
Improved Aptamers for the Diagnosis and Potential Treatment of HER2-Positive Cancer

 ,
,  and
and 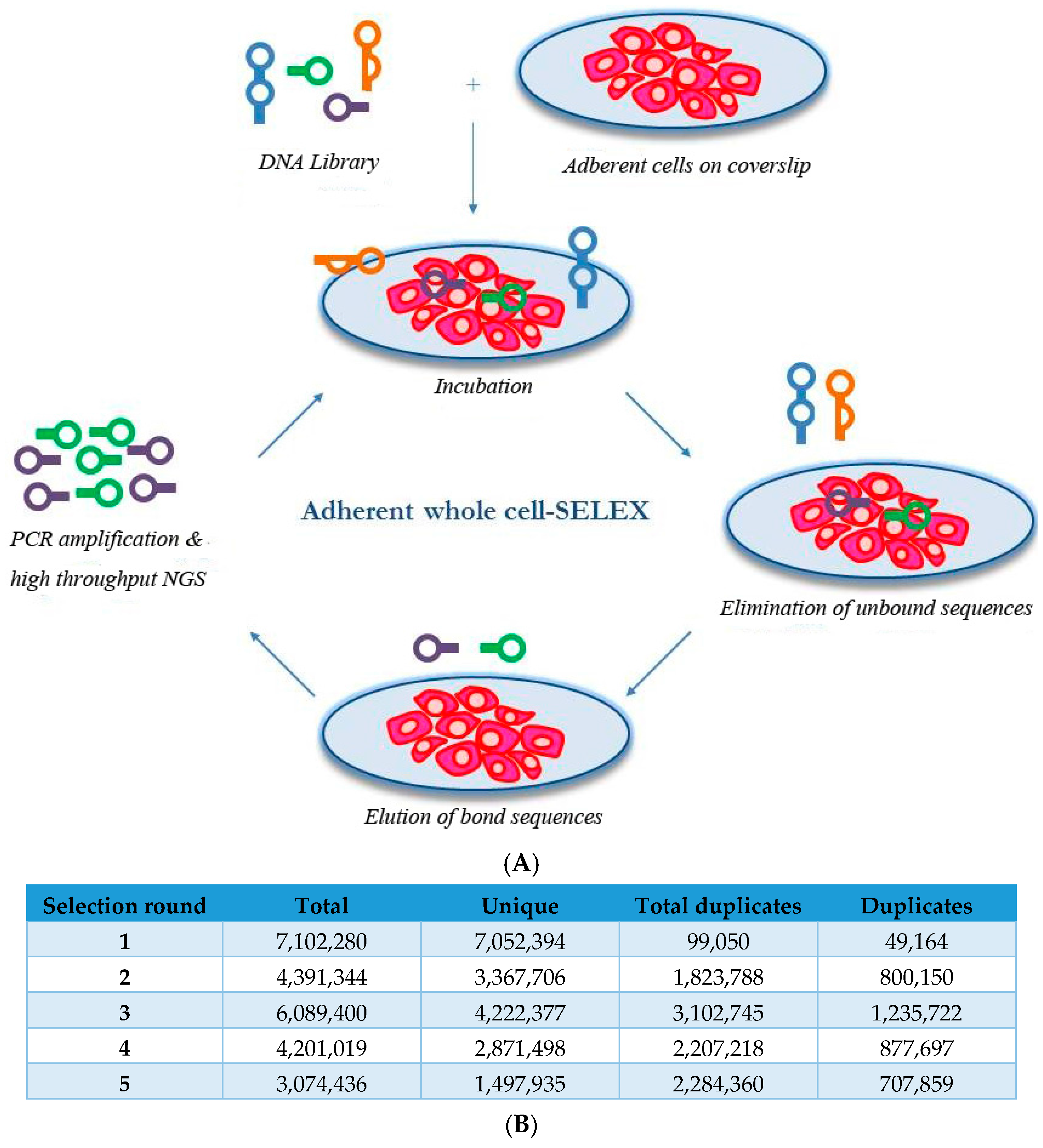{