
Dynamic Diversity of NLR Genes in Triticum and Mining of Promising NLR Alleles for Disease Resistance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of NLR Genes in Wheat
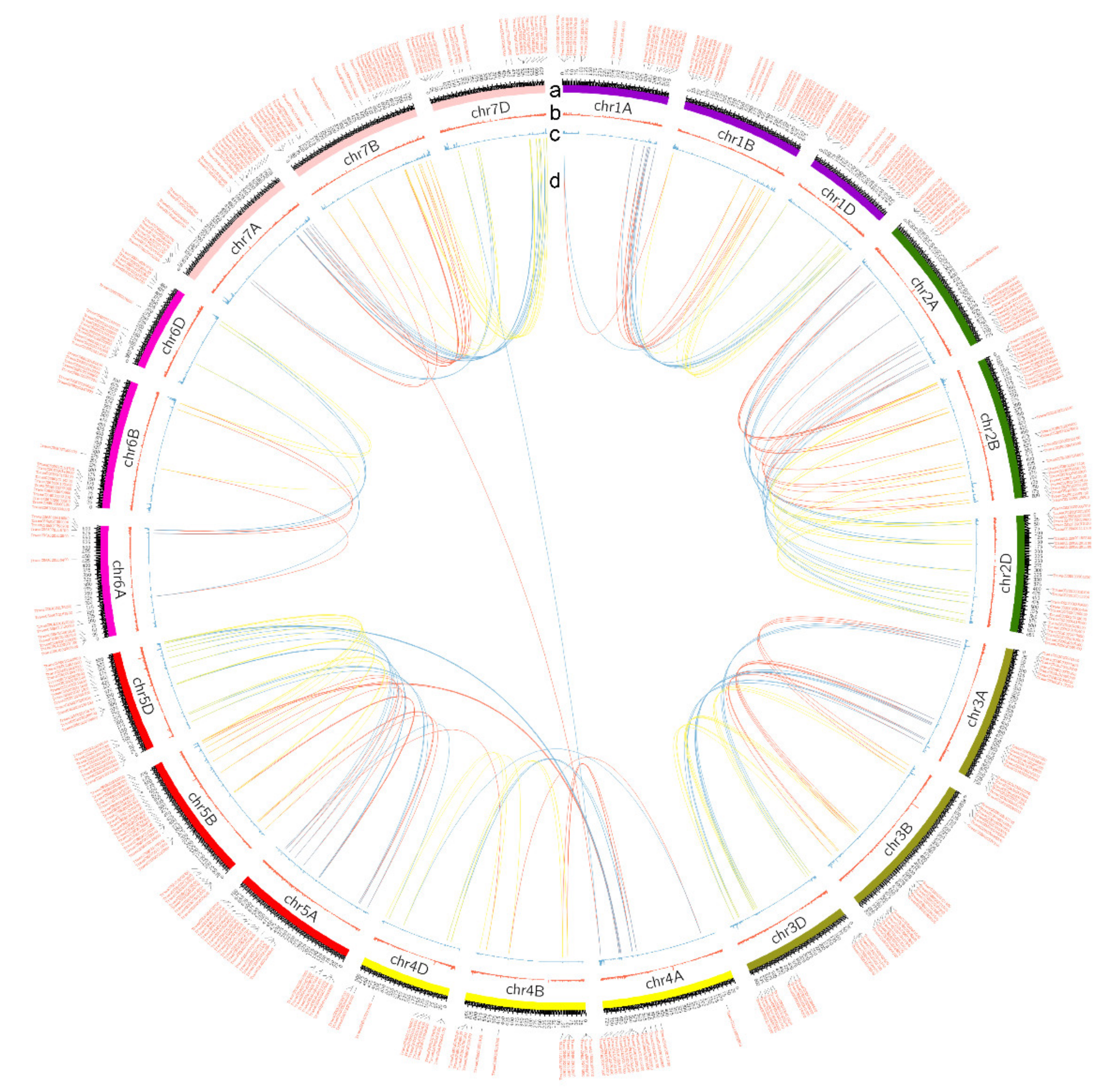
2.2. Chromosome Locations and Distribution of NLR Gene Clusters
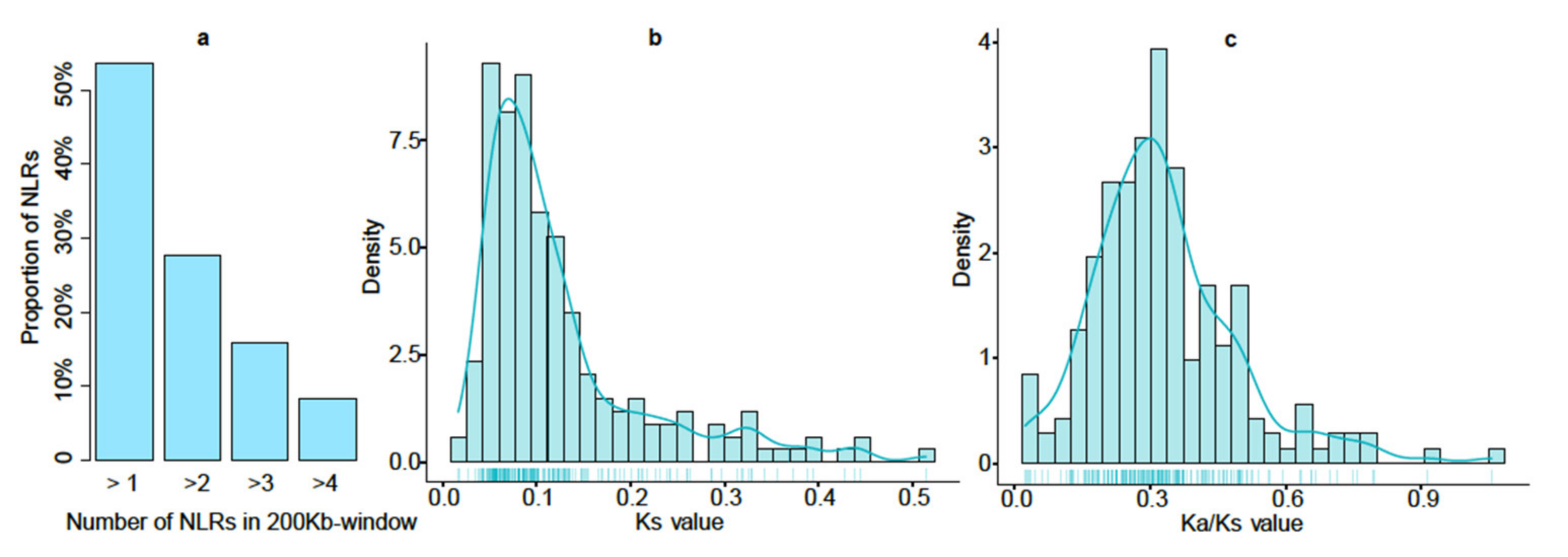
2.3. Identification of NLRs Synteny and Ka/Ks Evaluation
2.4. Evaluation of Dynamic Diversity of NLR Genes in Triticum and Aegilops
2.5. Detection of Selective Signal during Domestication and Improvement
2.6. Transcriptome Data from the Pathogen Inoculation Experiment
3. Results
3.1. Genome-Wide Identification of NLR Proteins in Wheat
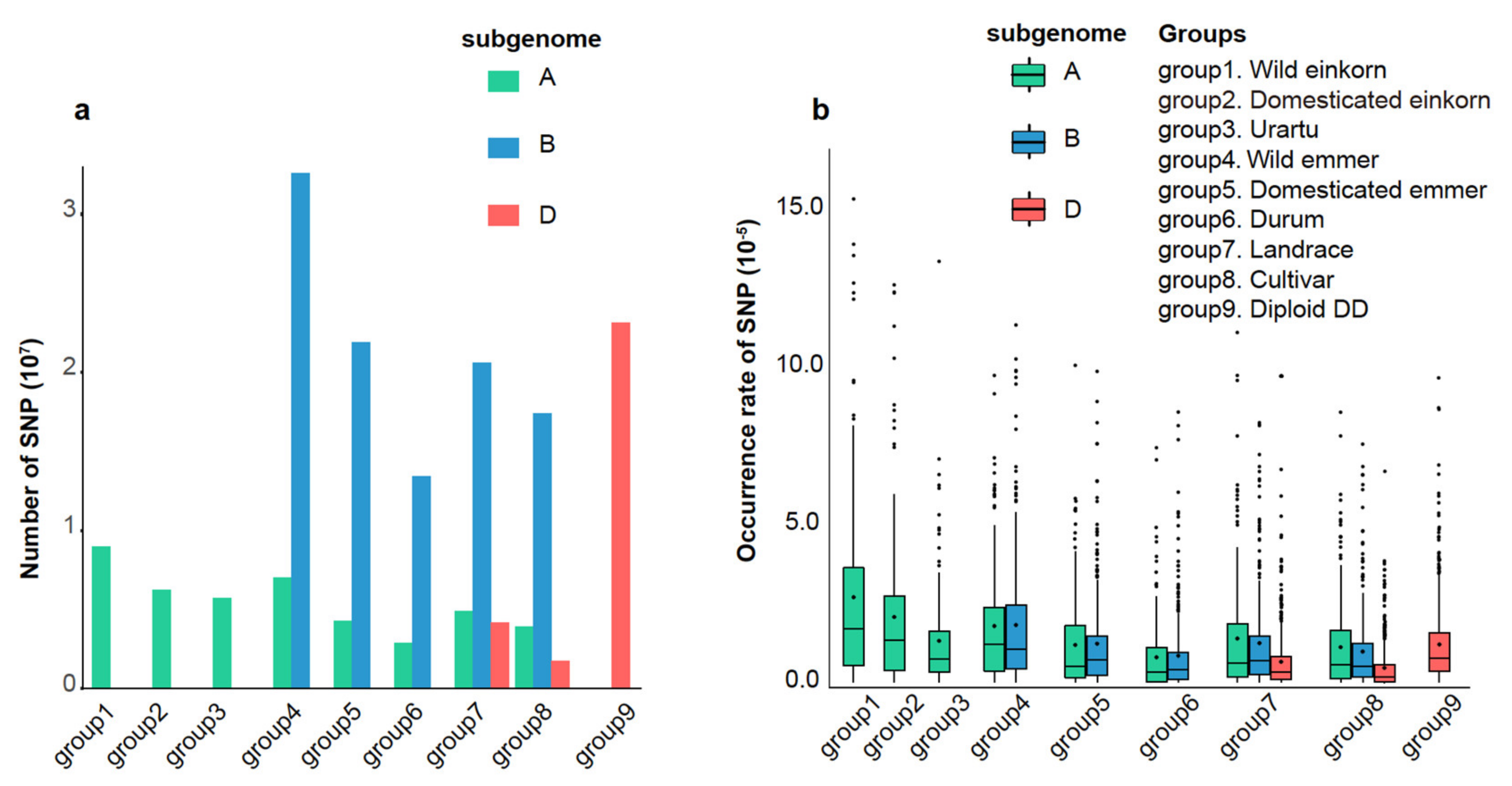
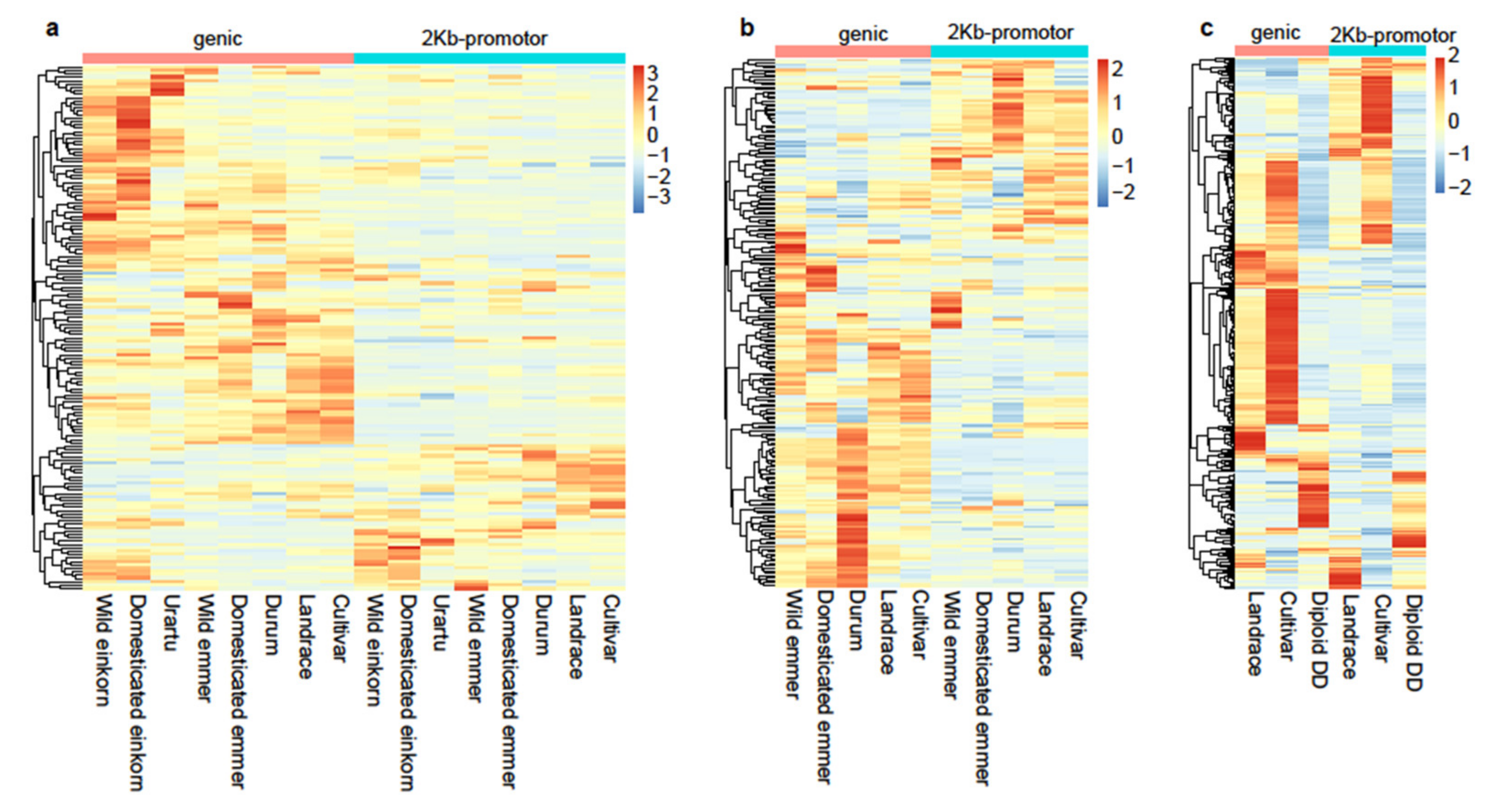
3.2. Dynamic Diversity of NLR Proteins in Evolution of Triticum and Aegilops
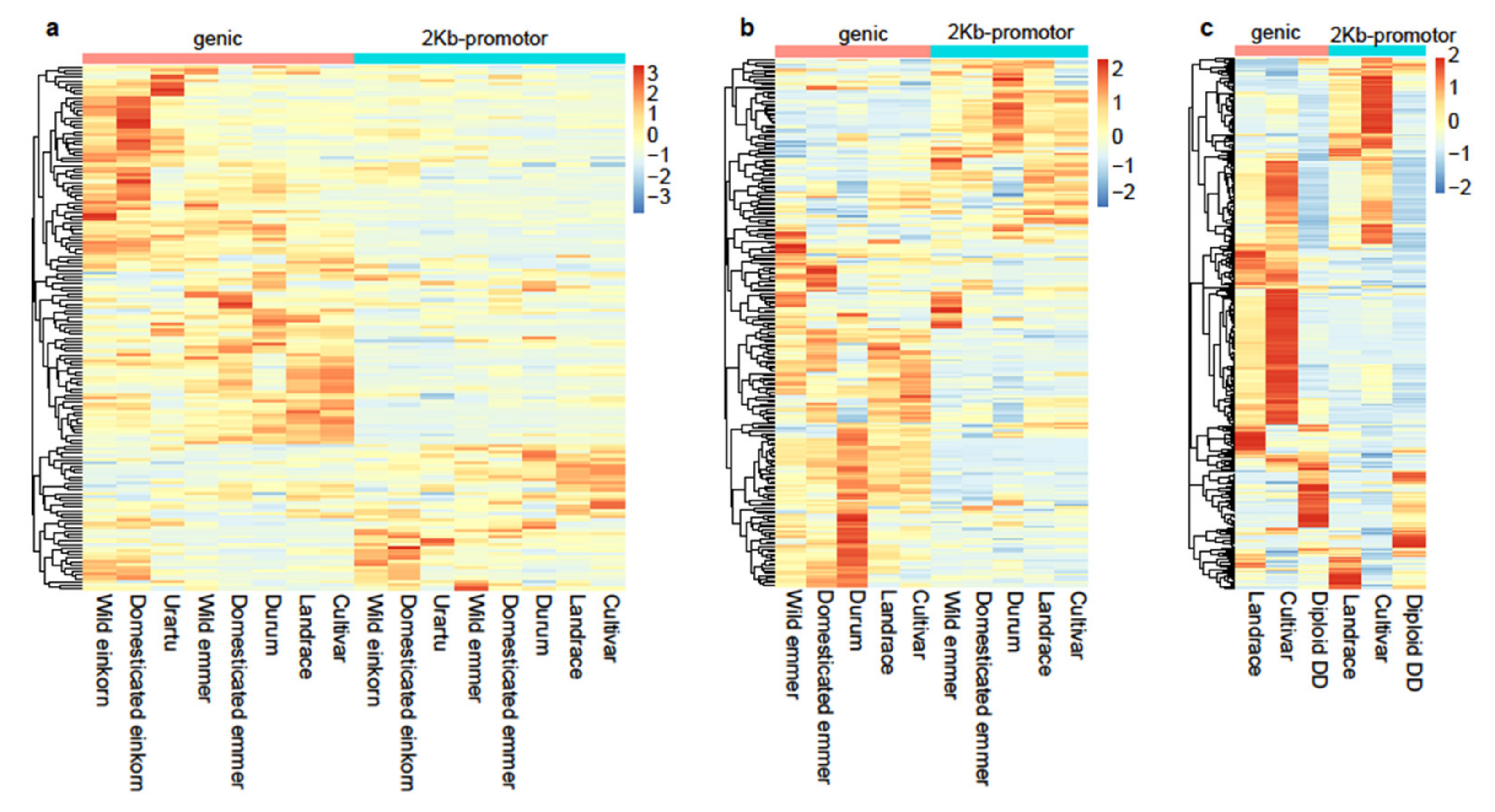
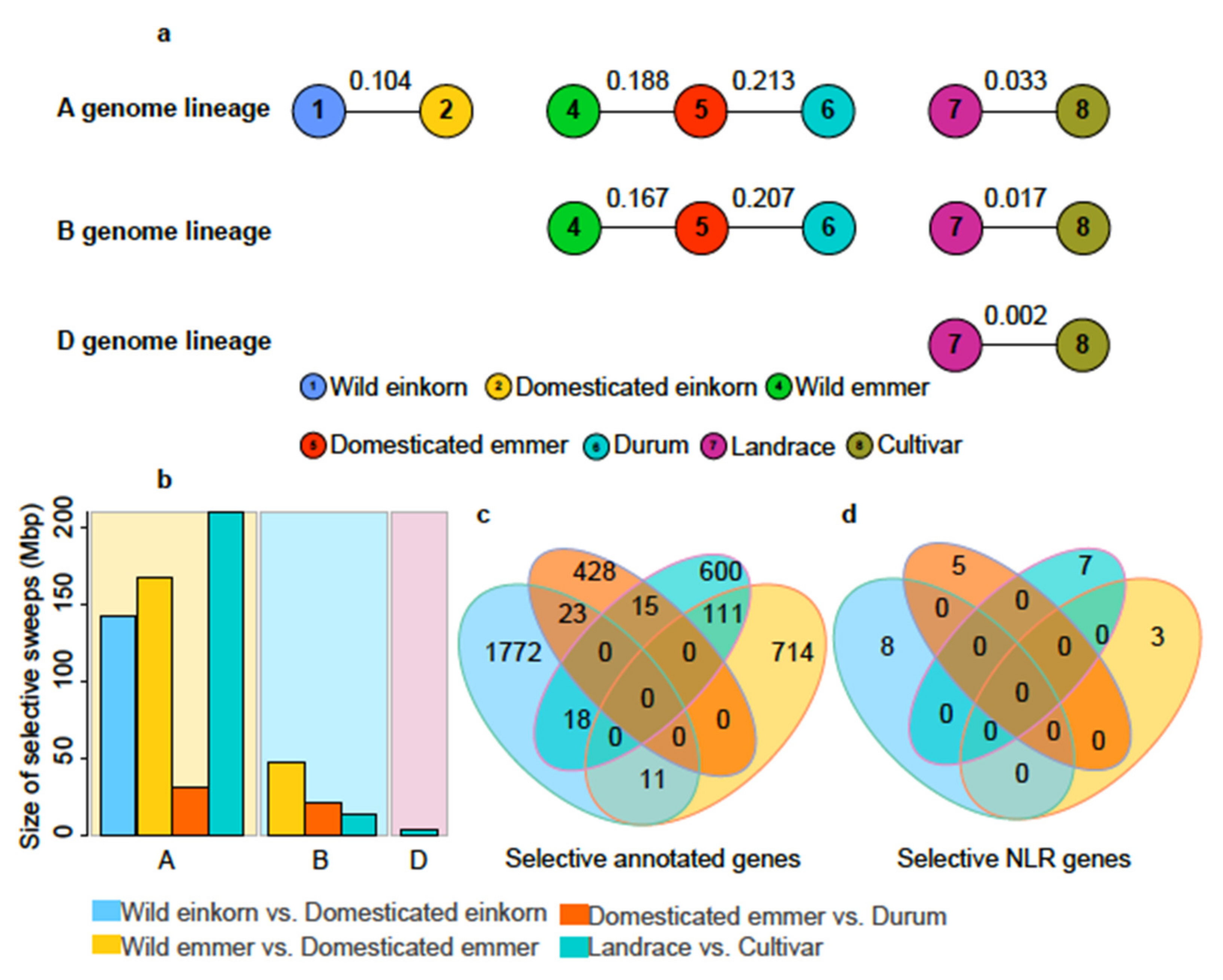
3.3. NLRs Carried Selective Signals during Wheat Domestication and Improvement
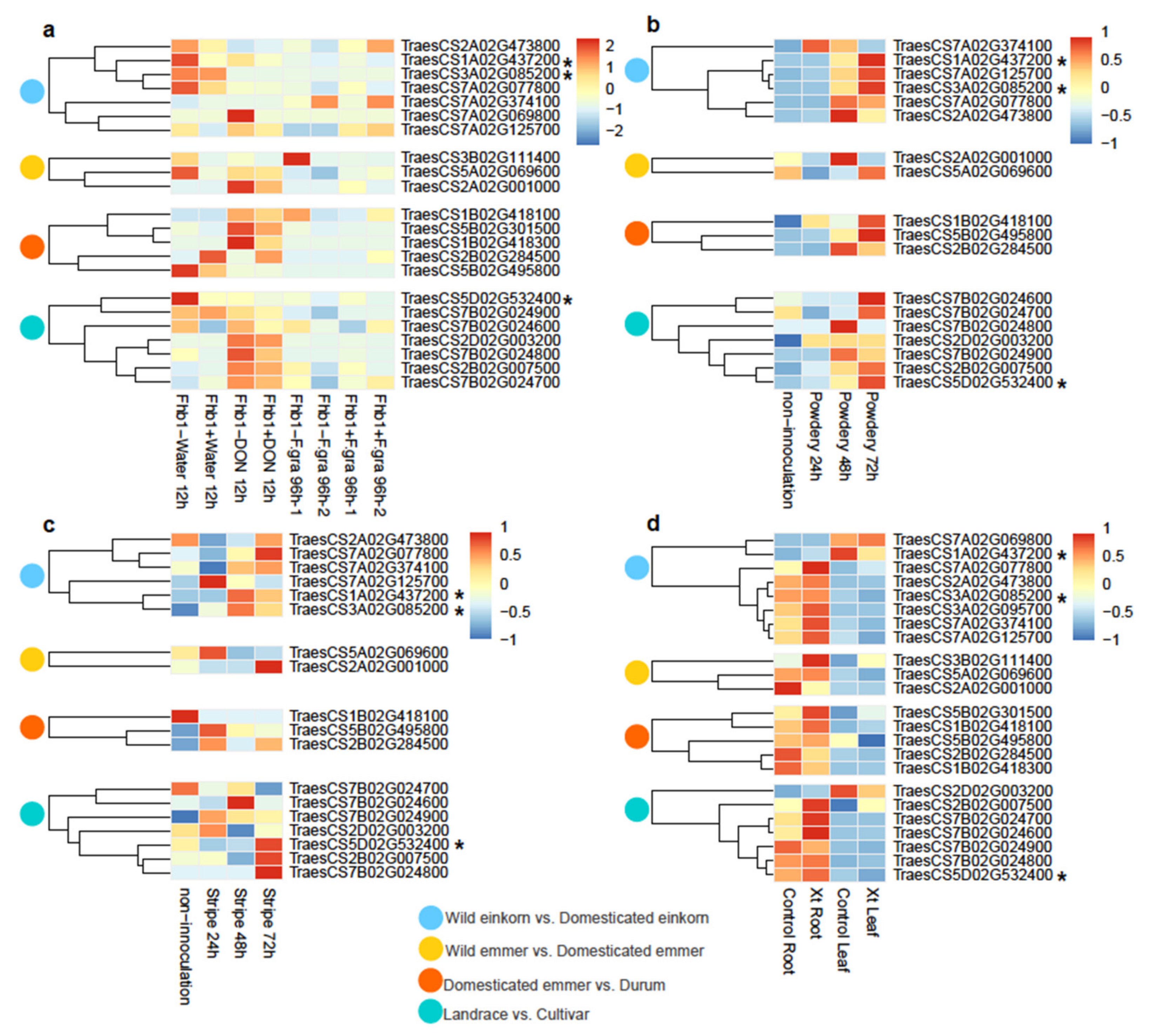
3.4. Transcriptome Profiles of the Beneficial NLR Alleles for Disease Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ning, Y.; Liu, W.; Wang, G.-L. Balancing Immunity and Yield in Crop Plants. Trends Plant. Sci. 2017, 22, 1069–1079. [Google Scholar] [CrossRef]
- Bailey-Serres, J.; Parker, J.E.; Ainsworth, E.A.; Oldroyd, G.E.D.; Schroeder, J.I. Genetic Strategies for Improving Crop Yields. Nature 2019, 575, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinton, J.; Uauy, C. A Reductionist Approach to Dissecting Grain Weight and Yield in Wheat. J. Integr. Plant. Biol. 2019, 61, 337–358. [Google Scholar] [CrossRef] [Green Version]
