
Genomic Fabrics of the Excretory System’s Functional Pathways Remodeled in Clear Cell Renal Cell Carcinoma



Abstract
1. Introduction
Limits of the Gene Biomarker Paradigm in Cancer Diagnostics and Therapy
2. Materials and Methods
2.1. The Best Choice of Tissue Samples
2.2. Data Filtering and Normalization
2.3. Independent Characteristics of Gene Expression
2.3.1. Normalized Average Expression Level
2.3.2. Relative Expression Variability
2.3.3. Expression Coordination
2.3.4. Topology of the Transcriptome and the Gene Master Regulator
2.4. Transcriptome Alteration in Cancer
2.4.1. Measures of Expression Regulation
2.4.2. Regulation of the Control of Transcript Abundance
2.4.3. Regulation of Expression Coordination
2.4.4. The Transcriptomic Distance
2.5. Functional Pathways
3. Results
3.1. The Global Picture
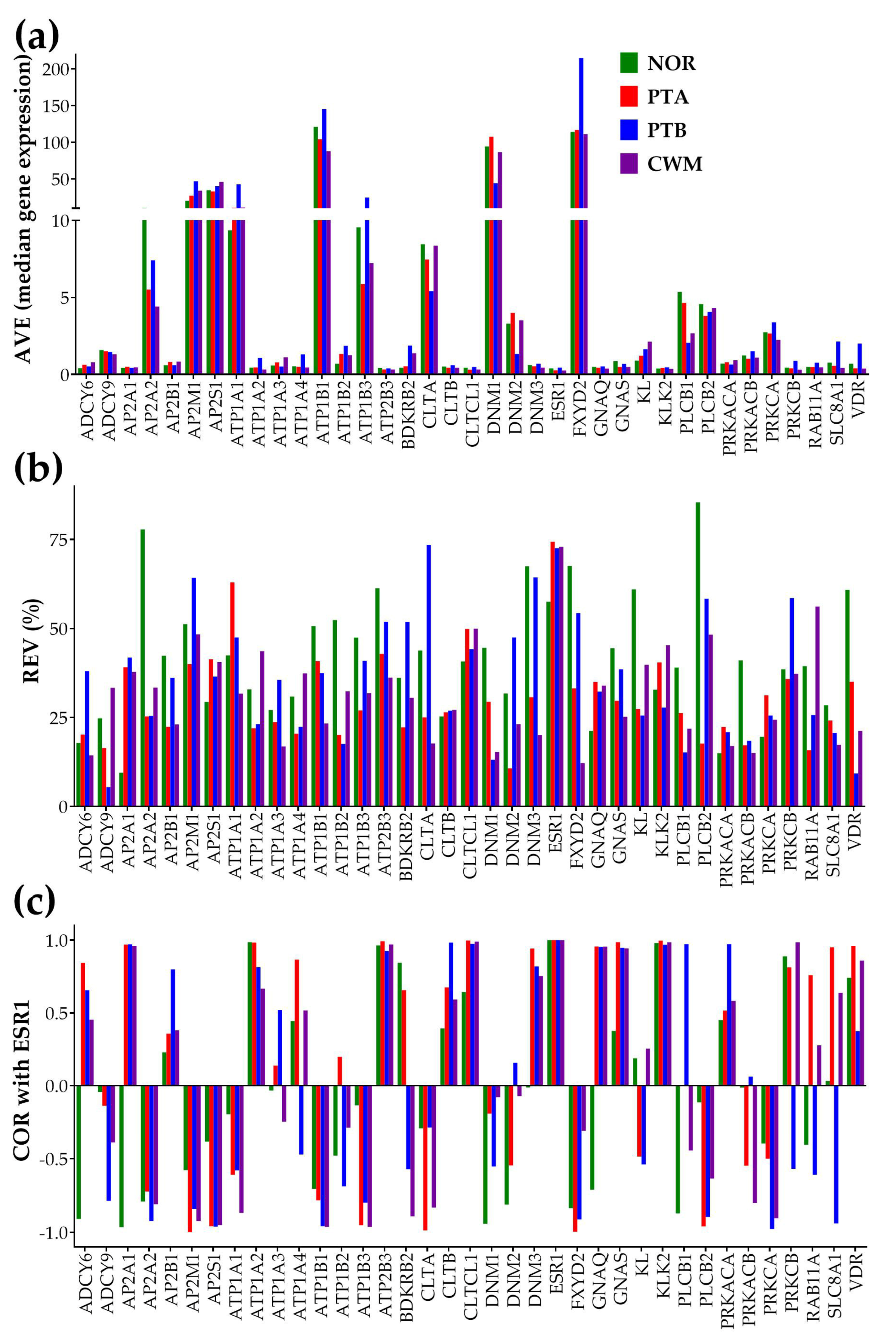
