
Comparative Genome Analysis Reveals Accumulation of Single-Nucleotide Repeats in Pathogenic Escherichia Lineages


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Detection of HPTs
2.3. Analysis of Intragenic HPTs
2.4. Analysis of Intergenic HPTs
2.5. Phylogenetic Analysis
2.6. Correlation Analysis between Pathogenic Factors and HPTs
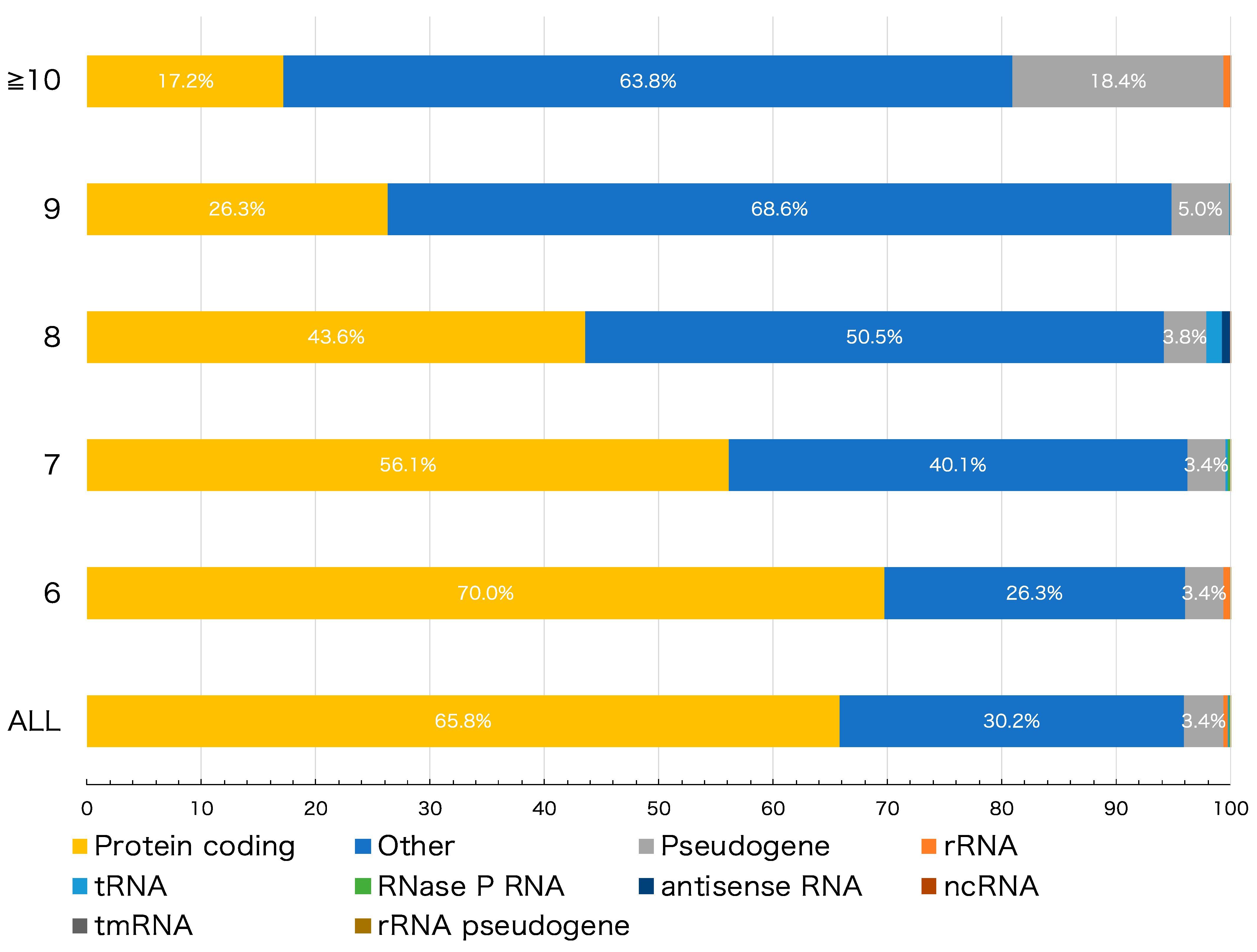
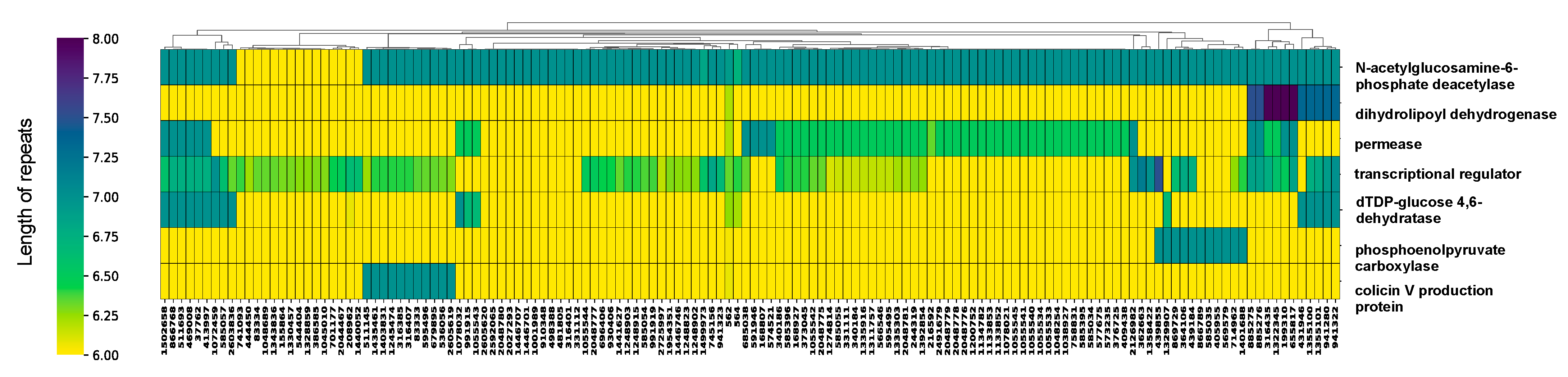
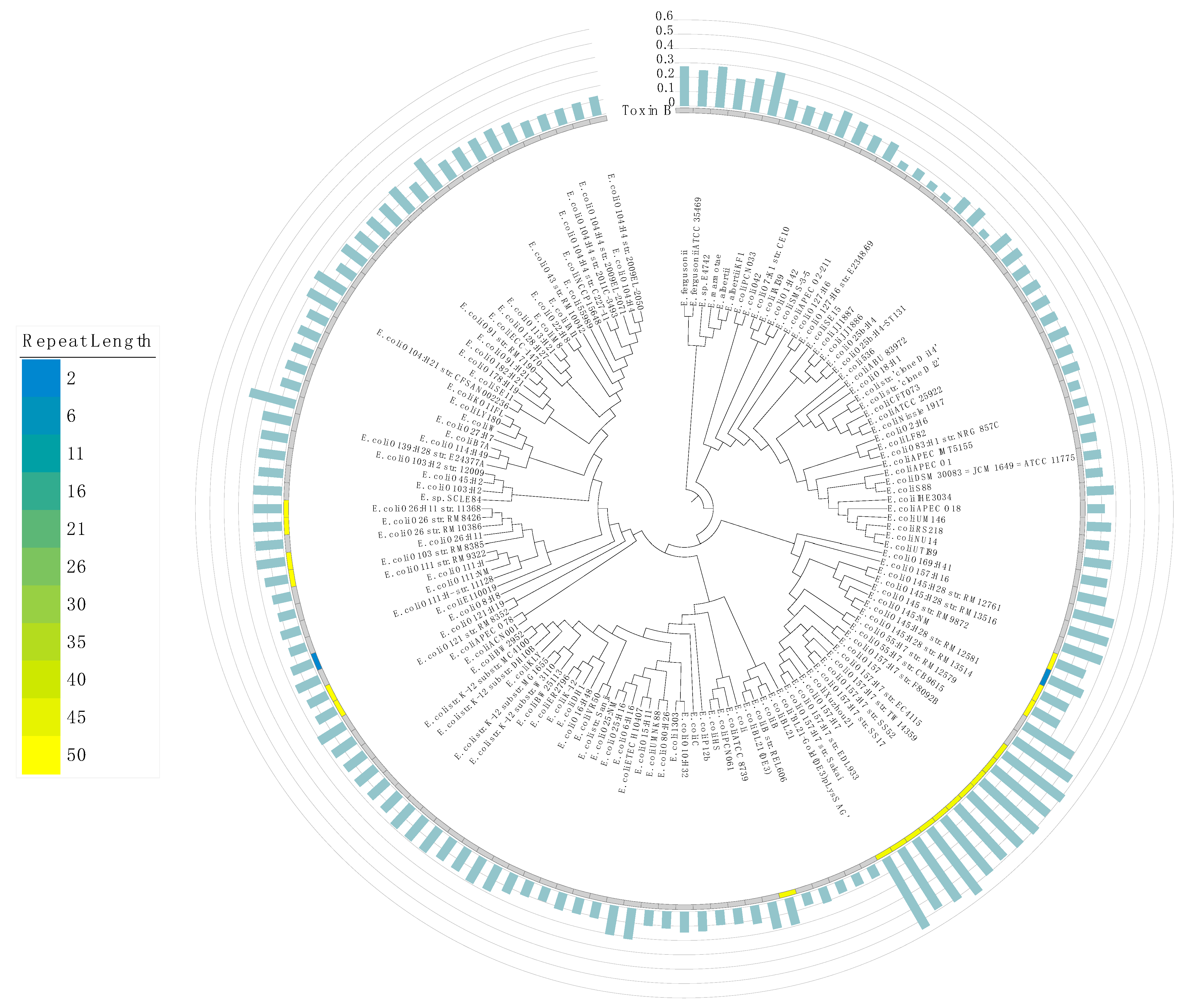
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stibitz, S.; Aaronson, W.; Monack, D.; Falkow, S. Phase variation in Bordetella pertussis by frameshift mutation in a gene for a novel two-component system. Nature 1989, 338, 266–269. [Google Scholar] [CrossRef]
- Park, S.F.; Purdy, D.; Leach, S. Localized reversible frameshift mutation in the flhA gene confers phase variability to flagellin gene expression in Campylobacter coli. J. Bacteriol. 2000, 182, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Saunders, N.J.; Jeffries, A.C.; Peden, J.F.; Hood, D.W.; Tettelin, H.; Rappuoli, R.; Moxon, E.R. Repeat-associated phase variable genes in the complete genome sequence of Neisseria meningitidis strain MC58. Mol. Microbiol. 2000, 37, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Kearns, D.B.; Chu, F.; Rudner, R.; Losick, R. Genes governing swarming in Bacillus subtilis and evidence for a phase variation mechanism controlling surface motility. Mol. Microbiol. 2004, 52, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Orsi, R.H.; Bowen, B.M.; Wiedmann, M. Homopolymeric tracts represent a general regulatory mechanism in prokaryotes. BMC Genom. 