- Godfray, H.C.J.; Beddington, J.R.; Crute, I.R.; Haddad, L.; Lawrence, D.; Muir, J.F.; Pretty, J.; Robinson, S.; Thomas, S.M.; Toulmin, C. Food Security: The Challenge of Feeding 9 Billion People. Science 2010, 327, 812–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadesse, W.; Sanchez-Garcia, M.; Assefa, S.G.; Amri, A.; Bishaw, Z.; Ogbonnaya, F.C.; Baum, M. Genetic Gains in Wheat Breeding and Its Role in Feeding the World. Crop. Breed. Genet. Genom. 2019, 1, e190005. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.P.; Singh, P.K.; Rutkoski, J.; Hodson, D.P.; He, X.; Jørgensen, L.N.; Hovmøller, M.S.; Huerta-Espino, J. Disease Impact on Wheat Yield Potential and Prospects of Genetic Control. Annu. Rev. Phytopathol. 2016, 54, 303–322. [Google Scholar] [CrossRef] [Green Version]
- Ficke, A.; Cowger, C.; Bergstrom, G.; Brodal, G. Understanding Yield Loss and Pathogen Biology to Improve Disease Management: Septoria Nodorum Blotch—A Case Study in Wheat. Plant Dis. 2018, 102, 696–707. [Google Scholar] [CrossRef] [Green Version]
- Van de Weyer, A.-L.; Monteiro, F.; Furzer, O.J.; Nishimura, M.T.; Cevik, V.; Witek, K.; Jones, J.D.G.; Dangl, J.L.; Weigel, D.; Bemm, F. A Species-Wide Inventory of NLR Genes and Alleles in Arabidopsis Thaliana. Cell 2019, 178, 1260–1272.e14. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.D.G.; Vance, R.E.; Dangl, J.L. Intracellular Innate Immune Surveillance Devices in Plants and Animals. Science 2016, 354, aaf6395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, F.; Nishimura, M.T. Structural, Functional, and Genomic Diversity of Plant NLR Proteins: An Evolved Resource for Rational Engineering of Plant Immunity. Annu. Rev. Phytopathol. 2018, 56, 243–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourelis, J.; van der Hoorn, R.A.L. Defended to the Nines: 25 Years of Resistance Gene Cloning Identifies Nine Mechanisms for R Protein Function. Plant Cell 2018, 30, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johal, G.; Briggs, S. Reductase Activity Encoded by the HM1 Disease Resistance Gene in Maize. Science 1992, 258, 985–987. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, M.; Singh, J.; Song, B.; Tang, Z.; Liu, Y.; Wang, R.; Qin, M.; Li, J.; Khan, A.; et al. Contrasting Genetic Variation and Positive Selection Followed the Divergence of NBS-Encoding Genes in Asian and European Pears. BMC Genom. 2020, 21, 809. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jiang, X.-M.; Shao, Z.-Q. Genome-Wide Analysis of NLR Disease Resistance Genes in an Updated Reference Genome of Barley. Front. Genet. 2021, 12, 694682. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Steuernagel, B.; Gaurav, K.; Chandramohan, S.; Long, Y.; Matny, O.; Johnson, R.; Enk, J.; Periyannan, S.; Singh, N.; et al. Resistance Gene Cloning from a Wild Crop Relative by Sequence Capture and Association Genetics. Nat. Biotechnol. 2019, 37, 139–143. [Google Scholar] [CrossRef]