3.2. Independent Characteristics of Gene Expression
3.3. ccRCC Changed the Gene Hierarchy
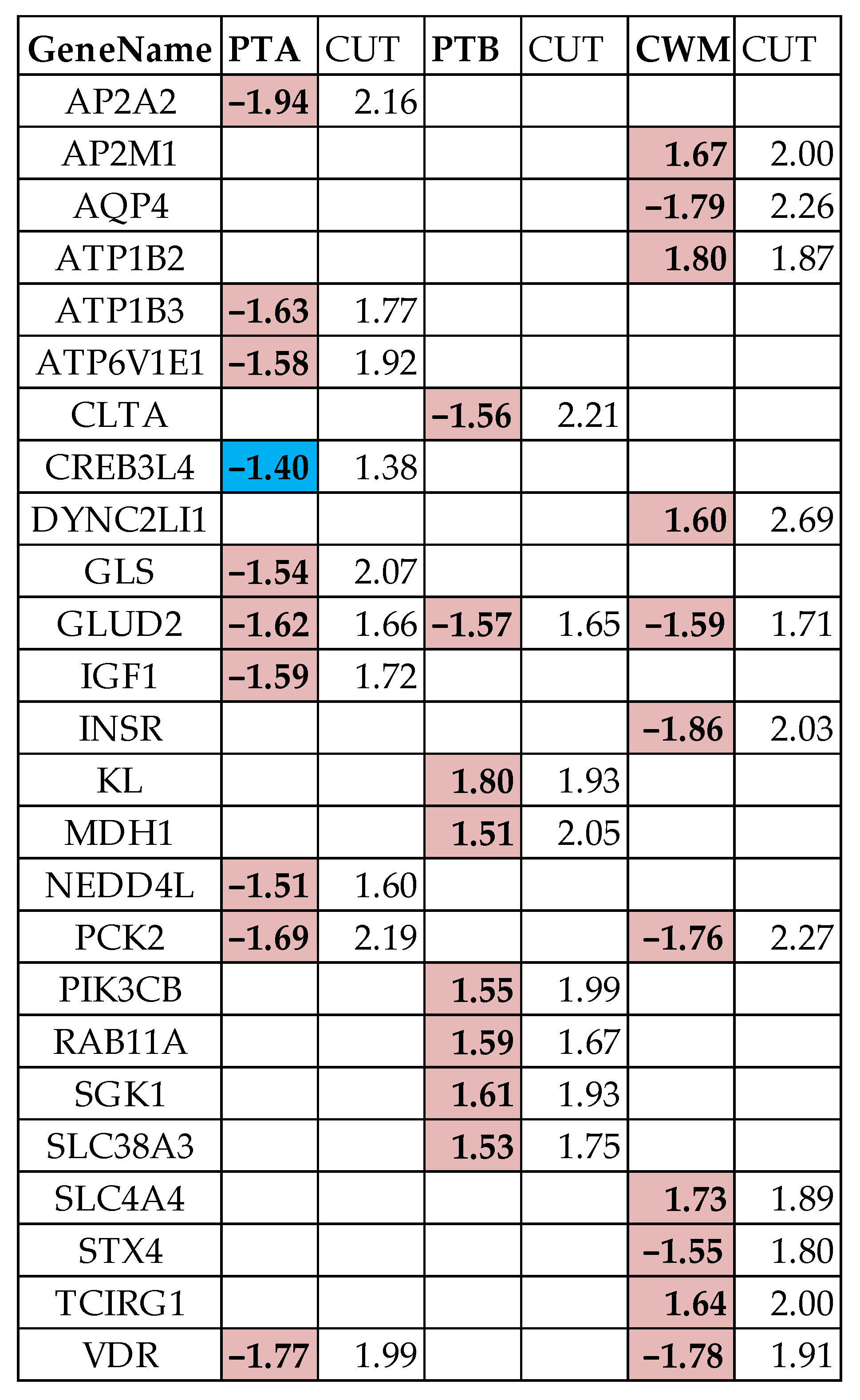
3.4. Measures of Individual Gene Regulation
3.5. Overall Regulation of the Excretory Pathways
3.6. False Hits
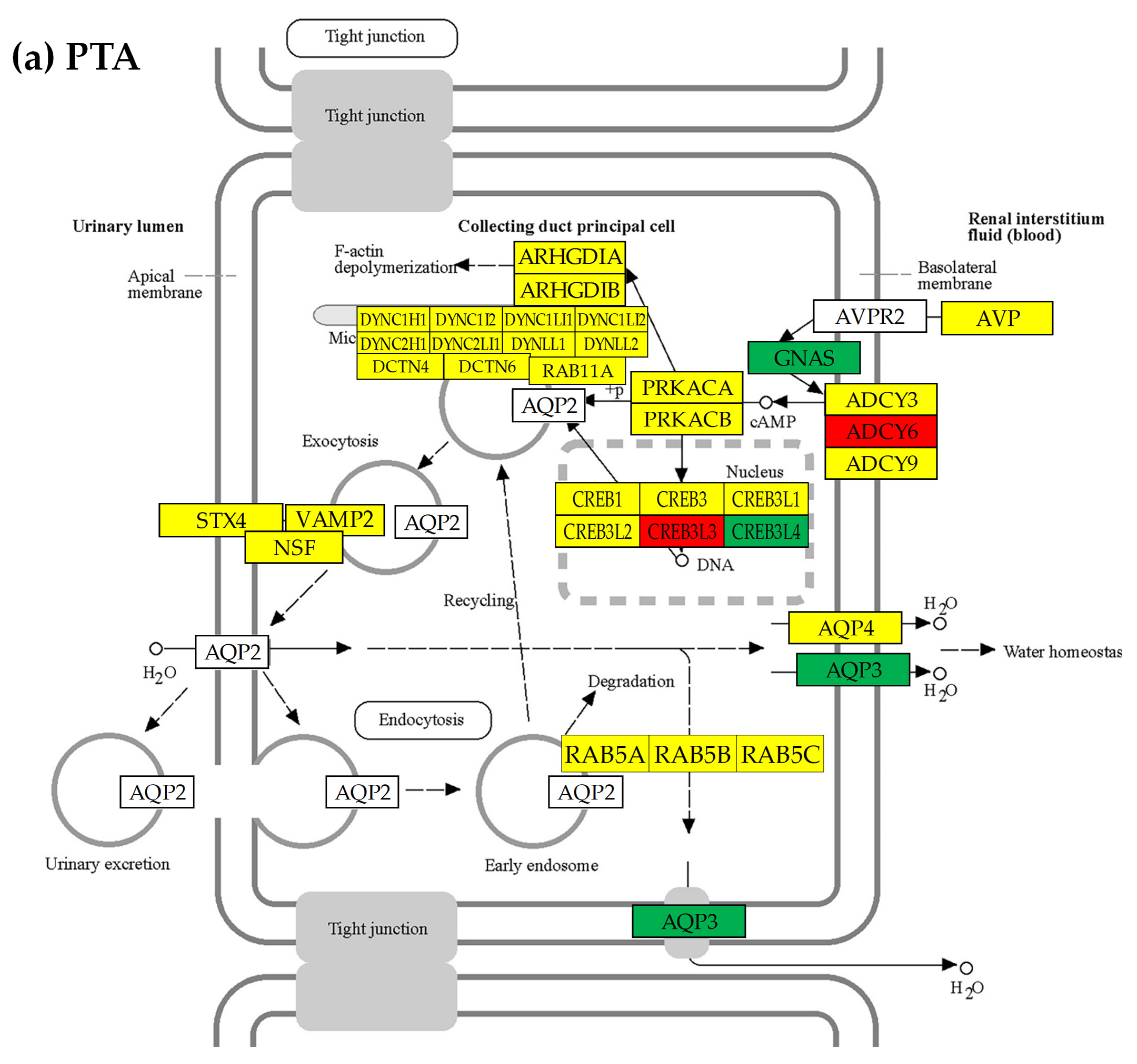
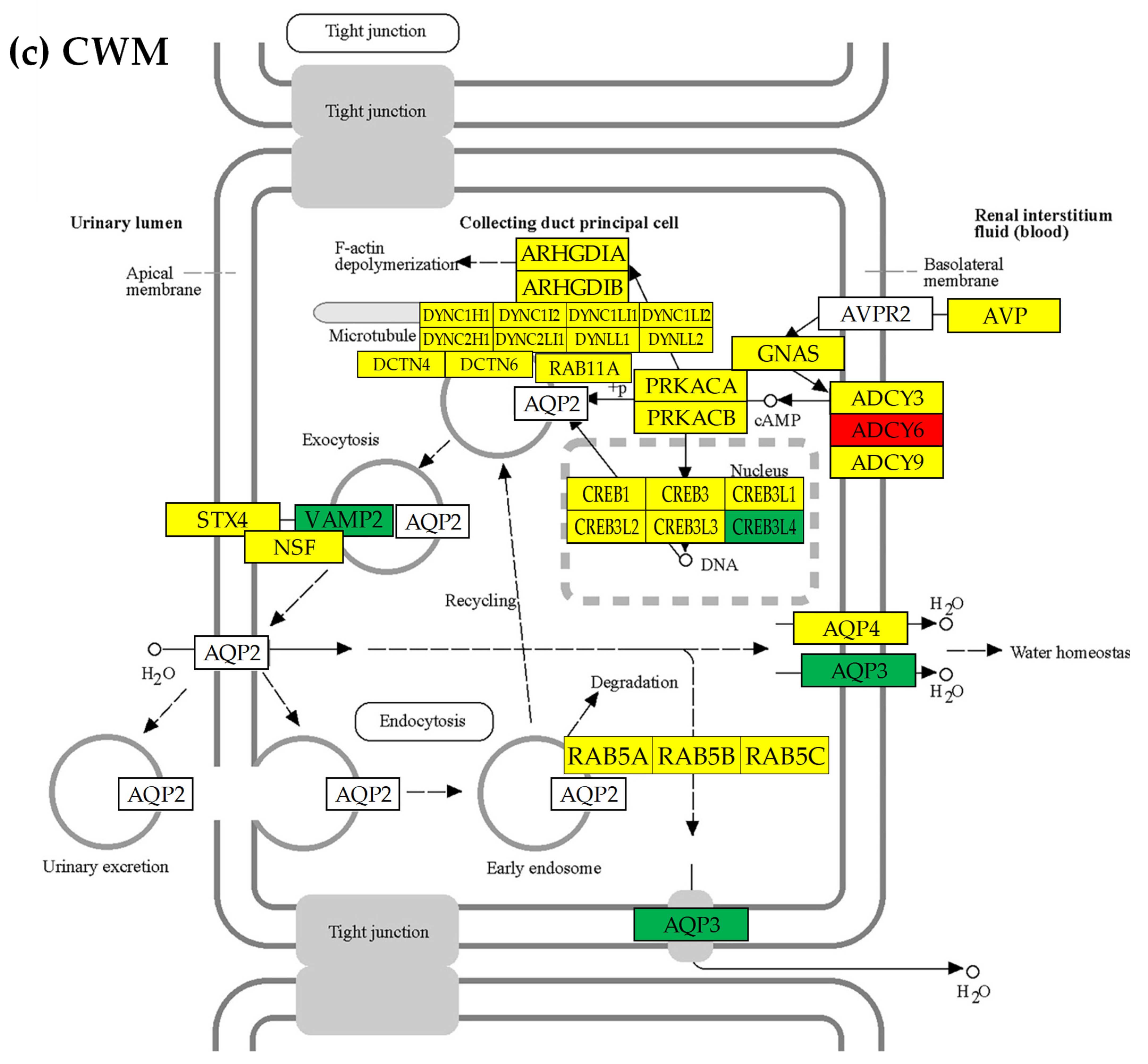
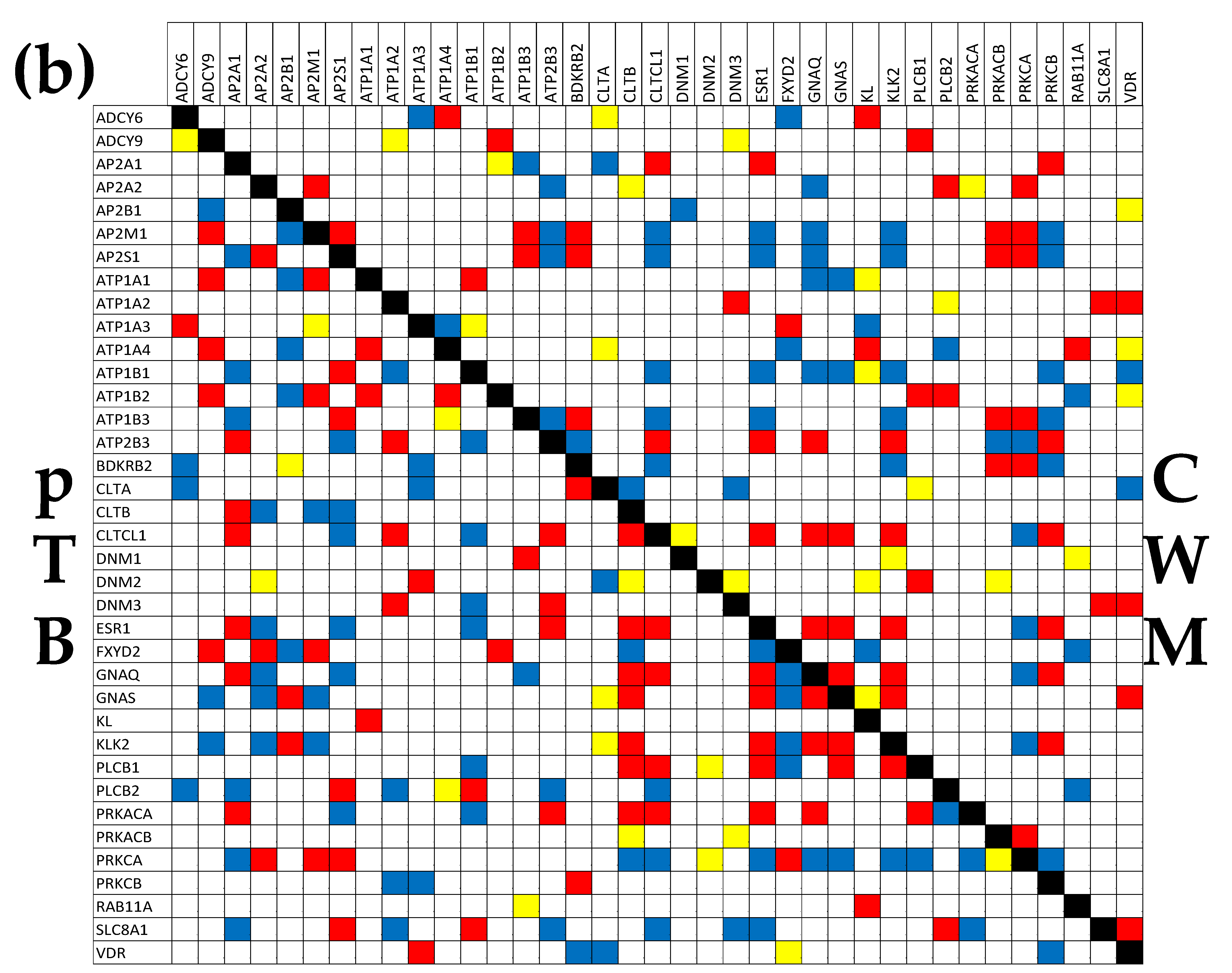
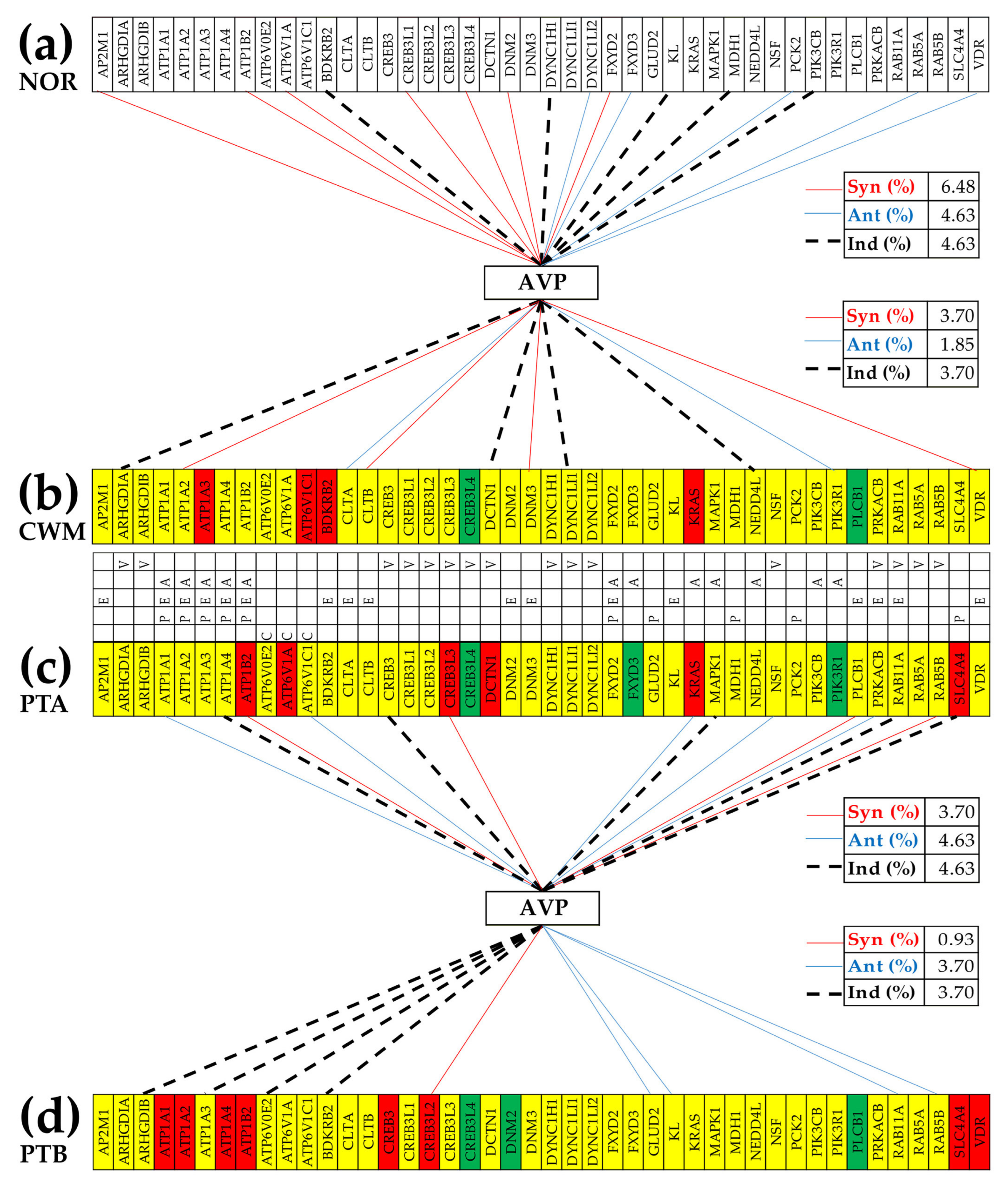
3.7. Location of the Regulated Genes in the Excretory System’s Functional Pathways
3.8. Tumor Heterogeneity of the Transcriptomic Networks
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
- 1.
- Normalized gene expression levels in the biological replica “k” in condition “c”:ai(c;k) is the sum of the net fluorescences of all microarray spots probing gene “i” in the biological replica “k” of condition “c”.
- 2.