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
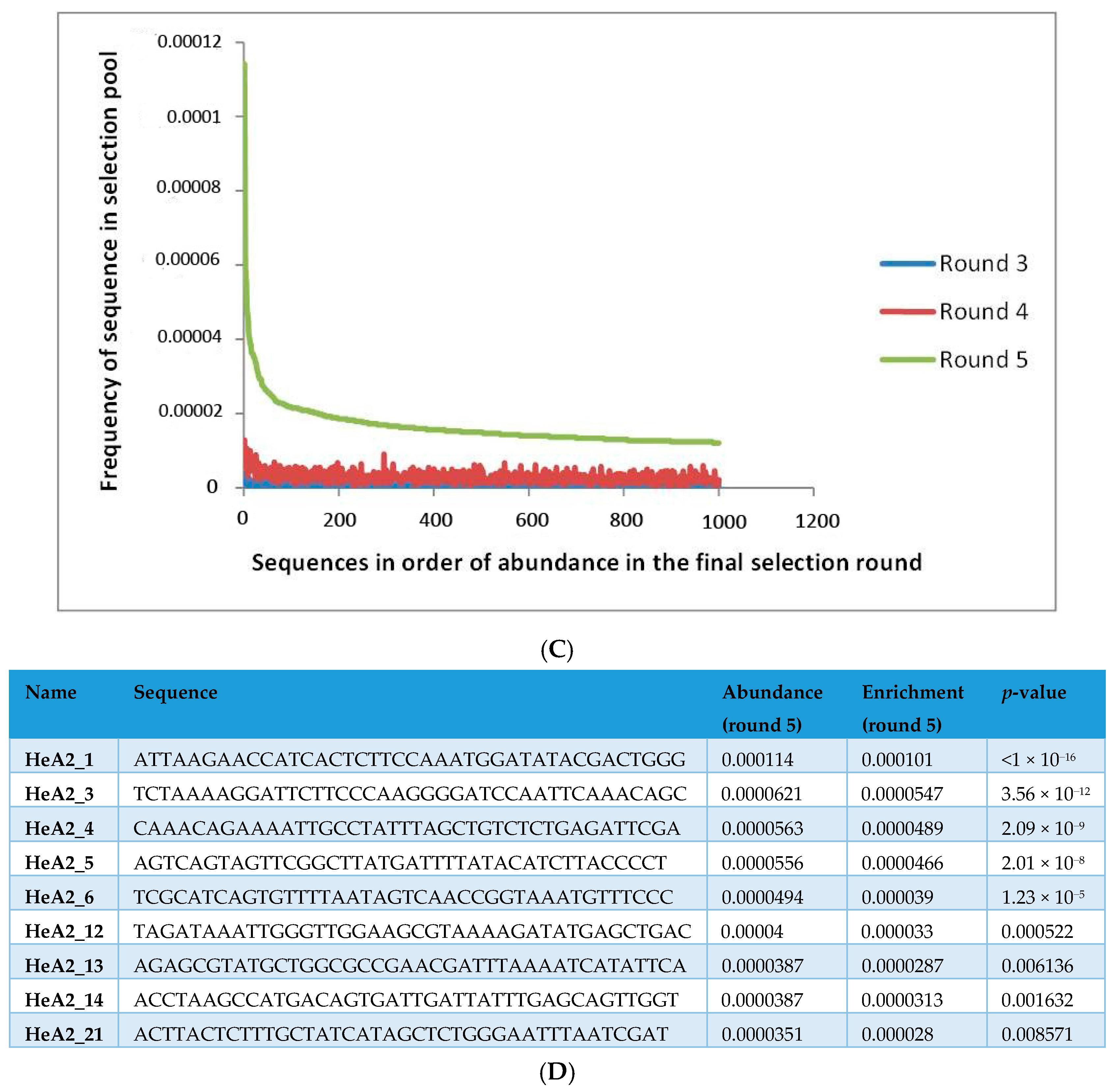