- The International Wheat Genome Sequencing Consortium (IWGSC); Appels, R.; Eversole, K.; Stein, N.; Feuillet, C.; Keller, B.; Rogers, J.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; et al. Shifting the Limits in Wheat Research and Breeding Using a Fully Annotated Reference Genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Liu, J.; Wen, J.; Nie, X.; Xu, L.; Chen, N.; Li, Z.; Wang, Q.; Zheng, Z.; Li, M.; et al. Frequent Intra- and Inter-Species Introgression Shapes the Landscape of Genetic Variation in Bread Wheat. Genome Biol. 2019, 20, 136. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Zhao, X.; Li, Y.; Xu, J.; Bi, A.; Kang, L.; Xu, D.; Chen, H.; Wang, Y.; Wang, Y.; et al. Triticum Population Sequencing Provides Insights into Wheat Adaptation. Nat. Genet. 2020, 52, 1412–1422. [Google Scholar] [CrossRef]
- Przewieslik-Allen, A.M.; Wilkinson, P.A.; Burridge, A.J.; Winfield, M.O.; Dai, X.; Beaumont, M.; King, J.; Yang, C.; Griffiths, S.; Wingen, L.U.; et al. The Role of Gene Flow and Chromosomal Instability in Shaping the Bread Wheat Genome. Nat. Plants 2021, 7, 172–183. [Google Scholar] [CrossRef]