2010, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orsi, R.H.; Ripoll, D.R.; Yeung, M.; Nightingale, K.K.; Wiedmann, M. Recombination and positive selection contribute to evolution of Listeria monocytogenes InlA. Microbiology 2007, 153, 2666–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernitzsch, S.R.; Tirier, S.M.; Beier, D.; Sharma, C.M. A variable homopolymeric G-repeat defines small RNA-mediated posttranscriptional regulation of a chemotaxis receptor in Helicobacter pylori. Proc. Natl. Acad. Sci. USA 2014, 111, E501–E510. [Google Scholar] [CrossRef] [Green Version]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar] [CrossRef] [Green Version]
- Treier, M.; Pfeifle, C.; Tautz, D. Comparison of the gap segmentation gene hunchback between Drosophila melanogaster and Drosophila virilis reveals novel modes of evolutionary change. EMBO J. 1989, 8, 1517–1525. [Google Scholar] [CrossRef]
- Hancock, J.M. The contribution of slippage-like processes to genome evolution. J. Mol. Evol. 1995, 41, 1038–1047. [Google Scholar] [CrossRef]
- Scholz, C.F.P.; Brüggemann, H.; Lomholt, H.B.; Tettelin, H.; Kilian, M. Genome stability of Propionibacterium acnes: A comprehensive study of indels and homopolymeric tracts. Sci. Rep. 2016, 6, 20662. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Reback, J.; McKinney, W.; Den Van Bossche, J.; Augspurger, T.; Cloud, P.; Klein, A.; Roeschke, M.; Hawkins, S.; Tratner, J.; She, C.; et al. pandas-dev/pandas: Pandas 1.0.3. Zenodo 2020. [Google Scholar] [CrossRef]
- Tukey, J.W. The Problem of Multiple Comparisons; Chapman & Hall, Princeton University: London, UK, 1953. [Google Scholar]
- Seabold, S.; Perktold, J. Statsmodels: Econometric and Statistical Modeling with Python. In Proceedings of the 9th Python in Science Conference (SCIPY 2010), Austin, TX, USA, 28 June–3 July 2010. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Ishiya, K.; Aburatani, S. Outlier detection for minor compositional variations in taxonomic abundance data. Appl. Sci. 2019, 9, 1355. [Google Scholar] [CrossRef] [Green Version]