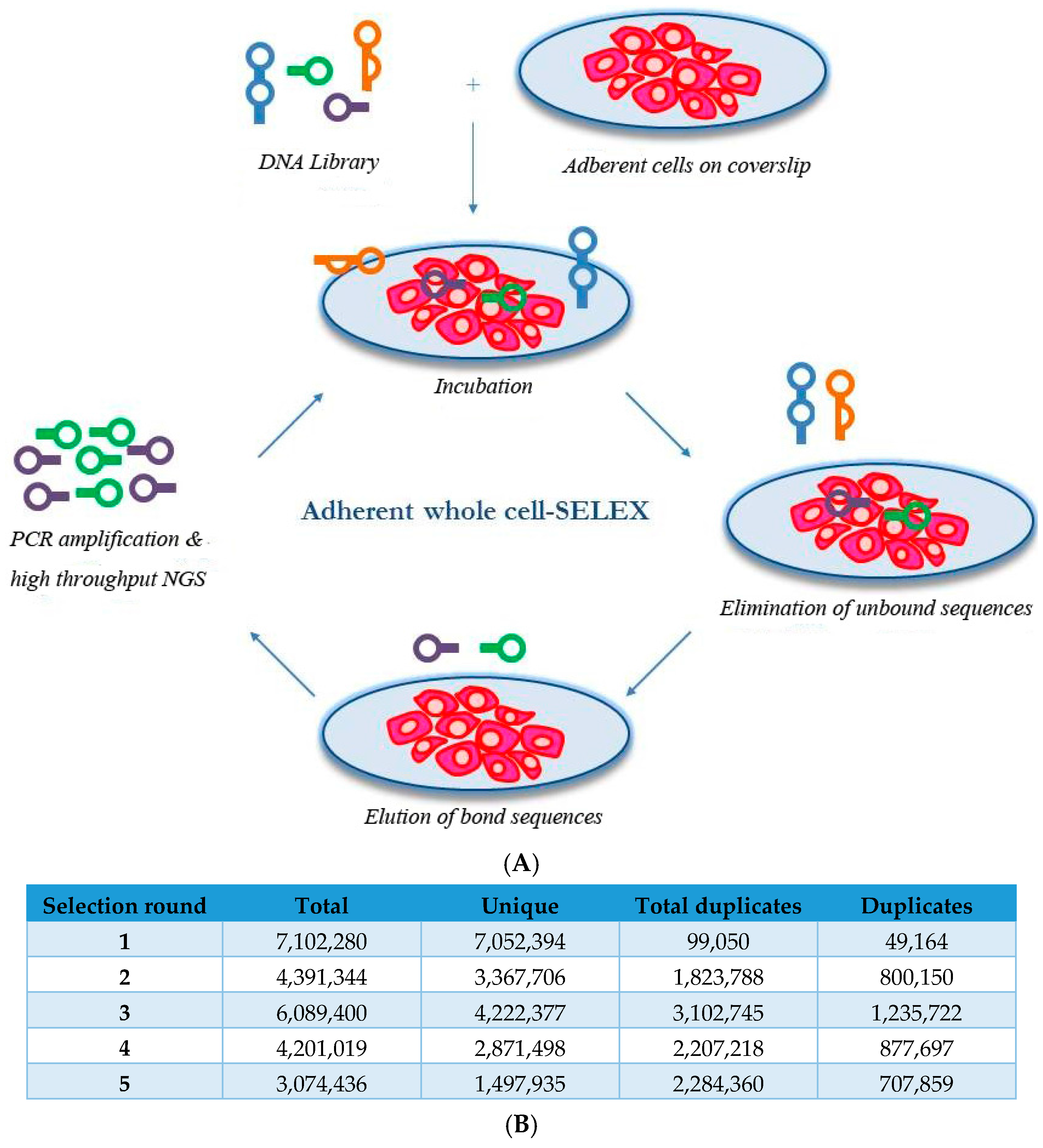
