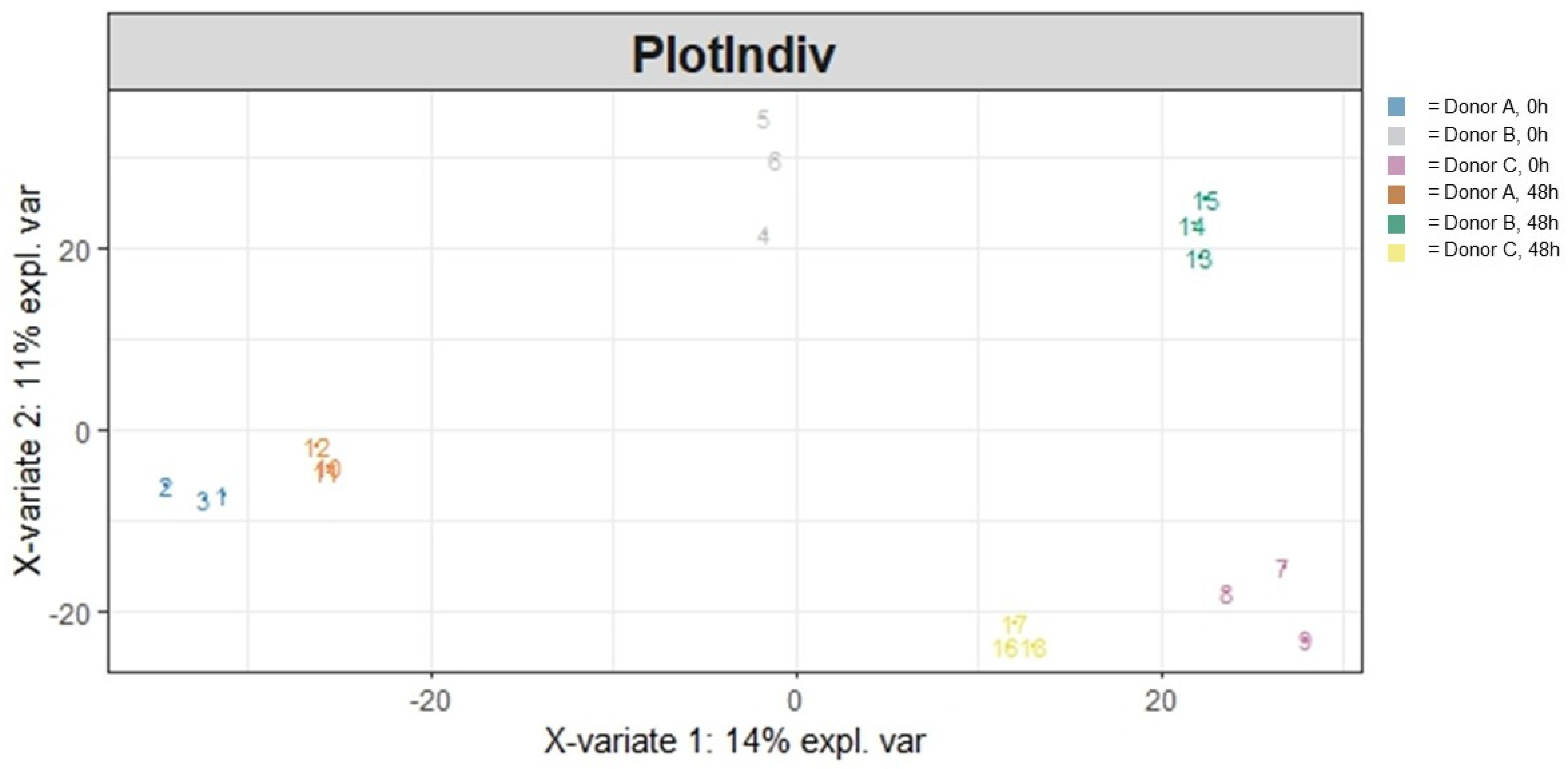
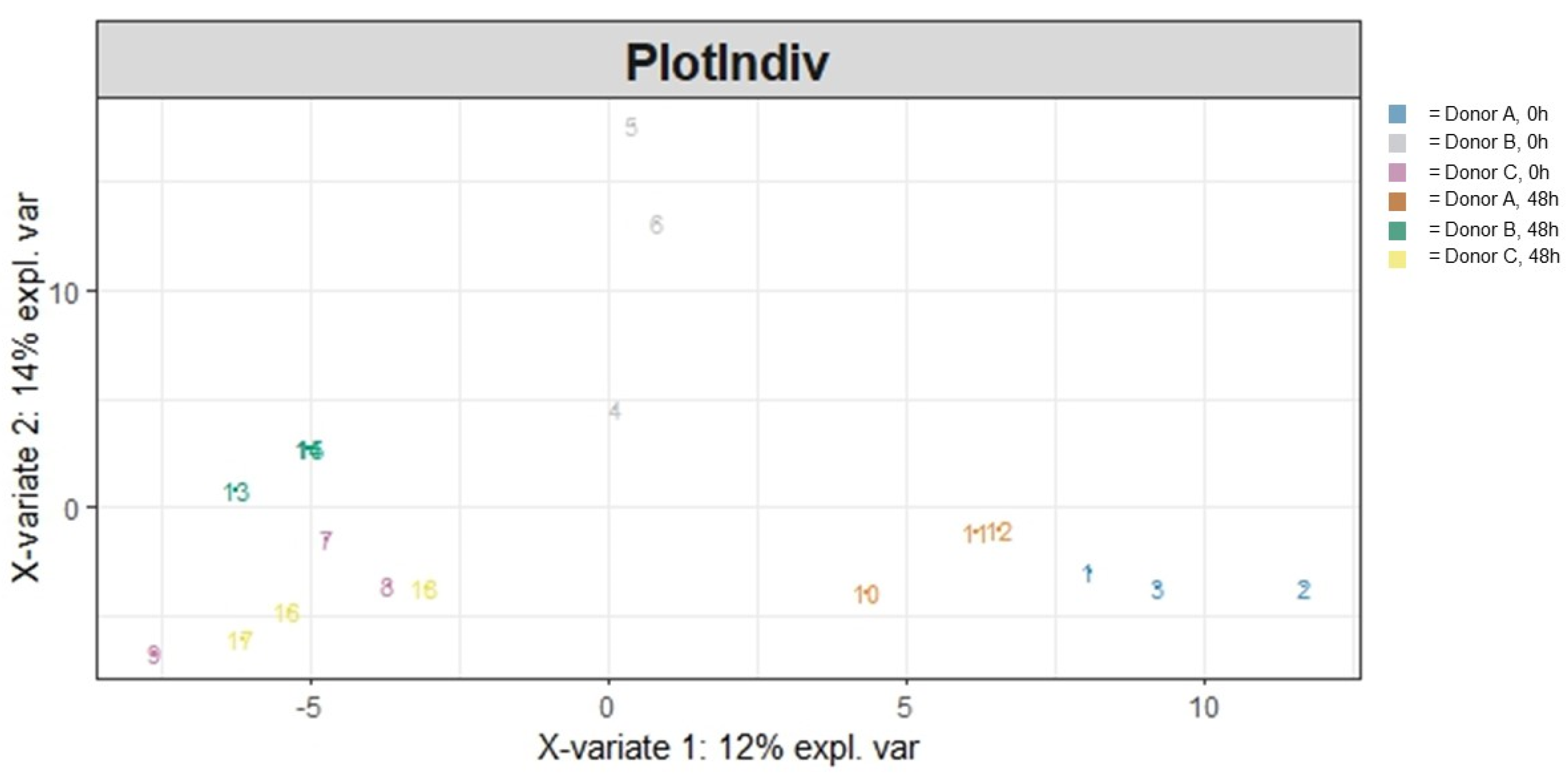
3.1.2. Beta Diversity
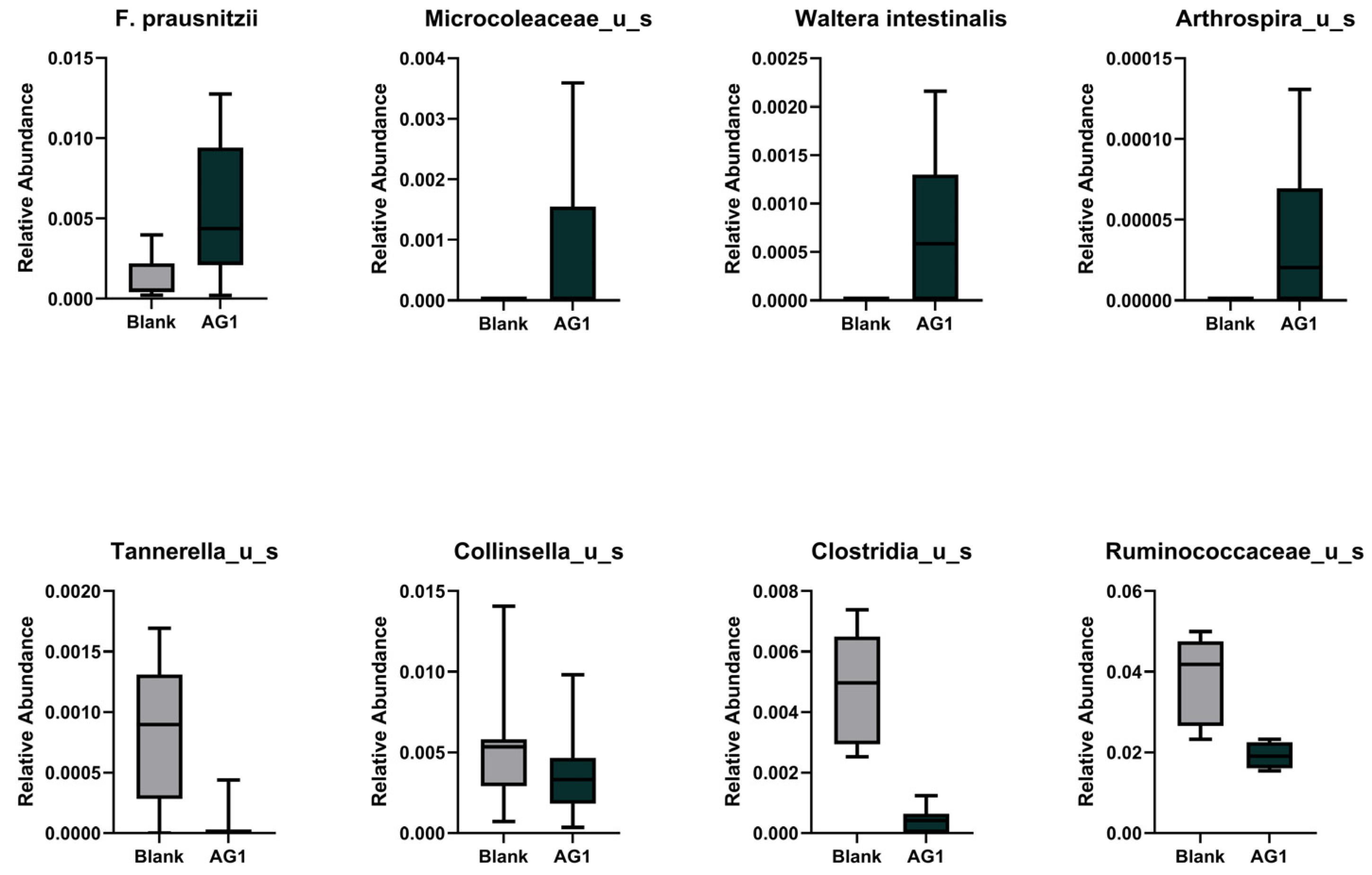
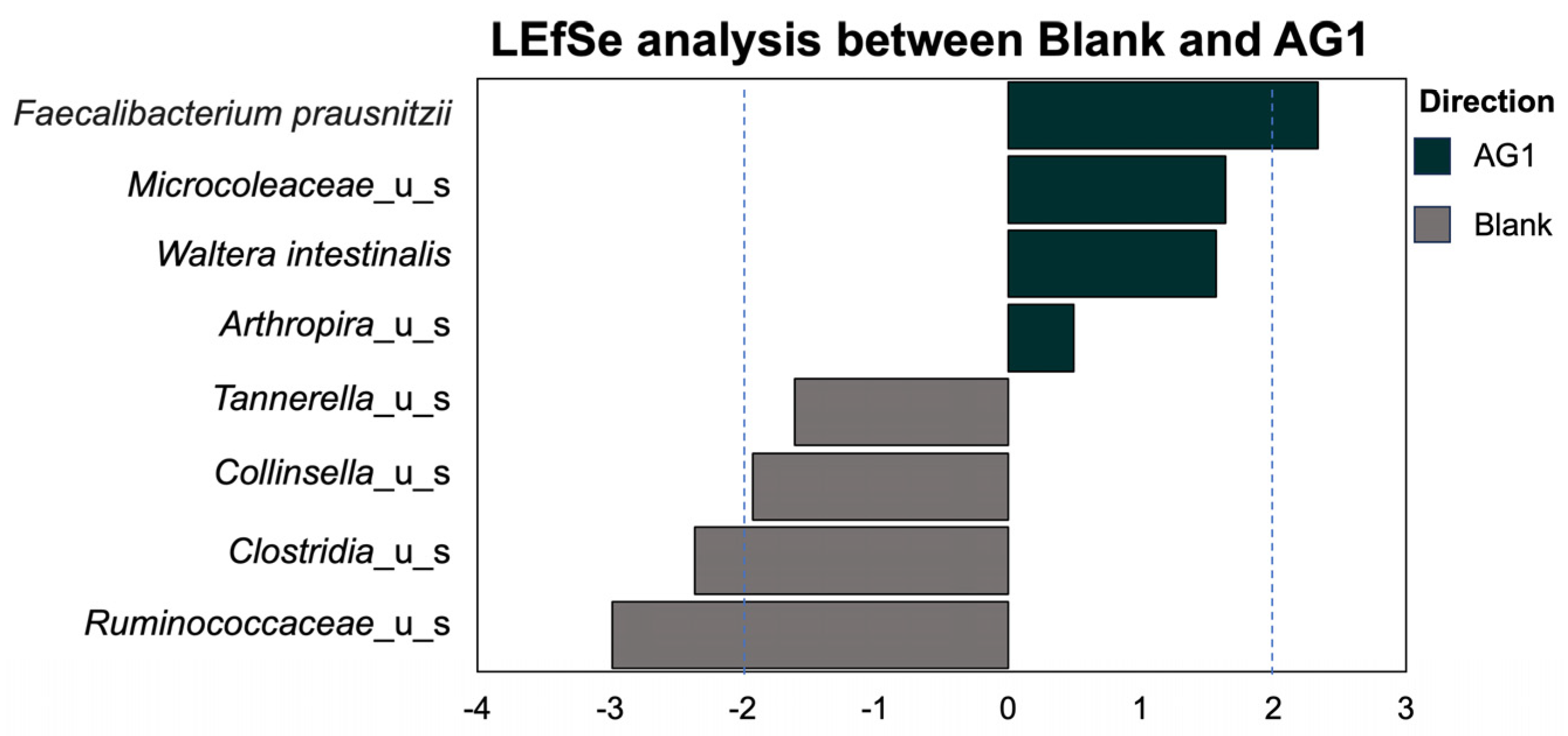
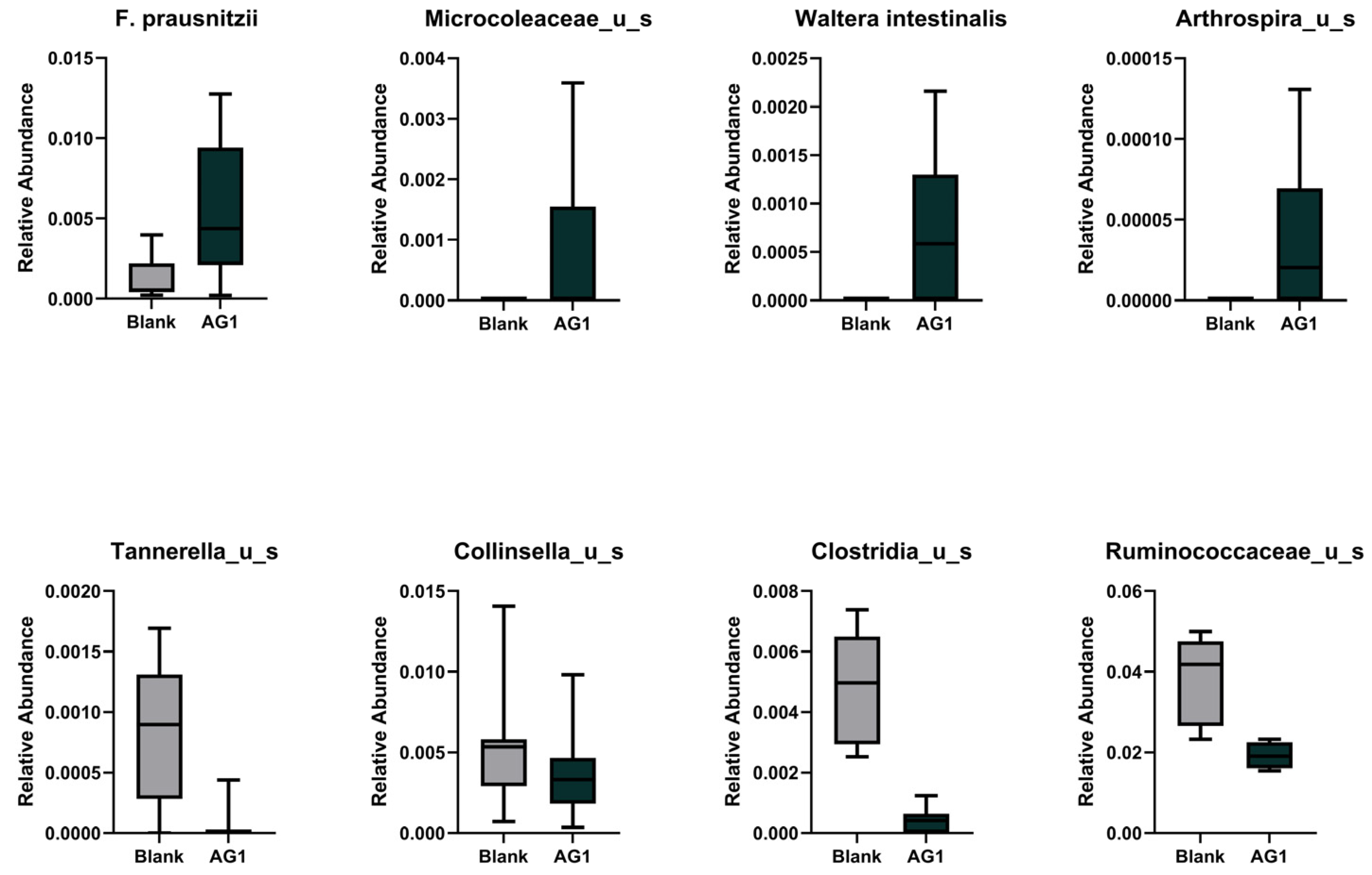
3.1.3. Specific Taxonomic Differences
3.2. Community Function
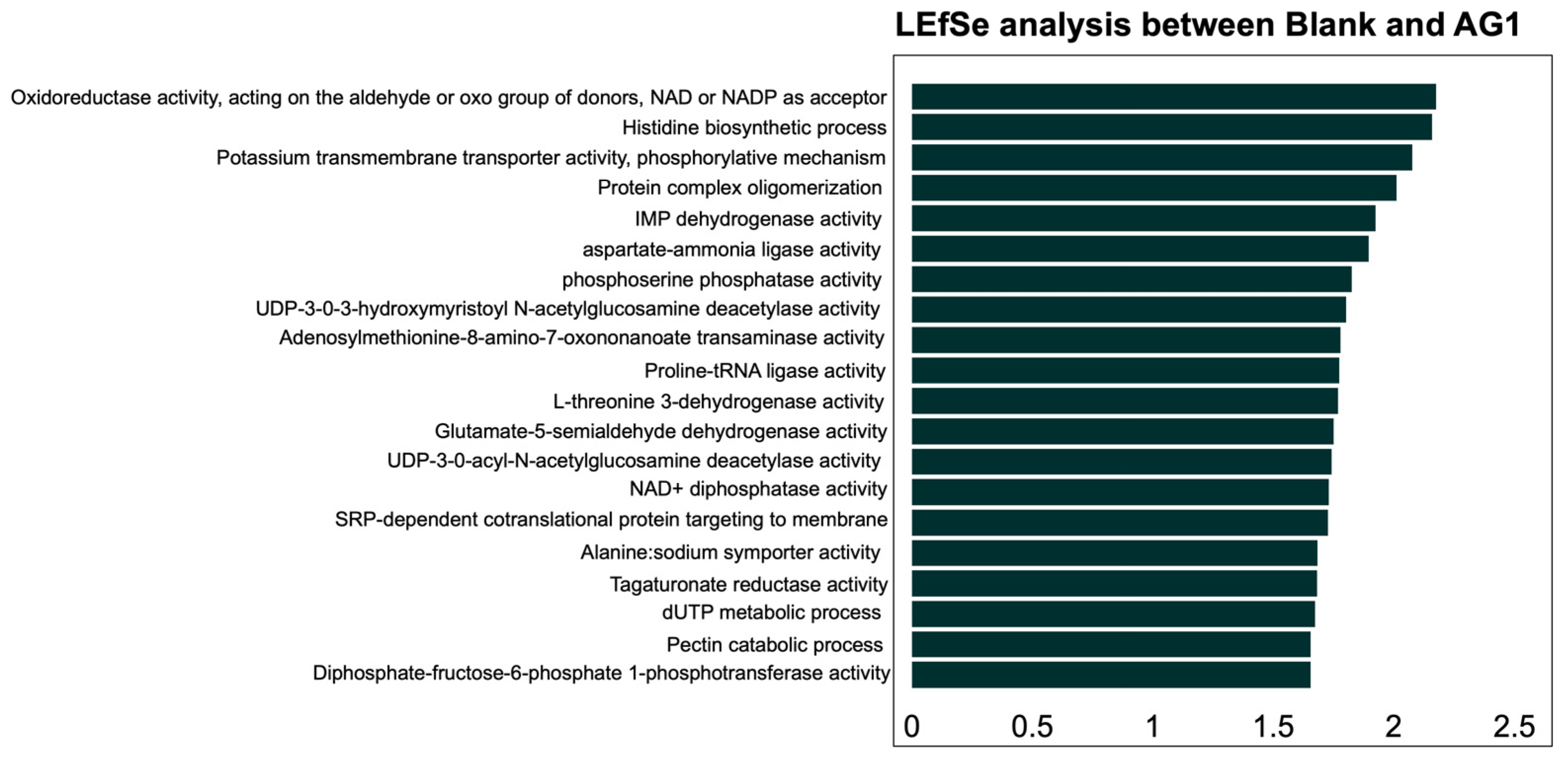
3.2.1. Community Functional Heterogeneity
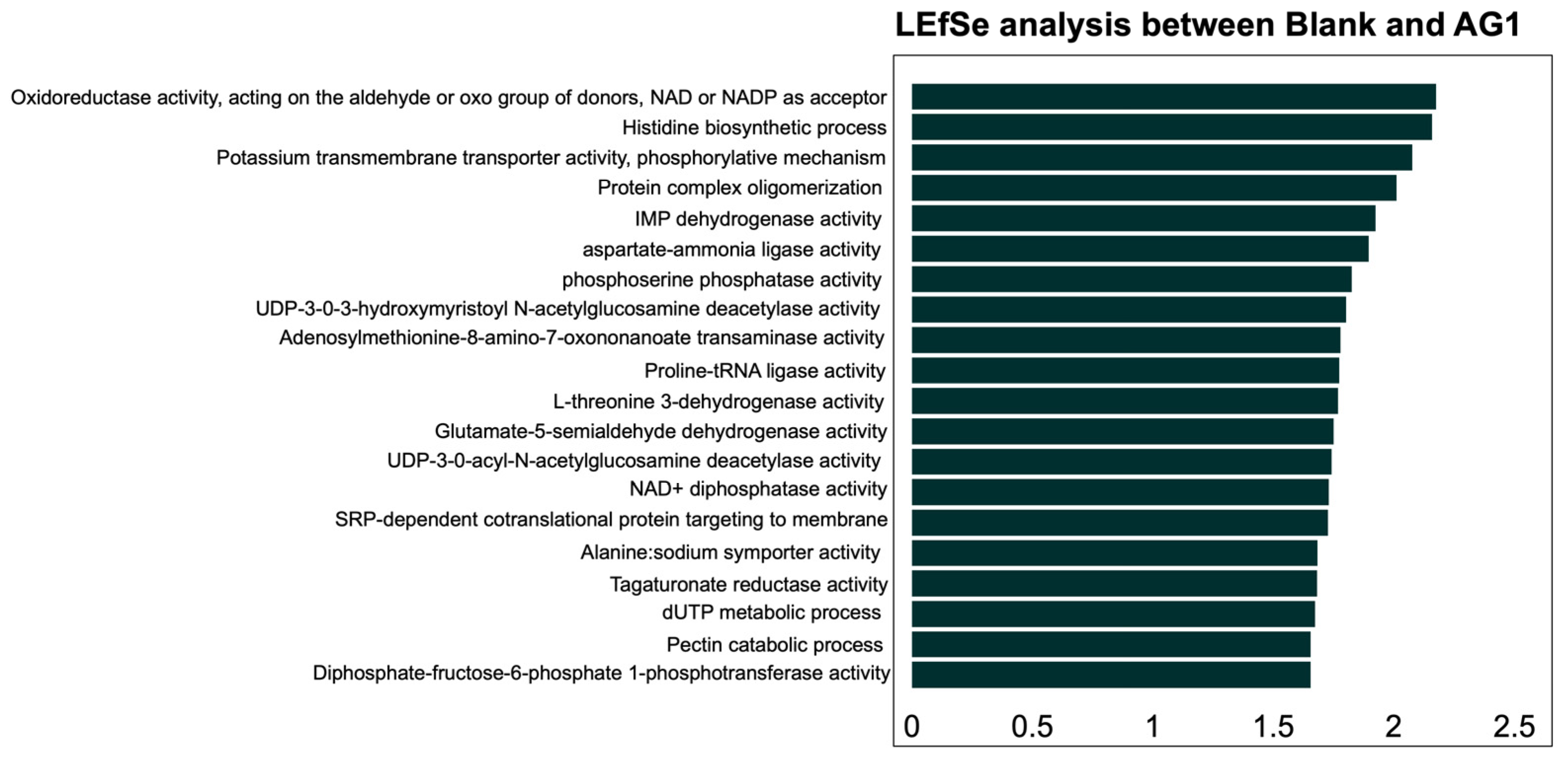
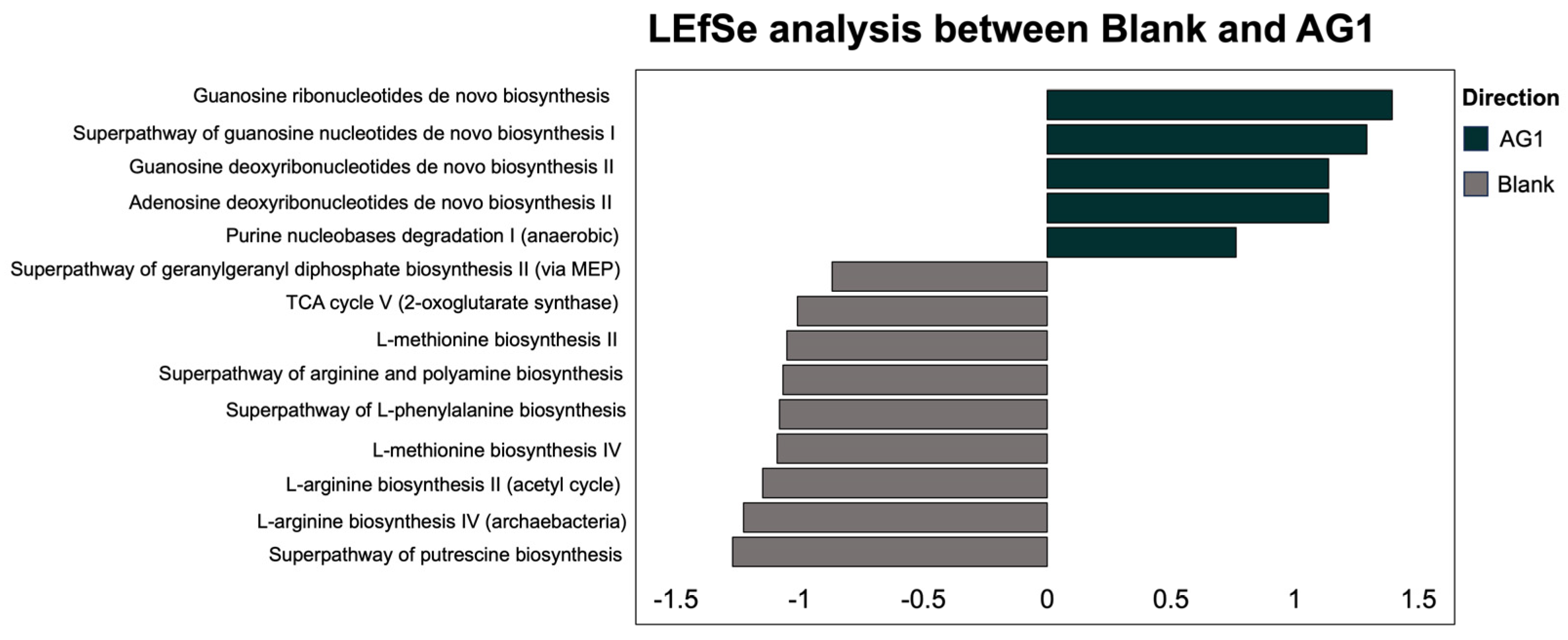
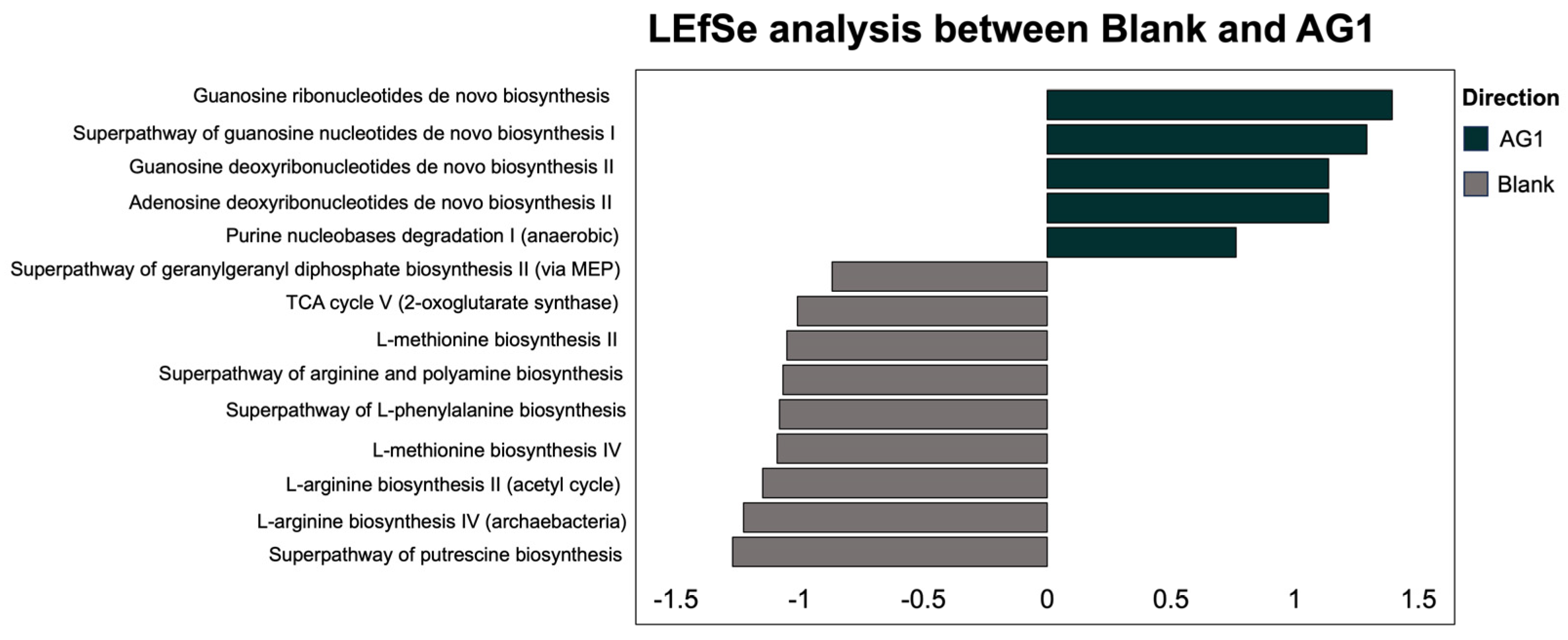
3.2.2. Specific Community Functional Pathways