- He, F.; Pasam, R.; Shi, F.; Kant, S.; Keeble-Gagnere, G.; Kay, P.; Forrest, K.; Fritz, A.; Hucl, P.; Wiebe, K.; et al. Exome Sequencing Highlights the Role of Wild-Relative Introgression in Shaping the Adaptive Landscape of the Wheat Genome. Nat. Genet. 2019, 51, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A. The Pfam Protein Families Database. Nucleic Acids Res. 2000, 28, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Song, W.; Xie, X.; Wang, Z.; Guan, P.; Peng, H.; Jiao, Y.; Ni, Z.; Sun, Q.; Guo, W. A Collinearity-Incorporating Homology Inference Strategy for Connecting Emerging Assemblies in the Triticeae Tribe as a Pilot Practice in the Plant Pangenomic Era. Molecular Plant 2020, 13, 1694–1708. [Google Scholar] [CrossRef]
- Hao, Z.; Lv, D.; Ge, Y.; Shi, J.; Weijers, D.; Yu, G.; Chen, J. RIdeogram: Drawing SVG Graphics to Visualize and Map Genome-Wide Data on the Idiograms. PeerJ Comput. Sci. 2020, 6, e251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. The Evolutionary Fate and Consequences of Duplicate Genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofstad, A.N.; Nussbaumer, T.; Akhunov, E.; Shin, S.; Kugler, K.G.; Kistler, H.C.; Mayer, K.F.X.; Muehlbauer, G.J. Examining the Transcriptional Response in Wheat Fhb1 Near-Isogenic Lines to Fusarium Graminearum Infection and Deoxynivalenol Treatment. Plant Genome 2016, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yang, Y.; Wang, C.; Liu, M.; Li, H.; Fu, Y.; Wang, Y.; Nie, Y.; Liu, X.; Ji, W. Large-Scale Transcriptome Comparison Reveals Distinct Gene Activations in Wheat Responding to Stripe Rust and Powdery Mildew. BMC Genom. 2014, 15, 898. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Seco, D.; Chiapello, M.; Bracale, M.; Pesce, C.; Bagnaresi, P.; Dubois, E.; Moulin, L.; Vannini, C.; Koebnik, R. Transcriptome and Proteome Analysis Reveal New Insight into Proximal and Distal Responses of Wheat to Foliar Infection by Xanthomonas translucens. Sci. Rep. 2017, 7, 10157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koebner, R.M.D.; Shepherd, K.W. Controlled Introgression to Wheat of Genes from Rye Chromosome Arm 1RS by Induction of Allosyndesis: 1. Isolation of Recombinants. Theor. Appl. Genet. 1986, 73, 197–208. [Google Scholar] [CrossRef]
- Ko, J.M.; Seo, B.B.; Suh, D.Y.; Do, G.S.; Park, D.S.; Kwack, Y.H. Production of a New Wheat Line Possessing the 1BL.1RS Wheat-Rye Translocation Derived from Korean Rye Cultivar Paldanghomil. Theor. Appl. Genet. 2002, 104, 171–176. [Google Scholar] [CrossRef]
- Holub, E.B. The Arms Race Is Ancient History in Arabidopsis, the Wildflower. Nat. Rev. Genet. 2001, 2, 516–527. [Google Scholar] [CrossRef]
- Ramírez-González, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; van Ex, F.; Pasha, A.; et al. The Transcriptional Landscape of Polyploid Wheat. Science 2018, 361, eaar6089. [Google Scholar] [CrossRef] [Green Version]
- The International Wheat Genome Sequencing Consortium (IWGSC); Mayer, K.F.X.; Rogers, J.; Dole el, J.; Pozniak, C.; Eversole, K.; Feuillet, C.; Gill, B.; Friebe, B.; Lukaszewski, A.J.; et al. A Chromosome-Based Draft Sequence of the Hexaploid Bread Wheat (Triticum aestivum) Genome. Science 2014, 345, 1251788. [Google Scholar] [CrossRef]