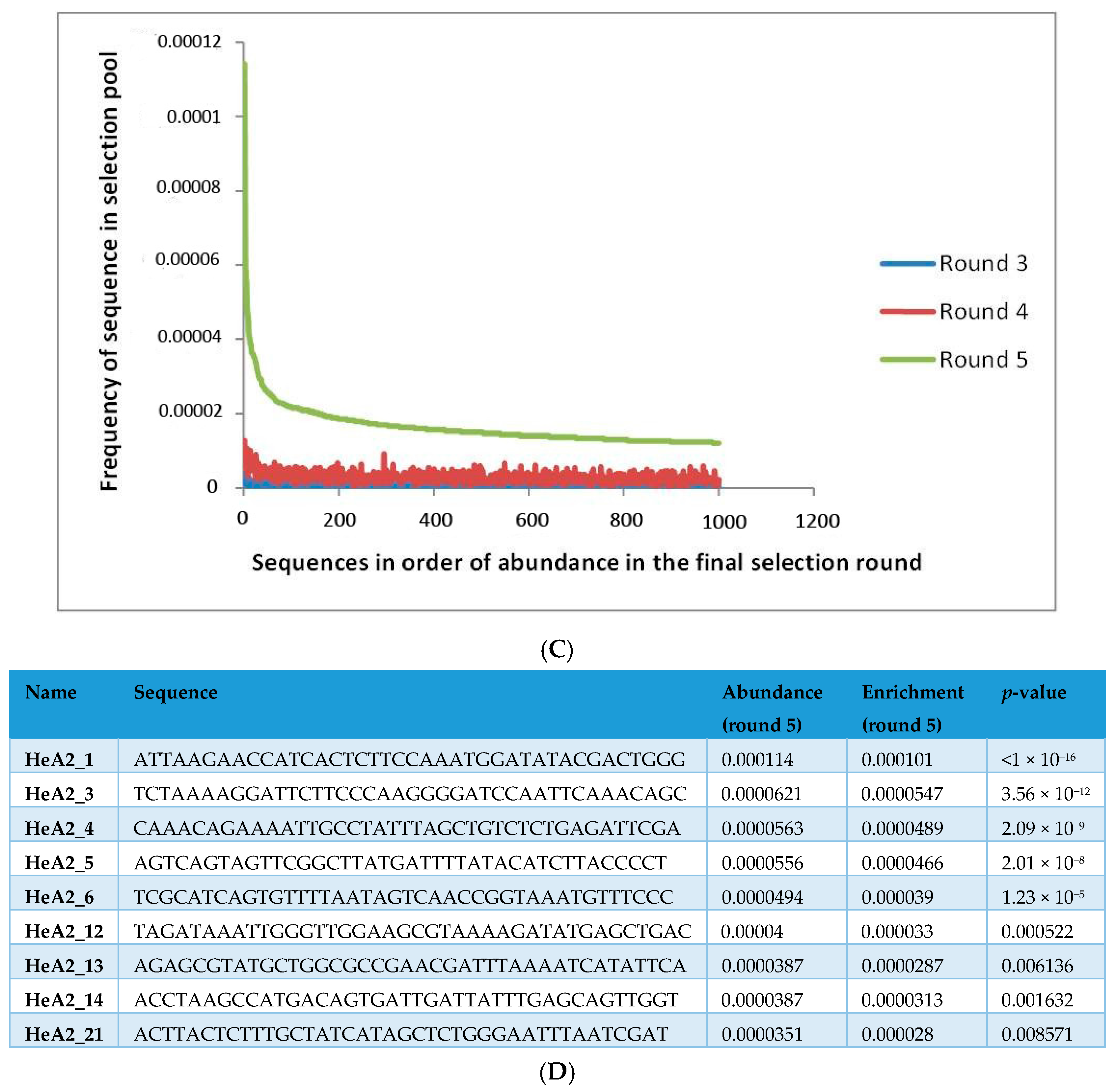
2.1. Whole-Cell SELEX on SKBR3 Breast Cancer Cells