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gibson, Y.; Roberfroid, M.B. Dietary Modulation of the Human Colonie Microbiota: Introducing the Concept of Prebiotics. J. Nutr. 1995, 125, 1401–1412. [Google Scholar] [CrossRef]
- Davani-Davari, D.; Negahdaripour, M.; Karimzadeh, I.; Seifan, M.; Mohkam, M.; Masoumi, S.J.; Berenjian, A.; Ghasemi, Y. Prebiotics: Definition, Types, Sources, Mechanisms, and Clinical Applications. Foods 2019, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Carrera-Quintanar, L.; Roa, R.I.L.; Quintero-Fabián, S.; Sánchez-Sánchez, M.A.; Vizmanos, B.; Ortuño-Sahagún, D. Phytochemicals That Influence Gut Microbiota as Prophylactics and for the Treatment of Obesity and Inflammatory Diseases. Mediat. Inflamm. 2018, 2018, 9734845. [Google Scholar] [CrossRef]
- Kan, J.; Wu, F.; Wang, F.; Zheng, J.; Cheng, J.; Li, Y.; Yang, Y.; Du, J. Phytonutrients: Sources, Bioavailability, Interaction with Gut Microbiota, and Their Impacts on Human Health. Front. Nutr. 2022, 9, 960309. [Google Scholar] [CrossRef] [PubMed]
- Plamada, D.; Vodnar, D.C. Polyphenols—Gut Microbiota Interrelationship: A Transition to a New Generation of Prebiotics. Nutrients 2022, 14, 137. [Google Scholar] [CrossRef] [PubMed]