- Waskom, M.; Gelbart, M.; Botvinnik, O.; Ostblom, J.; Hobson, P.; Lukauskas, S.; Gemperline, D.C.; Augspurger, T.; Halchenko, Y.; Warmenhoven, J.; et al. mwaskom/seaborn: v0.11.1. Zenodo 2020. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- De Nies, L.; Lopes, S.; Busi, S.B.; Galata, V.; Heintz-Buschart, A.; Laczny, C.C.; May, P.; Wilmes, P. PathoFact: A Pipeline for the Prediction of Virulence Factors and Antimicrobial Resistance Genes in Metagenomic Data. Microbiome 2021, 9, 49. [Google Scholar] [CrossRef]
- Dechering, K.J.; Cuelenaere, K.; Konings, R.N.; Leunissen, J.A. Distinct frequency-distributions of homopolymeric DNA tracts in different genomes. Nucleic Acids Res. 1998, 26, 4056–4062. [Google Scholar] [CrossRef]
- Josenhans, C.; Eaton, K.A.; Thevenot, T.; Suerbaum, S. Switching of flagellar motility in Helicobacter pylori by reversible length variation of a short homopolymeric sequence repeat in fliP, a gene encoding a basal body protein. Infect. Immun. 2000, 68, 4598–4603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogol, E.B.; Cummings, C.A.; Burns, R.C.; Relman, D.A. Phase variation and microevolution at homopolymeric tracts in Bordetella pertussis. BMC Genom. 2007, 8, 122. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.N.; Lubkowska, L.; Hui, M.; Court, C.; Chen, S.; Court, D.L.; Strathern, J.; Jin, D.J.; Kashlev, M. Isolation and characterization of RNA polymerase rpoB mutations that alter transcription slippage during elongation in Escherichia coli. J. Biol. Chem. 2013, 288, 2700–2710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Makino, K.; Ohnishi, M.; Kurokawa, K.; Ishii, K.; Yokoyama, K.; Han, C.G.; Ohtsubo, E.; Nakayama, K.; Murata, T.; et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001, 8, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikh, N.; Tarr, P.I. Escherichia coli O157:H7 Shiga toxin-encoding bacteriophages: Integrations, excisions, truncations, and evolutionary implications. J. Bacteriol. 