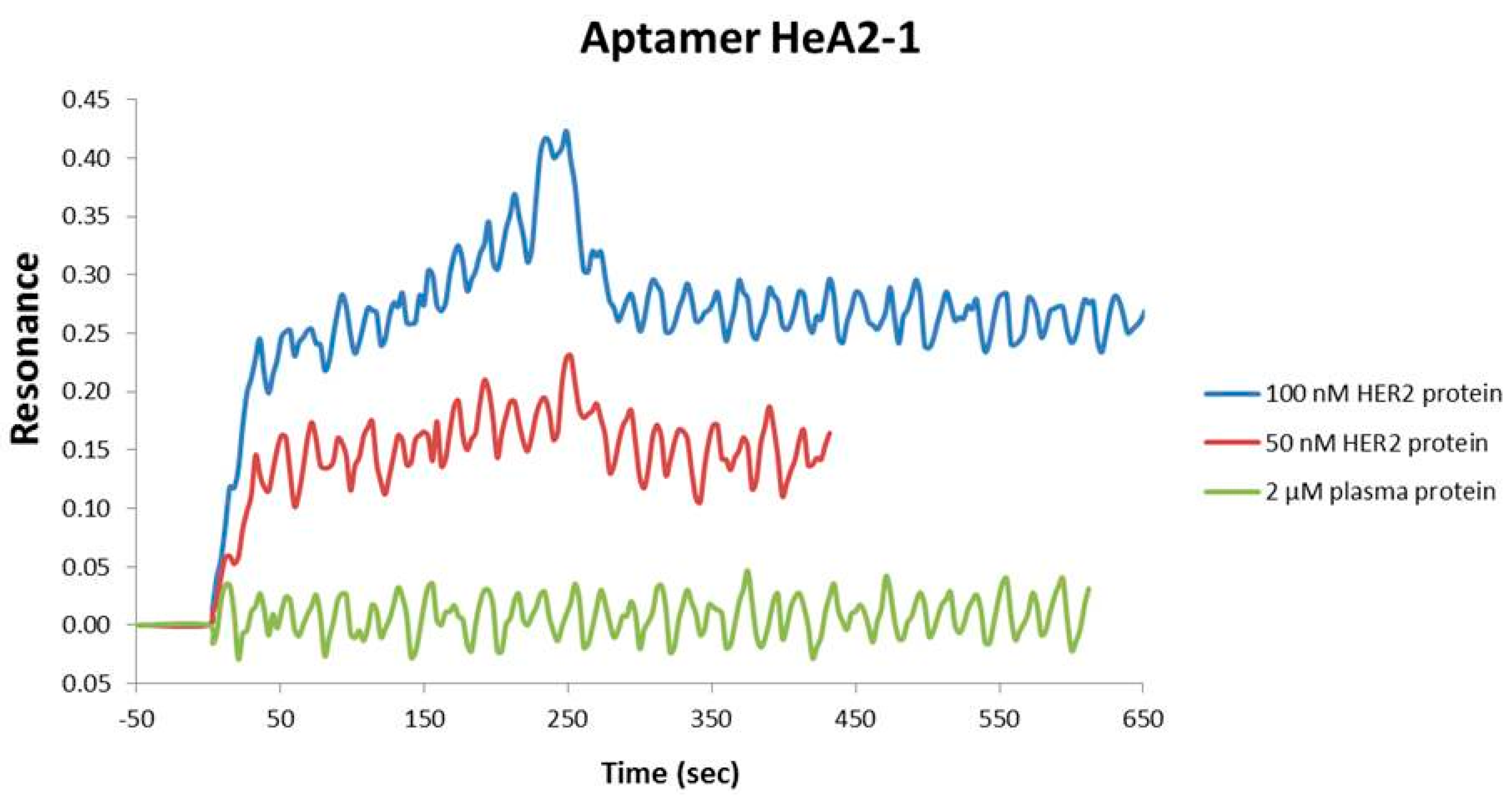
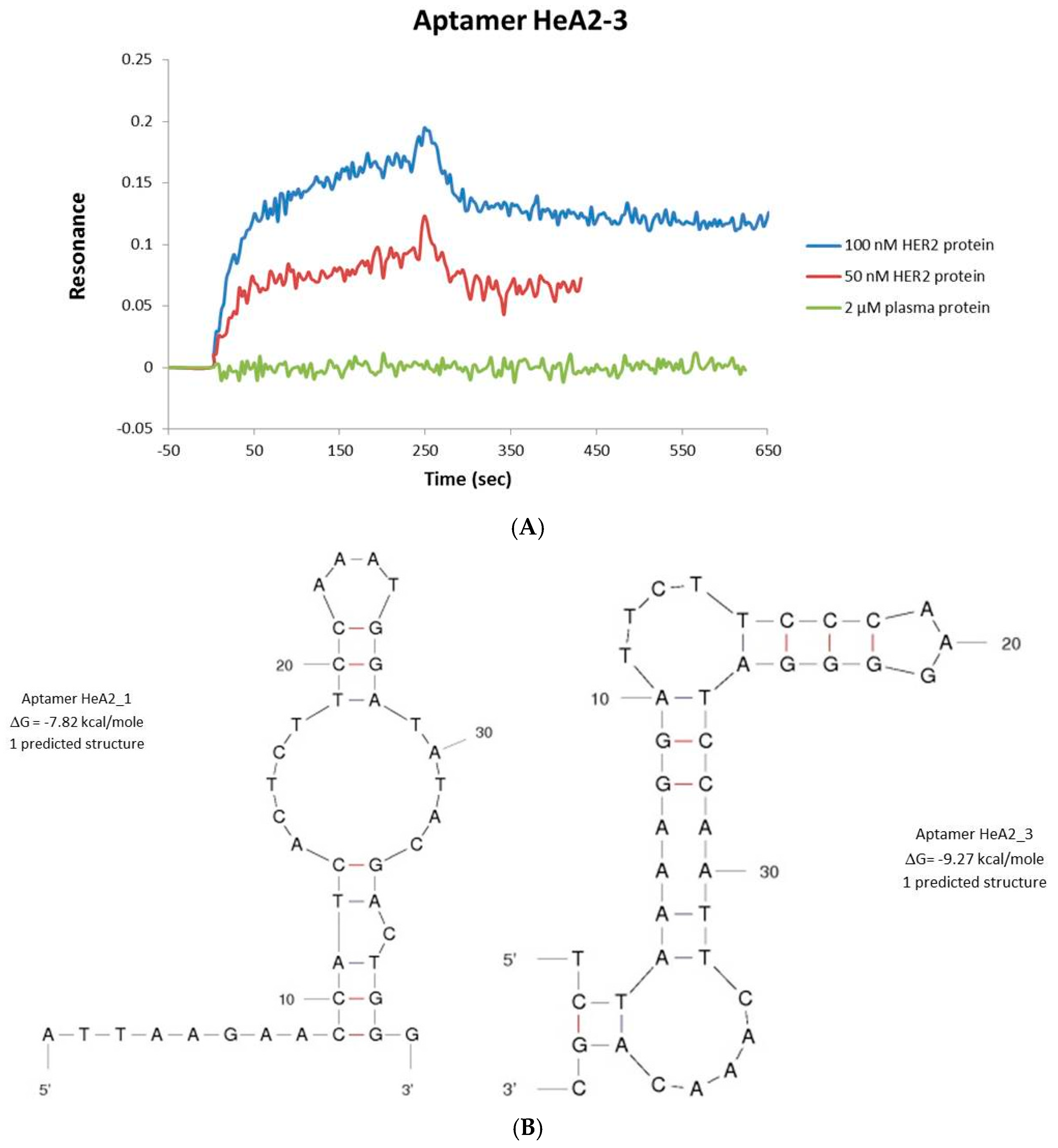