- Yang, Z.; Bielawski, J.P. Statistical Methods for Detecting Molecular Adaptation. Trends Ecol. Evol. 2000, 15, 496–503. [Google Scholar] [CrossRef]
- Yu, K.; Feng, M.; Yang, G.; Sun, L.; Qin, Z.; Cao, J.; Wen, J.; Li, H.; Zhou, Y.; Chen, X.; et al. Changes in Alternative Splicing in Response to Domestication and Polyploidization in Wheat. Plant Physiol. 2020, 184, 1955–1968. [Google Scholar] [CrossRef]
- Yang, G.; Liu, Z.; Gao, L.; Yu, K.; Feng, M.; Yao, Y.; Peng, H.; Hu, Z.; Sun, Q.; Ni, Z.; et al. Genomic Imprinting Was Evolutionarily Conserved during Wheat Polyploidization. Plant Cell 2018, 30, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, X.; Pritchard, J.K. Coregulation of Tandem Duplicate Genes Slows Evolution of Subfunctionalization in Mammals. Science 2016, 352, 1009–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuillet, C.; Langridge, P.; Waugh, R. Cereal Breeding Takes a Walk on the Wild Side. Trends Genet. 2008, 24, 24–32. [Google Scholar] [CrossRef]
- Hafeez, A.N.; Arora, S.; Ghosh, S.; Gilbert, D.; Bowden, R.L.; Wulff, B.B.H. Creation and Judicious Application of a Wheat Resistance Gene Atlas. Mol. Plant 2021, 14, 1053–1070. [Google Scholar] [CrossRef]