- Korsholm, A.S.; Kjær, T.N.; Ornstrup, M.J.; Pedersen, S.B. Comprehensive Metabolomic Analysis in Blood, Urine, Fat, and Muscle in Men with Metabolic Syndrome: A Randomized, Placebo-Controlled Clinical Trial on the Effects of Resveratrol after Four Months’ Treatment. Int. J. Mol. Sci. 2017, 18, 554. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Kuo, H.C.; Hudlikar, R.; Sargsyan, D.; Li, S.; Wang, L.; Wu, R.; Kong, A.N. Gut Microbiota, Dietary Phytochemicals, and Benefits to Human Health. Curr. Pharmacol. Rep. 2019, 5, 332–344. [Google Scholar] [CrossRef]
- Santhiravel, S.; Bekhit, A.E.D.A.; Mendis, E.; Jacobs, J.L.; Dunshea, F.R.; Rajapakse, N.; Ponnampalam, E.N. The Impact of Plant Phytochemicals on the Gut Microbiota of Humans for a Balanced Life. Int. J. Mol. Sci. 2022, 23, 8124. [Google Scholar] [CrossRef]
- Luo, B.; Wen, Y.; Ye, F.; Wu, Y.; Li, N.; Farid, M.S.; Chen, Z.; El-Seedi, H.R.; Zhao, C. Bioactive Phytochemicals and Their Potential Roles in Modulating Gut Microbiota. J. Agric. Food Res. 2023, 12, 100583. [Google Scholar] [CrossRef]
- Kirby, T.O.; Townsend, J.R.; Sapp, P.A.; Govaert, M.; Duysburgh, C.; Marshall, T.M.; Marzorati, M.; Esposito, R. The Novel Synbiotic, AG1®, Increases Short-Chained Fatty Acid Production in the Simulator of Human Intestinal Microbial Ecosystem (SHIME) Model®. Nutraceuticals 2023, 3, 489–498. [Google Scholar] [CrossRef]
- Athletic Greens International Ingredients. Available online: https://drinkag1.com/about-ag1/ingredients/ctr?gclid=Cj0KCQiAy9msBhD0ARIsANbk0A9N8AyOuI_gpdv_zFiMOvlxRHdyo_hcPdwZw8-xOrpQBypgj7XlOyMaAiK0EALw_wcB&gclsrc=aw.ds&gad_source=1 (accessed on 7 December 2023).