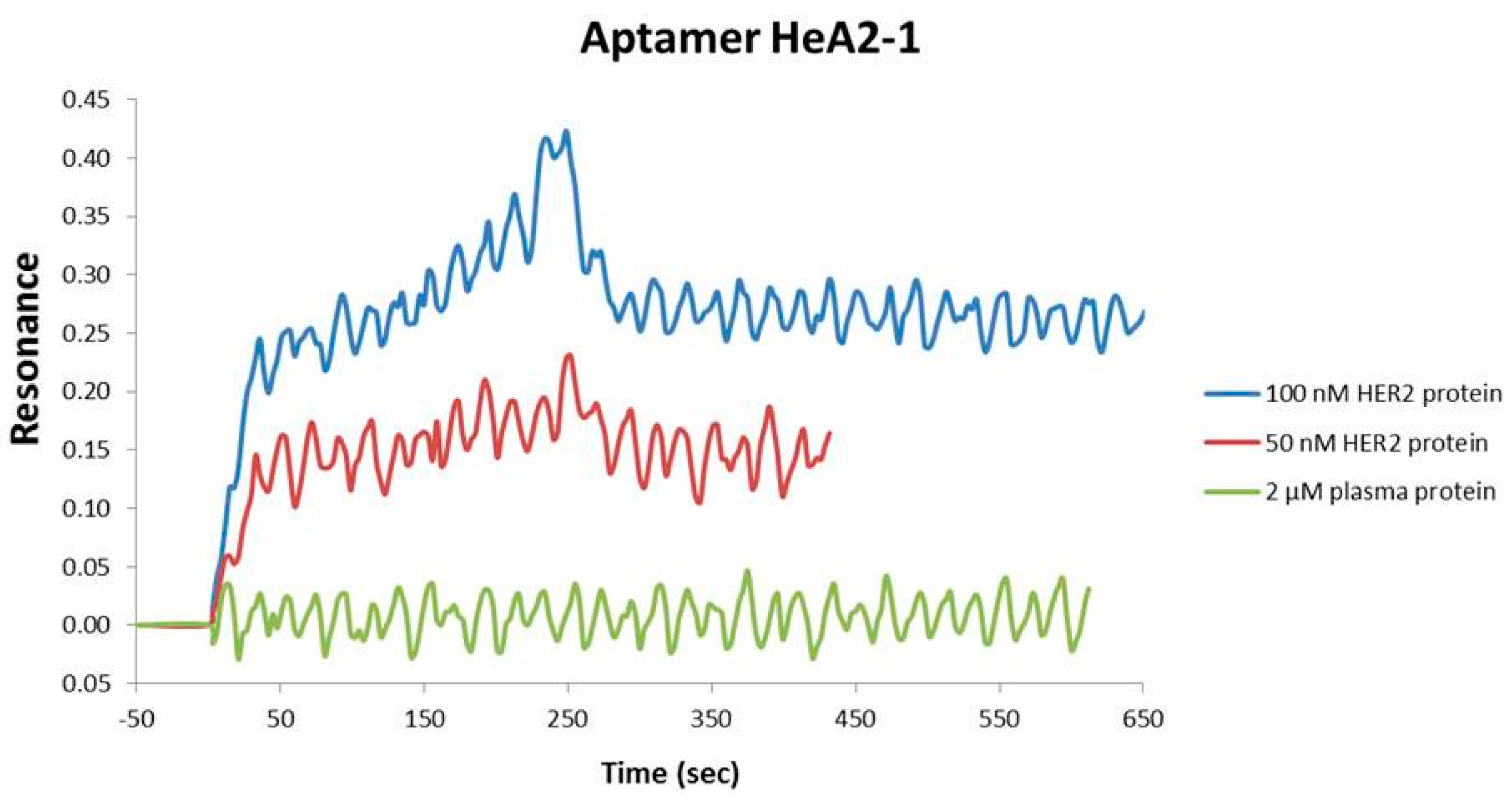