- Relative expression variation:and χ2 is the chi-square score with β probability for r degrees of freedom.
- 3.
- Transcriptome configuration function:where A1, …, AN are the probabilities of each configuration of gene clustering, and f2, f3, …, fN are distribution functions symmetrical to the permutation(s) of the genes. The above expansion satisfies the following conditions:
- 4.
- Pair-wise correlation of gene expression levels:
- 5.
- Gene Commanding Height:
- 6.
- Statistically significant regulation of the expression level:
- 7.
- Weighted Individual (Gene) Regulation:
- 8.
- Weighted Pathway Regulation:
- 9.
- Relative Expression Control:
- 10.
- Regulation of the Expression Control:
- 11.
- Regulation of the Expression Coordination:
- 12.
- Transcriptomic Distance:
References
- Cancer Treatment Options at Houston Methodist Organization. Available online: https://www.houstonmethodist.org/cancer/treatment-options/ (accessed on 3 May 2023).
- Pecoraro, A.; Campi, R.; Marchioni, M. European Association of Urology Young Academic Urologists Renal Cancer Working Group. Techniques and outcomes of percutaneous tumour ablation for small renal masses. Curr. Opin. Urol. 2023, 33, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Lanza, C.; Carriero, S.; Ascenti, V.; Tintori, J.; Ricapito, F.; Lavorato, R.; Biondetti, P.; Angileri, S.A.; Piacentino, F.; Fontana, F.; et al. Percutaneous Application of High Power Microwave Ablation with 150 W for the Treatment of Tumors in Lung, Liver, and Kidney: A Preliminary Experience. Technol. Cancer Res. Treat. 2023, 22, 15330338231185277. [Google Scholar] [CrossRef] [PubMed]
- Key Statistics about Kidney Cancer. Available online: https://www.cancer.org/cancer/kidney-cancer.html (accessed on 6 September 2023).