- Walkowiak, S.; Gao, L.; Monat, C.; Haberer, G.; Kassa, M.T.; Brinton, J.; Ramirez-Gonzalez, R.H.; Kolodziej, M.C.; Delorean, E.; Thambugala, D.; et al. Multiple Wheat Genomes Reveal Global Variation in Modern Breeding. Nature 2020, 588, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bai, S.; Li, H.; Sun, G.; Zhang, D.; Ma, F.; Zhao, X.; Nie, F.; Li, J.; Chen, L.; et al. Introgressing the Aegilops Tauschii Genome into Wheat as a Basis for Cereal Improvement. Nat. Plants 2021, 7, 774–786. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, S.; Ge, W.; Zhao, L.; Hou, B.; Wang, K.; Lyu, Z.; Chen, L.; Xu, S.; Guo, J.; et al. Horizontal Gene Transfer of Fhb7 from Fungus Underlies Fusarium Head Blight Resistance in Wheat. Science 2020, 368, eaba5435. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Cheng, S. Dynamic Diversity of NLR Genes in Triticum and Mining of Promising NLR Alleles for Disease Resistance. Curr. Issues Mol. Biol. 2021, 43, 965-977. https://doi.org/10.3390/cimb43020069
Li X, Cheng S. Dynamic Diversity of NLR Genes in Triticum and Mining of Promising NLR Alleles for Disease Resistance. Current Issues in Molecular Biology. 2021; 43(2):965-977. https://doi.org/10.3390/cimb43020069
Chicago/Turabian StyleLi, Xiaolong, and Shifeng Cheng. 2021. "Dynamic Diversity of NLR Genes in Triticum and Mining of Promising NLR Alleles for Disease Resistance" Current Issues in Molecular Biology 43, no. 2: 965-977. https://doi.org/10.3390/cimb43020069
APA StyleLi, X., & Cheng, S. (2021). Dynamic Diversity of NLR Genes in Triticum and Mining of Promising NLR Alleles for Disease Resistance. Current Issues in Molecular Biology, 43(2), 965-977. https://doi.org/10.3390/cimb43020069