2003, 185, 3596–3605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yara, D.A.; Greig, D.R.; Gally, D.L.; Dallman, T.J.; Jenkins, C. Comparison of Shiga toxin-encoding bacteriophages in highly pathogenic strains of Shiga toxin-producing Escherichia coli O157:H7 in the UK. Microb. Genom. 2020, 6, e000334. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Count a | Mean b | SD c | Min d | Max e | |
|---|---|---|---|---|---|
| A | 6,606,099 | 6.27 | 0.55 | 6.00 | 56 |
| C | 569,850 | 6.22 | 0.57 | 6.00 | 70 |
| G | 567,499 | 6.22 | 0.58 | 6.00 | 68 |
| T | 6,592,065 | 6.27 | 0.56 | 6.00 | 108 |
| All | 14,335,513 | 6.27 | 0.55 | 6.00 | 108 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ishiya, K.; Nakashima, N. Comparative Genome Analysis Reveals Accumulation of Single-Nucleotide Repeats in Pathogenic Escherichia Lineages. Curr. Issues Mol. Biol. 2022, 44, 498-504. https://doi.org/10.3390/cimb44020034
Ishiya K, Nakashima N. Comparative Genome Analysis Reveals Accumulation of Single-Nucleotide Repeats in Pathogenic Escherichia Lineages. Current Issues in Molecular Biology. 2022; 44(2):498-504. https://doi.org/10.3390/cimb44020034
Chicago/Turabian StyleIshiya, Koji, and Nobutaka Nakashima. 2022. "Comparative Genome Analysis Reveals Accumulation of Single-Nucleotide Repeats in Pathogenic Escherichia Lineages" Current Issues in Molecular Biology 44, no. 2: 498-504. https://doi.org/10.3390/cimb44020034
APA StyleIshiya, K., & Nakashima, N. (2022). Comparative Genome Analysis Reveals Accumulation of Single-Nucleotide Repeats in Pathogenic Escherichia Lineages. Current Issues in Molecular Biology, 44(2), 498-504. https://doi.org/10.3390/cimb44020034