- Mieville, V.; Griffioen, A.W.; Benamran, D.; Nowak-Sliwinska, P. Advanced in vitro models for renal cell carcinoma therapy design. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2023, 1878, 188942. [Google Scholar] [CrossRef] [PubMed]
- Dahle, D.O.; Skauby, M.; Langberg, C.W.; Brabrand, K.; Wessel, N.; Midtvedt, K. Renal Cell Carcinoma and Kidney Transplantation: A Narrative Review. Transplantation 2022, 106, e52–e63. [Google Scholar] [CrossRef]
- Gong, Y.; Pang, H.; Yu, Z.; Wang, X.; Li, P.; Zhang, Q. Construction of inflammatory associated risk gene prognostic model of NSCLC and its correlation with chemotherapy sensitivity. Ann. Med. 2023, 55, 2200034. [Google Scholar] [CrossRef]
- Hara, Y.; Shiba, N.; Yoshida, K.; Yamato, G.; Kaburagi, T.; Shiraishi, Y.; Ohki, K.; Shiozawa, Y.; Kawamura, M.; Kawasaki, H.; et al. TP53 and RB1 alterations characterize poor prognostic subgroups in pediatric acute myeloid leukemia. Genes Chromosomes Cancer 2023, 62, 412–422. [Google Scholar] [CrossRef]
- Cancel-Tassin, G.; Koutros, S. Use of genomic markers to improve epidemiologic and clinical research in urology. Curr. Opin. Urol. 2023, 33, 414–420. [Google Scholar] [CrossRef]
- Wu, F.; Ning, H.; Sun, Y.; Wu, H.; Lyu, J. Integrative exploration of the mutual gene signatures and immune microenvironment between benign prostate hyperplasia and castration-resistant prostate cancer. Aging Male 2023, 26, 2183947. [Google Scholar] [CrossRef]
- Yerukala Sathipati, S.; Tsai, M.J.; Shukla, S.K.; Ho, S.Y. Artificial intelligence-driven pan-cancer analysis reveals miRNA signatures for cancer stage prediction. HGG Adv. 2023, 4, 100190. [Google Scholar] [CrossRef]
- Yang, L.; Yang, M.; Cui, C.; Long, X.; Li, Y.; Dai, W.; Lang, T.; Zhou, Q. The myo-inositol biosynthesis rate-limiting enzyme ISYNA1 suppresses the stemness of ovarian cancer via Notch1 pathway. Cell. Signal. 2023, 107, 110688. [Google Scholar] [CrossRef]
- Aveta, A.; Cilio, S.; Contieri, R.; Spena, G.; Napolitano, L.; Manfredi, C.; Franco, A.; Crocerossa, F.; Cerrato, C.; Ferro, M.; et al. Urinary MicroRNAs as Biomarkers of Urological Cancers: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 10846. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Shao, J.; Xu, X.; Hong, W.; Yu, W.; Zheng, S.; Ge, X. WTAP regulates autophagy in colon cancer cells by inhibiting FLNA through N6-methyladenosine. Cell Adhes. Migr. 2023, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.Y.; Tan, J.Y.; Liu, Z.; Shen, X.L.; Hu, Y.J. Lappaol F regulates the cell cycle by activating CDKN1C/p57 in human colorectal cancer cells. Pharm. Biol. 2023, 61, 337–344. [Google Scholar] [CrossRef]
- Yavuz, M.; Takanlou, L.S.; Avcı, Ç.B.; Demircan, T. A selective androgen receptor modulator, S4, displays robust anti-cancer activity on hepatocellular cancer cells by negatively regulating PI3K/AKT/mTOR signalling pathway. Gene 2023, 869, 147390. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, M.; Fukushige, T.; Iwasaki, H. Establishment and Characterization of a TFE3-rearranged Renal Cell Carcinoma Cell Line (FU-UR-2) with the PRCC-TFE3 Fusion Transcript. Anticancer Res. 2023, 43, 3463–3470. [Google Scholar] [CrossRef] [PubMed]
- Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Tataru, O.S.; Autorino, R.; Battaglia, M.; Ditonno, P.; et al. Cellular and Molecular Players in the Tumor Microenvironment of Renal Cell Carcinoma. J. Clin. Med. 2023, 12, 3888. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Iacobas, S.; Stout, R.F.; Spray, D.C. Cellular Environment Remodels the Genomic Fabrics of Functional Pathways in Astrocytes. Genes 2020, 11, 520. [Google Scholar] [CrossRef]
- NIH-National Cancer Institute Genomic Data Commons Data Portal. Available online: https://portal.gdc.cancer.gov/ (accessed on 20 June 2023).
- Lasorsa, F.; Rutigliano, M.; Milella, M.; Ferro, M.; Pandolfo, S.D.; Crocetto, F.; Autorino, R.; Battaglia, M.; Ditonno, P.; Lucarelli, G. Cancer Stem Cells in Renal Cell Carcinoma: Origins and Biomarkers. Int. J. Mol. Sci. 2023, 24, 13179. [Google Scholar] [CrossRef]
- Sarkar, O.S.; Donninger, H.; Al Rayyan, N.; Chew, L.C.; Stamp, B.; Zhang, X.; Whitt, A.; Li, C.; Hall, M.; Mitchell, R.A.; et al. Monocytic MDSCs exhibit superior immune suppression via adenosine and depletion of adenosine improves efficacy of immunotherapy. Sci. Adv. 2023, 9, eadg3736. [Google Scholar] [CrossRef]
- Liu, C.L.; Huang, W.C.; Cheng, S.P.; Chen, M.J.; Lin, C.H.; Chang, S.C.; Chang, Y.C. Characterization of Mammary Tumors Arising from MMTV-PyVT Transgenic Mice. Curr. Issues Mol. Biol. 2023, 45, 4518–4528. [Google Scholar] [CrossRef]
- Aggen, D.H.; Ager, C.R.; Obradovic, A.Z.; Chowdhury, N.; Ghasemzadeh, A.; Mao, W.; Chaimowitz, M.G.; Lopez-Bujanda, Z.A.; Spina, C.S.; Hawley, J.E.; et al. Blocking IL1 Beta Promotes Tumor Regression and Remodeling of the Myeloid Compartment in a Renal Cell Carcinoma Model: Multidimensional Analyses. Clin. Cancer Res. 2021, 27, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.Y.; Kuzmuk, V.; Hunter, S.; Lay, A.; Hayes, B.; Beesley, M.; Rollason, R.; Hurcombe, J.A.; Barrington, F.; Masson, C.; et al. Adeno-associated virus gene therapy prevents progression of kidney disease in genetic models of nephrotic syndrome. Sci. Transl. Med. 2023, 15, eabc8226. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Iacobas, S.; Urban-Maldonado, M.; Spray, D.C. Sensitivity of the brain transcriptome to connexin ablation. Biochim. Biophys. Acta (BBA)-Biomembr. 2005, 1711, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Iacobas, S.; Spray, D.C. Connexin-dependent transcellular transcriptomic networks in mouse brain. Prog. Biophys. Mol. Biol. 2007, 94, 168–184. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Iacobas, S.; Urban-Maldonado, M.; Scemes, E.; Spray, D.C. Similar transcriptomic alterations in Cx43 knock-down and knock-out astrocytes. Cell Commun. Adhes. 2008, 15, 195–206. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Iacobas, S.; Iacobas, D.A.; Spray, D.C.; Scemes, E. The connexin43 transcriptome during brain development: Importance of genetic background. Brain Res. 2012, 1487, 131–139. [Google Scholar] [CrossRef]