- Wang, Y.; Wen, L.; Tang, H.; Qu, J.; Rao, B. Probiotics and Prebiotics as Dietary Supplements for the Adjunctive Treatment of Type 2 Diabetes. Pol. J. Microbiol. 2023, 72, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Latif, A.; Shehzad, A.; Niazi, S.; Zahid, A.; Ashraf, W.; Iqbal, M.W.; Rehman, A.; Riaz, T.; Aadil, R.M.; Khan, I.M.; et al. Probiotics: Mechanism of Action, Health Benefits and Their Application in Food Industries. Front. Microbiol. 2023, 14, 1216674. [Google Scholar] [CrossRef] [PubMed]
- Barone, M.; D’Amico, F.; Brigidi, P.; Turroni, S. Gut Microbiome–Micronutrient Interaction: The Key to Controlling the Bioavailability of Minerals and Vitamins? BioFactors 2022, 48, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cobas, A.E.; Gomez-Valero, L.; Buchrieser, C. Metagenomic Approaches in Microbial Ecology: An Update on Whole-Genome and Marker Gene Sequencing Analyses. Microb. Genom. 2020, 6, mgen000409. [Google Scholar] [CrossRef] [PubMed]
- Travis, J.; Lattimore, L.G.; Harvey, M.; Frey, T. NSF International’s Role in the Dietary Supplements and Nutraceuticals Industries. In Nutraceutical and Functional Food Regulations in the United States and around the World; Elsevier Inc.: Amsterdam, The Netherlands, 2019; pp. 147–158. [Google Scholar] [CrossRef]
- van de Wiele, T.; van den Abbeele, P.; Ossieur, W.; Possemiers, S.; Marzorati, M. The Simulator of the Human Intestinal Microbial Ecosystem (SHIME®). In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Springer International Publishing: Berlin/Heidelberg, Germany, 2015; pp. 305–317. ISBN 9783319161044. [Google Scholar]
- Barbier, F.F.; Chabikwa, T.G.; Ahsan, M.U.; Cook, S.E.; Powell, R.; Tanurdzic, M.; Beveridge, C.A. A Phenol/Chloroform-Free Method to Extract Nucleic Acids from Recalcitrant, Woody Tropical Species for Gene Expression and Sequencing. Plant Methods 2019, 15, 62. [Google Scholar] [CrossRef]
- Joint Genome Institute BBDuk Guide. 2021. Available online: https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbduk-guide/ (accessed on 7 December 2023).
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-Level Functional Profiling of Metagenomes and Metatranscriptomes. Nat. Methods 2018, 15, 962–968. [Google Scholar] [CrossRef]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; et al. The MetaCyc Database of Metabolic Pathways and Enzymes and the BioCyc Collection of Pathway/Genome Databases. Nucleic Acids Res. 2008, 36, D623–D631. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 15 November 2023).
- Cryan, J.F.; O’Riordan, K.J.; Sandhu, K.; Peterson, V.; Dinan, T.G. The Gut Microbiome in Neurological Disorders. Lancet Neurol. 2020, 19, 179–194. [Google Scholar] [CrossRef]
- Denslow, J.S. Patterns of Plant Species Diversity during Succession under Different Disturbance Regimes. Oecologia 1980, 46, 18–21. [Google Scholar] [CrossRef]
- Li, Y.; Sun, X.; Zhang, M.; Khan, A.; Sun, W. Dominant Role of Rare Bacterial Taxa Rather than Abundant Taxa in Driving the Tailing Primary Succession. J. Hazard. Mater. 2024, 462, 132807. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.; Yue, M.; Liu, X.; Guo, Y.; Wang, M.; Xu, J.; Zhang, C.; Chen, Y.; Zhang, L.; Zhang, R. Patterns of Taxonomic, Phylogenetic Diversity during a Long-Term Succession of Forest on the Loess Plateau, China: Insights into Assembly Process. Sci. Rep. 2016, 6, 27087. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yin, A.; Li, H.; Wang, R.; Wu, G.; Shen, J.; Zhang, M.; Wang, L.; Hou, Y.; Ouyang, H.; et al. Dietary Modulation of Gut Microbiota Contributes to Alleviation of Both Genetic and Simple Obesity in Children. EBioMedicine 2015, 2, 968–984. [Google Scholar] [CrossRef] [PubMed]
- Matsuki, T.; Tajima, S.; Hara, T.; Yahagi, K.; Ogawa, E.; Kodama, H. Infant Formula with Galacto-Oligosaccharides (OM55N) Stimulates the Growth of Indigenous Bifidobacteria in Healthy Term Infants. Benef. Microbes 2016, 7, 453–461. [Google Scholar] [CrossRef]
- Mahalak, K.K.; Firrman, J.; Narrowe, A.B.; Hu, W.; Jones, S.M.; Bittinger, K.; Moustafa, A.M.; Liu, L.S. Fructooligosaccharides (FOS) Differentially Modifies the in Vitro Gut Microbiota in an Age-Dependent Manner. Front. Nutr. 2023, 9, 1058910. [Google Scholar] [CrossRef]
- You, S.; Ma, Y.; Yan, B.; Pei, W.; Wu, Q.; Ding, C.; Huang, C. The Promotion Mechanism of Prebiotics for Probiotics: A Review. Front. Nutr. 2022, 9, 1000517. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Fan, C.; Zhang, C.; von Gadow, K.; Fan, X. How Beta Diversity and the Underlying Causes Vary with Sampling Scales in the Changbai Mountain Forests. Ecol. Evol. 2017, 7, 10116–10123. [Google Scholar] [CrossRef]
- Fitzgerald, C.B.; Shkoporov, A.N.; Sutton, T.D.S.; Chaplin, A.V.; Velayudhan, V.; Ross, R.P.; Hill, C. Comparative Analysis of Faecalibacterium Prausnitzii Genomes Shows a High Level of Genome Plasticity and Warrants Separation into New Species-Level Taxa. BMC Genom. 2018, 19, 931. [Google Scholar] [CrossRef]
- Bai, Z.; Zhang, N.; Jin, Y.; Chen, L.; Mao, Y.; Sun, L.; Fang, F.; Liu, Y.; Han, M.; Li, G. Comprehensive Analysis of 84 Faecalibacterium Prausnitzii Strains Uncovers Their Genetic Diversity, Functional Characteristics, and Potential Risks. Front. Cell. Infect. Microbiol. 2022, 12, 919701. [Google Scholar] [CrossRef]
- Parsaei, M.; Sarafraz, N.; Moaddab, S.Y.; Ebrahimzadeh Leylabadlo, H. The Importance of Faecalibacterium Prausnitzii in Human Health and Diseases. New Microbes New Infect. 2021, 43, 100928. [Google Scholar] [CrossRef]