- Li, F.; Aljahdali, I.A.M.; Zhang, R.; Nastiuk, K.L.; Krolewski, J.J.; Ling, X. Kidney cancer biomarkers and targets for therapeutics: Survivin (BIRC5), XIAP, MCL-1, HIF1α, HIF2α, NRF2, MDM2, MDM4, p53, KRAS and AKT in renal cell carcinoma. J. Exp. Clin. Cancer Res. 2021, 40, 254. [Google Scholar] [CrossRef]
- Tu, S.-M.; Zhang, M.; Wood, C.G.; Pisters, L.L. Stem Cell Theory of Cancer: Origin of Tumor Heterogeneity and Plasticity. Cancers 2021, 13, 4006. [Google Scholar] [CrossRef]
- Li, H.; Zhao, S.; Fan, H.Y.; Li, Y.; Wu, X.P.; Miao, Y.P. The Effect of Histogram Analysis of DCE-MRI Parameters on Differentiating Renal Tumors. Clin. Lab. 2023, 69, 2201–2207. [Google Scholar] [CrossRef]
- Brady, L.; Kriner, M.; Coleman, I.; Morrissey, C.; Roudier, M.; True, L.D.; Gulati, R.; Plymate, S.R.; Zhou, Z.; Birditt, B.; et al. Inter and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat. Commun. 2021, 12, 1426. [Google Scholar] [CrossRef]
- Iacobas, S.; Iacobas, D.A. Personalized 3-Gene Panel for Prostate Cancer Target Therapy. Curr. Issues Mol. Biol. 2022, 44, 360–382. [Google Scholar] [CrossRef] [PubMed]
- de Vries-Brilland, M.; Rioux-Leclercq, N.; Meylan, M.; Dauvé, J.; Passot, C.; Spirina-Menand, E.; Flippot, R.; Fromont, G.; Gravis, G.; Geoffrois, L.; et al. Comprehensive analyses of immune tumor microenvironment in papillary renal cell carcinoma. J. Immunother. Cancer 2023, 11, e006885. [Google Scholar] [CrossRef] [PubMed]
- Gui, Z.; Du, J.; Wu, N.; Shen, N.; Yang, Z.; Yang, H.; Wang, X.; Zhao, N.; Zeng, Z.; Wei, R.; et al. Immune regulation and prognosis indicating ability of a newly constructed multi-genes containing signature in clear cell renal cell carcinoma. BMC Cancer 2023, 23, 649. [Google Scholar] [CrossRef] [PubMed]
- Qiagen Ingenuity Pathway Analysis. Available online: https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ (accessed on 12 July 2023).
- DAVID Functional Annotation Bioinformatics Microarray Analysis. Available online: https://david.ncifcrf.gov (accessed on 12 July 2023).
- Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.kegg.jp/kegg/pathway.html (accessed on 12 July 2023).
- Iacobas, D.A.; Mgbemena, V.; Iacobas, S.; Menezes, K.M.; Wang, H.; Saganti, P.B. Genomic fabric remodeling in metastatic clear cell renal cell carcinoma (ccRCC): A new paradigm and proposal for a personalized gene therapy approach. Cancers 2020, 12, 3678. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A.; Tuli, N.; Iacobas, S.; Rasamny, J.K.; Moscatello, A.; Geliebter, J.; Tiwari, R.M. Gene master regulators of papillary and anaplastic thyroid cancer phenotypes. Oncotarget 2018, 9, 2410–2424. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, S.; Iacobas, D.A. A Personalized Genomics Approach of the Prostate Cancer. Cells 2021, 10, 1644. [Google Scholar] [CrossRef] [PubMed]
- Remodeling of Major Genomic Fabrics and Their Interplay in Metastatic Clear Cell Renal Carcinoma. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE72304 (accessed on 6 October 2023).
- Iacobas, D.A.; Iacobas, S.; Lee, P.R.; Cohen, J.E.; Fields, R.D. Coordinated Activity of Transcriptional Networks Responding to the Pattern of Action Potential Firing in Neurons. Genes 2019, 10, 754. [Google Scholar] [CrossRef] [PubMed]
- Ebbing, D.; Gammon, S.D. General Chemistry—Standalone Book; Student, Ed.; Cengage Learning: Boston, MA, USA, 2015; pp. 88–100. [Google Scholar]
- Hansen, J.-P.; McDonald, I.R. Chapter 7—Time-dependent Correlation and Response Functions. In Theory of Simple Liquids, 4th ed.; Academic Press: London, UK, 2013; pp. 265–310. [Google Scholar]
- Eisen, M.; Spellman, P.; Brown, P.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef]
- Butte, A.J.; Tamayo, P.; Slonim, D.; Golub, T.R.; Kohane, I.S. Discovering functional relationships between RNA expression and chemotherapeutic susceptibility using relevance networks. Proc. Natl. Acad. Sci. USA 2000, 97, 12182–12186. [Google Scholar] [CrossRef]
- Horvath, S.; Dong, J. Geometric Interpretation of Gene Coexpression Network Analysis. PLoS Comput. Biol. 2008, 4, e1000117. [Google Scholar] [CrossRef]
- Oldham, M.C.; Langfelder, P.; Horvath, S. Network methods for describing sample relationships in genomic datasets: Application to Huntington’s disease. BMC Syst. Biol. 2012, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Costello, J.C.; Küner, R.; Vega, N.M.; Prill, R.J.; Camacho, D.M.; Allison, K.R.; Aderhold, A.; Bonneau, R.; Chen, Y.; et al. Wisdom of the crowds for robust gene network inference. Nat. Methods 2012, 9, 796–804. [Google Scholar] [CrossRef] [PubMed]
- P Value from Pearson (R) Calculator. Available online: https://www.socscistatistics.com/pvalues/pearsondistribution.aspx (accessed on 1 September 2023).
- Mathew, R.; Iacobas, S.; Huang, J.; Iacobas, D.A. Metabolic Deregulation in Pulmonary Hypertension. Curr. Issues Mol. Biol. 2023, 45, 4850–4874. [Google Scholar] [CrossRef]
- Iacobas, S.; Ede, N.; Iacobas, D.A. The Gene Master Regulators (GMR) Approach Provides Legitimate Targets for Personalized, Time-Sensitive Cancer Gene Therapy. Genes 2019, 10, 560. [Google Scholar] [CrossRef] [PubMed]
- Iacobas, D.A. Biomarkers, Master Regulators and Genomic Fabric Remodeling in a Case of Papillary Thyroid Carcinoma. Genes 2020, 11, 1030. [Google Scholar] [CrossRef]
- Iacobas, D.A.; Xi, L. Theory and Applications of the (Cardio) Genomic Fabric Approach to Post-Ischemic and Hypoxia-Induced Heart Failure. J. Pers. Med. 2022, 12, 1246. [Google Scholar] [CrossRef]
- Aldosterone-Regulated Sodium Reabsorption. Available online: https://www.genome.jp/pathway/hsa04960 (accessed on 1 September 2023).
- Collecting Duct Acid Secretion. Available online: https://www.genome.jp/pathway/hsa04966 (accessed on 1 September 2023).
- Endocrine and Other Factor-Regulated Calcium Reabsorption. Available online: https://www.genome.jp/pathway/hsa04961 (accessed on 1 September 2023).
- Proximal Tubule Bicarbonate Reclamation. Available online: https://www.genome.jp/kegg-bin/show_pathway?hsa04964 (accessed on 9 January 2023).
- Vasopressin-Regulated Water Reabsorption. Available online: https://www.genome.jp/kegg-bin/show_pathway?hsa04962 (accessed on 1 September 2023).
- Thomas, W.; Harvey, B.J. Estrogen-induced signalling and the renal contribution to salt and water homeostasis. Steroids 2023, 199, 109299. [Google Scholar] [CrossRef]
- Xu, W.H.; Xu, Y.; Tian, X.; Anwaier, A.; Liu, W.R.; Wang, J.; Zhu, W.K.; Cao, D.L.; Wang, H.K.; Shi, G.H.; et al. Large-scale transcriptome profiles reveal robust 20-signatures metabolic prediction models and novel role of G6PC in clear cell renal cell carcinoma. J. Cell. Mol. Med. 2020, 24, 9012–9027. [Google Scholar] [CrossRef]
- Li, Z.; Xu, H.; Yu, L.; Wang, J.; Meng, Q.; Mei, H.; Cai, Z.; Chen, W.; Huang, W. Patient-derived renal cell carcinoma organoids for personalized cancer therapy. Clin. Transl. Med. 2022, 12, e970. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892, Erratum in: N. Engl. J. Med. 2012, 367, 976. [Google Scholar] [CrossRef]
- Muto, Y.; Dixon, E.E.; Yoshimura, Y.; Wu, H.; Omachi, K.; Ledru, N.; Wilson, P.C.; King, A.J.; Eric Olson, N.; Gunawan, M.G.; et al. Defining cellular complexity in human autosomal dominant polycystic kidney disease by multimodal single cell analysis. Nat. Commun. 2022, 13, 6497. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, Y.; Wang, Z.; Zhang, Y.; Hou, Y.; Wang, X. Anoikis-related genes signature development for clear cell renal cell carcinoma prognosis and tumor microenvironment. Sci. Rep. 2023, 13, 18909. [Google Scholar] [CrossRef]
- Chen, W.; Lin, W.; Wu, L.; Xu, A.; Liu, C.; Huang, P. A Novel Prognostic Predictor of Immune Microenvironment and Therapeutic Response in Kidney Renal Clear Cell Carcinoma based on Necroptosis-related Gene Signature. Int. J. Med. Sci. 2022, 19, 377–392. [Google Scholar] [CrossRef]
- Clark, D.J.; Dhanasekaran, S.M.; Petralia, F.; Pan, J.; Song, X.; Hu, Y.; da Veiga Leprevost, F.; Reva, B.; Lih, T.M.; Clinical Proteomic Tumor Analysis Consortium; et al. Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 2019, 179, 964–983.e31, Erratum in Cell 2020, 180, 207. [Google Scholar] [CrossRef]
- Li, L.; Tan, H.; Zhou, J.; Hu, F. Predicting response of immunotherapy and targeted therapy and prognosis characteristics for renal clear cell carcinoma based on m1A methylation regulators. Sci. Rep. 2023, 13, 12645. [Google Scholar] [CrossRef]
- Wang, X.X.; Myakala, K.; Libby, A.E.; Krawczyk, E.; Panov, J.; Jones, B.A.; Bhasin, K.; Shults, N.; Qi, Y.; Krausz, K.W.; et al. Estrogen-Related Receptor Agonism Reverses Mitochondrial Dysfunction and Inflammation in the Aging Kidney. Am. J. Pathol. 2023, 193, 1969–1987. [Google Scholar] [CrossRef]
- Adamopoulos, P.G.; Kontos, C.K.; Diamantopoulos, M.A.; Scorilas, A. Molecular cloning of novel transcripts of the adaptor-related protein complex 2 alpha 1 subunit (AP2A1) gene, using Next-Generation Sequencing. Gene 2018, 678, 55–64. [Google Scholar] [CrossRef]
- Pleasance, E.; Titmuss, E.; Williamson, L.; Kwan, H.; Culibrk, L.; Zhao, E.Y.; Dixon, K.; Fan, K.; Bowlby, R.; Jones, M.R.; et al. Pan-cancer analysis of advanced patient tumors reveals interactions between therapy and genomic landscapes. Nat. Cancer 2020, 1, 452–468. [Google Scholar] [CrossRef]
- Zhang, G.; Xi, M.; Li, Y.; Wang, L.; Gao, L.; Zhang, L.; Yang, Z.; Shi, H. The ADCY9 genetic variants are associated with glioma susceptibility and patient prognosis. Genomics 2021, 113, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wang, T.; Zhang, A.; Zhu, J.; Zhou, T.; Zhou, Y.L.; Shi, J. ADCY9 functions as a novel cancer suppressor gene in lung adenocarcinoma. J. Thorac. Dis. 2023, 15, 1018–1035. [Google Scholar] [CrossRef] [PubMed]
- Chao, X.; Jia, Y.; Feng, X.; Wang, G.; Wang, X.; Shi, H.; Zhao, F.; Jiang, C. A Case-Control Study of ADCY9 Gene Polymorphisms and the Risk of Hepatocellular Carcinoma in the Chinese Han Population. Front. Oncol. 2020, 10, 1450. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Liu, J.; Sun, Y.; Wu, J.; Xiong, Z.; Zhang, Y.; Li, B.; Jin, T. Assessment of ADCY9 polymorphisms and colorectal cancer risk in the Chinese Han population. J. Gene Med. 2021, 23, e3298. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Cai, L.; Liang, J. Construction of prognosis model of bladder cancer based on transcriptome. J. Zhejiang Univ. (Med. Sci.) 2022, 51, 79–86. (In English) [Google Scholar] [CrossRef]
- Lee, Y.H.; Gyu Song, G. Genome-wide pathway analysis in pancreatic cancer. J. Buon 2015, 20, 1565–1575. [Google Scholar] [PubMed]
- Guo, R.; Liu, T.; Shasaltaneh, M.D.; Wang, X.; Imani, S.; Wen, Q. Targeting Adenylate Cyclase Family: New Concept of Targeted Cancer Therapy. Front. Oncol. 2022, 12, 829212. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, W.; Ding, Z.; Xu, T.; Zhang, X.; Xu, K. Comprehensive exploration of the expression and prognostic value of AQPs in clear cell renal cell carcinoma. Medicine 2022, 101, e29344. [Google Scholar] [CrossRef] [PubMed]
- Oncomine Solutions For Next-Generation Sequencing. Available online: http://www.oncomine.org (accessed on 1 October 2023).