- Murakami, R.; Hashikura, N.; Yoshida, K.; Xiao, J.-Z.; Odamaki, T. Growth-Promoting Effect of Alginate on Faecalibacterium Prausnitzii through Cross-Feeding with Bacteroides. Food Res. Int. 2021, 144, 110326. [Google Scholar] [CrossRef] [PubMed]
- Wylensek, D.; Hitch, T.C.A.; Riedel, T.; Afrizal, A.; Kumar, N.; Wortmann, E.; Liu, T.; Devendran, S.; Lesker, T.R.; Hernández, S.B.; et al. A Collection of Bacterial Isolates from the Pig Intestine Reveals Functional and Taxonomic Diversity. Nat. Commun. 2020, 11, 6389. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Hu, T.; Chen, J.; Liang, H.; Zhou, J.; Wu, Z.; Ye, C.; Jin, X.; Xu, X.; Zhang, W.; et al. The Genomic Landscape of Reference Genomes of Cultivated Human Gut Bacteria. Nat. Commun. 2023, 14, 1663. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.L.; Wilkins, T.D. Clostridia: Sporeforming Anaerobic Bacilli. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Lv, X.-C.; Chen, M.; Huang, Z.-R.; Guo, W.-L.; Ai, L.-Z.; Bai, W.-D.; Yu, X.-D.; Liu, Y.-L.; Rao, P.-F.; Ni, L. Potential Mechanisms Underlying the Ameliorative Effect of Lactobacillus Paracasei FZU103 on the Lipid Metabolism in Hyperlipidemic Mice Fed a High-Fat Diet. Food Res. Int. 2021, 139, 109956. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Arango, L.F.; Barrett, H.L.; Wilkinson, S.A.; Callaway, L.K.; McIntyre, H.D.; Morrison, M.; Dekker Nitert, M. Low Dietary Fiber Intake Increases Collinsella Abundance in the Gut Microbiota of Overweight and Obese Pregnant Women. Gut Microbes 2018, 9, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fåk, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Bäckhed, F.; Nielsen, J. Symptomatic Atherosclerosis Is Associated with an Altered Gut Metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An Expansion of Rare Lineage Intestinal Microbes Characterizes Rheumatoid Arthritis. Genome Med. 2016, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Cammann, D.; Lu, Y.; Cummings, M.J.; Zhang, M.L.; Cue, J.M.; Do, J.; Ebersole, J.; Chen, X.; Oh, E.C.; Cummings, J.L.; et al. Genetic Correlations between Alzheimer’s Disease and Gut Microbiome Genera. Sci. Rep. 2023, 13, 5258. [Google Scholar] [CrossRef]
- Oluwagbemigun, K.; Schnermann, M.E.; Schmid, M.; Cryan, J.F.; Nöthlings, U. A Prospective Investigation into the Association between the Gut Microbiome Composition and Cognitive Performance among Healthy Young Adults. Gut Pathog. 2022, 14, 15. [Google Scholar] [CrossRef]
- Rinninella, E.; Raoul, P.; Cintoni, M.; Franceschi, F.; Miggiano, G.A.D.; Gasbarrini, A.; Mele, M.C. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms 2019, 7, 14. [Google Scholar] [CrossRef]
- Davenport, E.R.; Mizrahi-Man, O.; Michelini, K.; Barreiro, L.B.; Ober, C.; Gilad, Y. Seasonal Variation in Human Gut Microbiome Composition. PLoS ONE 2014, 9, 90731. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the Adult Digestive Tract Bacterial Microbiome Based on Seven Mouth Surfaces, Tonsils, Throat and Stool Samples. Genome Biol. 2012, 13, R42. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.J.; Zheng, J.J.; Kang, J.W.; Saboe, A.; Knights, D.; Zivkovic, A.M. A Guide to Diet-Microbiome Study Design. Front. Nutr. 2020, 7, 79. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Sum of Squares | R2 | F | Adjusted p-Value |
|---|---|---|---|---|
| Treatment | 0.2342 | 0.06964 | 14.4451 | <0.001 |
| Donor | 2.6596 | 0.79093 | 82.0295 | <0.001 |
| Treatment/Donor | 0.2743 | 0.08158 | 8.4608 | <0.001 |
| Residual | 0.4689 | 0.13943 |
| Variable | Sum of Squares | R2 | F | Adjusted p-Value |
|---|---|---|---|---|
| Treatment | 0.017889 | 0.07066 | 2.2808 | 0.034 |
| Donor | 0.110552 | 0.43667 | 7.0477 | <0.001 |
| Treatment/Donor | 0.030612 | 0.12092 | 1.9515 | 0.025 |
| Residual | 0.094118 | 0.37176 |
| Variable | Sum of Squares | R2 | F | Adjusted p-Value |
|---|---|---|---|---|
| Treatment | 0.12389 | 0.08555 | 1.7761 | 0.051 |
| Donor | 0.35297 | 0.24375 | 2.5301 | <0.001 |
| Treatment/Donor | 0.13418 | 0.09266 | 0.9618 | 0.496 |
| Residual | 0.83704 | 0.57803 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirby, T.O.; Sapp, P.A.; Townsend, J.R.; Govaert, M.; Duysburgh, C.; Marzorati, M.; Marshall, T.M.; Esposito, R. AG1® Induces a Favorable Impact on Gut Microbial Structure and Functionality in the Simulator of Human Intestinal Microbial Ecosystem® Model. Curr. Issues Mol. Biol. 2024, 46, 557-569. https://doi.org/10.3390/cimb46010036
Kirby TO, Sapp PA, Townsend JR, Govaert M, Duysburgh C, Marzorati M, Marshall TM, Esposito R. AG1® Induces a Favorable Impact on Gut Microbial Structure and Functionality in the Simulator of Human Intestinal Microbial Ecosystem® Model. Current Issues in Molecular Biology. 2024; 46(1):557-569. https://doi.org/10.3390/cimb46010036
Chicago/Turabian StyleKirby, Trevor O., Philip A. Sapp, Jeremy R. Townsend, Marlies Govaert, Cindy Duysburgh, Massimo Marzorati, Tess M. Marshall, and Ralph Esposito. 2024. "AG1® Induces a Favorable Impact on Gut Microbial Structure and Functionality in the Simulator of Human Intestinal Microbial Ecosystem® Model" Current Issues in Molecular Biology 46, no. 1: 557-569. https://doi.org/10.3390/cimb46010036
APA StyleKirby, T. O., Sapp, P. A., Townsend, J. R., Govaert, M., Duysburgh, C., Marzorati, M., Marshall, T. M., & Esposito, R. (2024). AG1® Induces a Favorable Impact on Gut Microbial Structure and Functionality in the Simulator of Human Intestinal Microbial Ecosystem® Model. Current Issues in Molecular Biology, 46(1), 557-569. https://doi.org/10.3390/cimb46010036