- The University of ALabama at Birmingham CANcer Data Analysis Portal. Available online: http://ualcan.path.uab.edu (accessed on 1 October 2023).
- Sinha, S.; Dwivedi, N.; Tao, S.; Jamadar, A.; Kakade, V.R.; Neil, M.O.; Weiss, R.H.; Enders, J.; Calvet, J.P.; Thomas, S.M.; et al. Targeting the vasopressin type-2 receptor for renal cell carcinoma therapy. Oncogene 2020, 39, 1231–1245. [Google Scholar] [CrossRef]
- Baltzer, S.; Bulatov, T.; Schmied, C.; Krämer, A.; Berger, B.T.; Oder, A.; Walker-Gray, R.; Kuschke, C.; Zühlke, K.; Eichhorst, J.; et al. Aurora Kinase A Is Involved in Controlling the Localization of Aquaporin-2 in Renal Principal Cells. Int. J. Mol. Sci. 2022, 23, 763. [Google Scholar] [CrossRef]
- KEGG-Constructed cAMP Signaling Pathway. Available online: https://www.genome.jp/pathway/hsa04024+109 (accessed on 1 October 2023).
- Kim, T.H.; Park, J.M.; Kim, M.Y.; Ahn, Y.H. The role of CREB3L4 in the proliferation of prostate cancer cells. Sci. Rep. 2017, 7, 45300. [Google Scholar] [CrossRef]
- Giménez-Bachs, J.M.; Salinas-Sánchez, A.S.; Serrano-Oviedo, L.; Nam-Cha, S.H.; Rubio-Del Campo, A.; Sánchez-Prieto, R. Carbonic anhydrase IX as a specific biomarker for clear cell renal cell carcinoma: Comparative study of Western blot and immunohistochemistry and implications for diagnosis. Scand. J. Urol. Nephrol. 2012, 46, 358–364. [Google Scholar] [CrossRef]
- Tostain, J.; Li, G.; Gentil-Perret, A.; Gigante, M. Carbonic anhydrase 9 in clear cell renal cell carcinoma: A marker for diagnosis, prognosis and treatment. Eur. J. Cancer 2010, 46, 3141–3148. [Google Scholar] [CrossRef]
- Ferro, M.; Falagario, U.G.; Barone, B.; Maggi, M.; Crocetto, F.; Busetto, G.M.; Giudice, F.D.; Terracciano, D.; Lucarelli, G.; Lasorsa, F.; et al. Artificial Intelligence in the Advanced Diagnosis of Bladder Cancer-Comprehensive Literature Review and Future Advancement. Diagnostics 2023, 13, 2308. [Google Scholar] [CrossRef]
- Ferro, M.; Crocetto, F.; Barone, B.; Del Giudice, F.; Maggi, M.; Lucarelli, G.; Busetto, G.M.; Autorino, R.; Marchioni, M.; Cantiello, F.; et al. Artificial intelligence and radiomics in evaluation of kidney lesions: A comprehensive literature review. Ther. Adv. Urol. 2023, 15, 17562872231164803. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Site | # of Cases | # of Genes | Protein Coding | # of Mutations | Primary Site | # of Cases | # of Genes | Protein Coding | # of Mutations |
|---|---|---|---|---|---|---|---|---|---|
| Bladder | 1725 | 20,183 | 19,692 | 114,662 | Lung | 12,262 | 21,318 | 19,790 | 443,974 |
| Bone marrow | 11,027 | 21,474 | 19,705 | 163,756 | Ovary | 3381 | 20,266 | 19,673 | 64,142 |
| Brain | 1452 | 20,343 | 19,729 | 93,128 | Pancreas | 2776 | 19,874 | 19,502 | 36,676 |
| Breast | 9121 | 20,454 | 19,727 | 113,777 | Prostate | 2387 | 19,638 | 19,402 | 27,468 |
| Colorectal | 8140 | 21,060 | 19,794 | 337,634 | Skin | 2893 | 20,739 | 19,770 | 353,213 |
| Head & neck | 2792 | 20,535 | 19,712 | 116,274 | Stomach | 1631 | 20,336 | 19,739 | 182,493 |
| Kidney | 3501 | 20,129 | 19,631 | 65,471 | Uterus | 2803 | 21,471 | 19,781 | 769,622 |
| PTA | PTB | CWM | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Path | Genes | %Up | %Down | WPR | %Up | %Down | WPR | %Up | %Down | WPR |
| ALDO | 26/37 | 11.54 | 11.54 | 1.12 | 30.77 | 0.00 | 8.19 | 11.54 | 11.54 | 2.32 |
| COLL | 16/27 | 6.25 | 12.50 | 5.20 | 12.50 | 0.00 | 16.36 | 12.50 | 0.00 | 9.69 |
| ENDO | 37/53 | 5.41 | 2.70 | 0.88 | 18.92 | 5.41 | 7.68 | 8.11 | 5.41 | 2.15 |
| PROX | 18/23 | 16.67 | 0.00 | 0.96 | 38.89 | 0.00 | 9.04 | 11.11 | 0.00 | 2.12 |
| VASO | 36/44 | 8.33 | 11.11 | 0.69 | 11.11 | 5.56 | 0.93 | 2.78 | 8.33 | 0.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iacobas, D.A.; Obiomon, E.A.; Iacobas, S. Genomic Fabrics of the Excretory System’s Functional Pathways Remodeled in Clear Cell Renal Cell Carcinoma. Curr. Issues Mol. Biol. 2023, 45, 9471-9499. https://doi.org/10.3390/cimb45120594
Iacobas DA, Obiomon EA, Iacobas S. Genomic Fabrics of the Excretory System’s Functional Pathways Remodeled in Clear Cell Renal Cell Carcinoma. Current Issues in Molecular Biology. 2023; 45(12):9471-9499. https://doi.org/10.3390/cimb45120594
Chicago/Turabian StyleIacobas, Dumitru Andrei, Ehiguese Alade Obiomon, and Sanda Iacobas. 2023. "Genomic Fabrics of the Excretory System’s Functional Pathways Remodeled in Clear Cell Renal Cell Carcinoma" Current Issues in Molecular Biology 45, no. 12: 9471-9499. https://doi.org/10.3390/cimb45120594
APA StyleIacobas, D. A., Obiomon, E. A., & Iacobas, S. (2023). Genomic Fabrics of the Excretory System’s Functional Pathways Remodeled in Clear Cell Renal Cell Carcinoma. Current Issues in Molecular Biology, 45(12), 9471-9499. https://doi.org/10.3390/cimb45120594