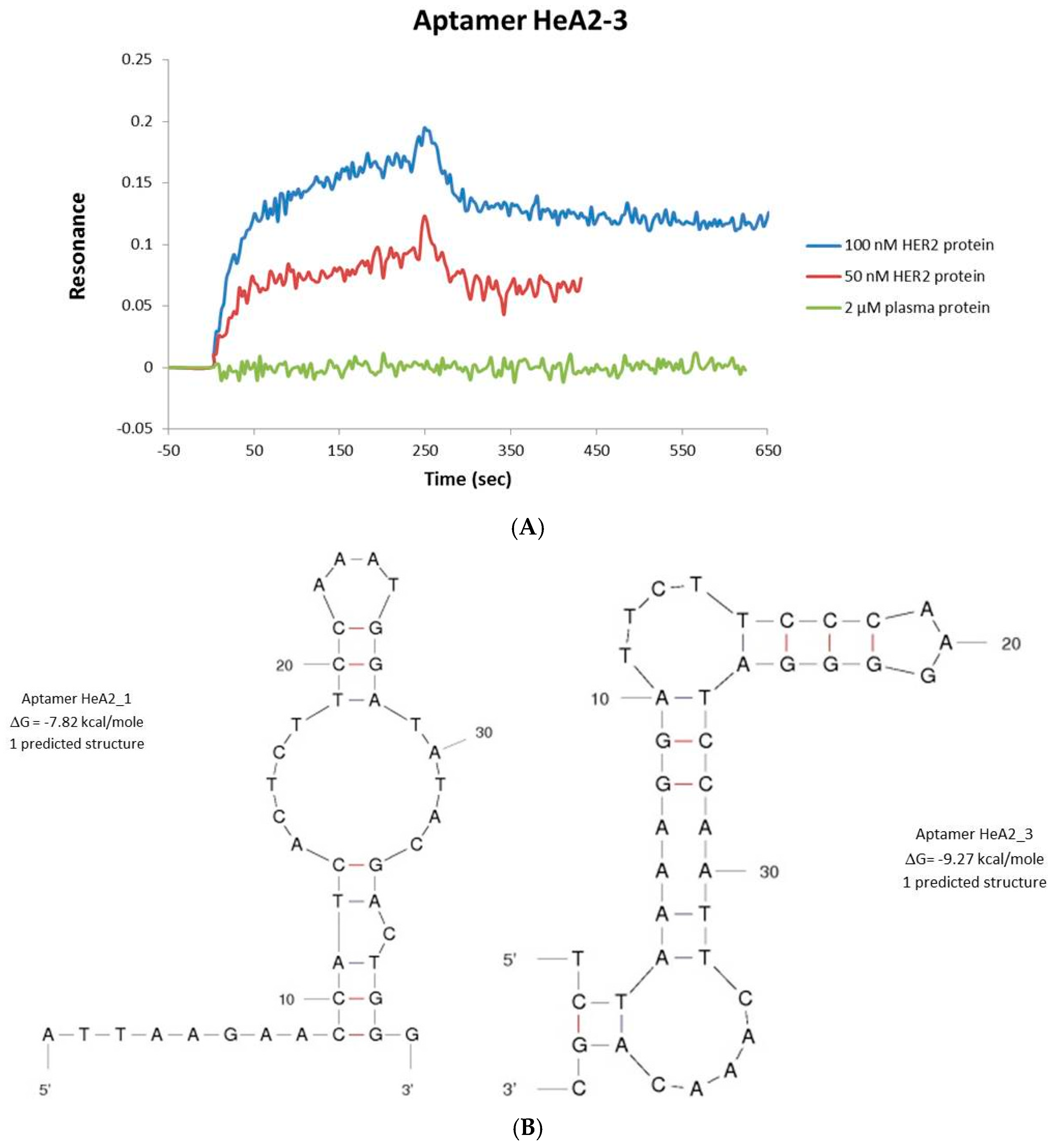
2.2. Aptamer Protein Binding and Binding Affinity
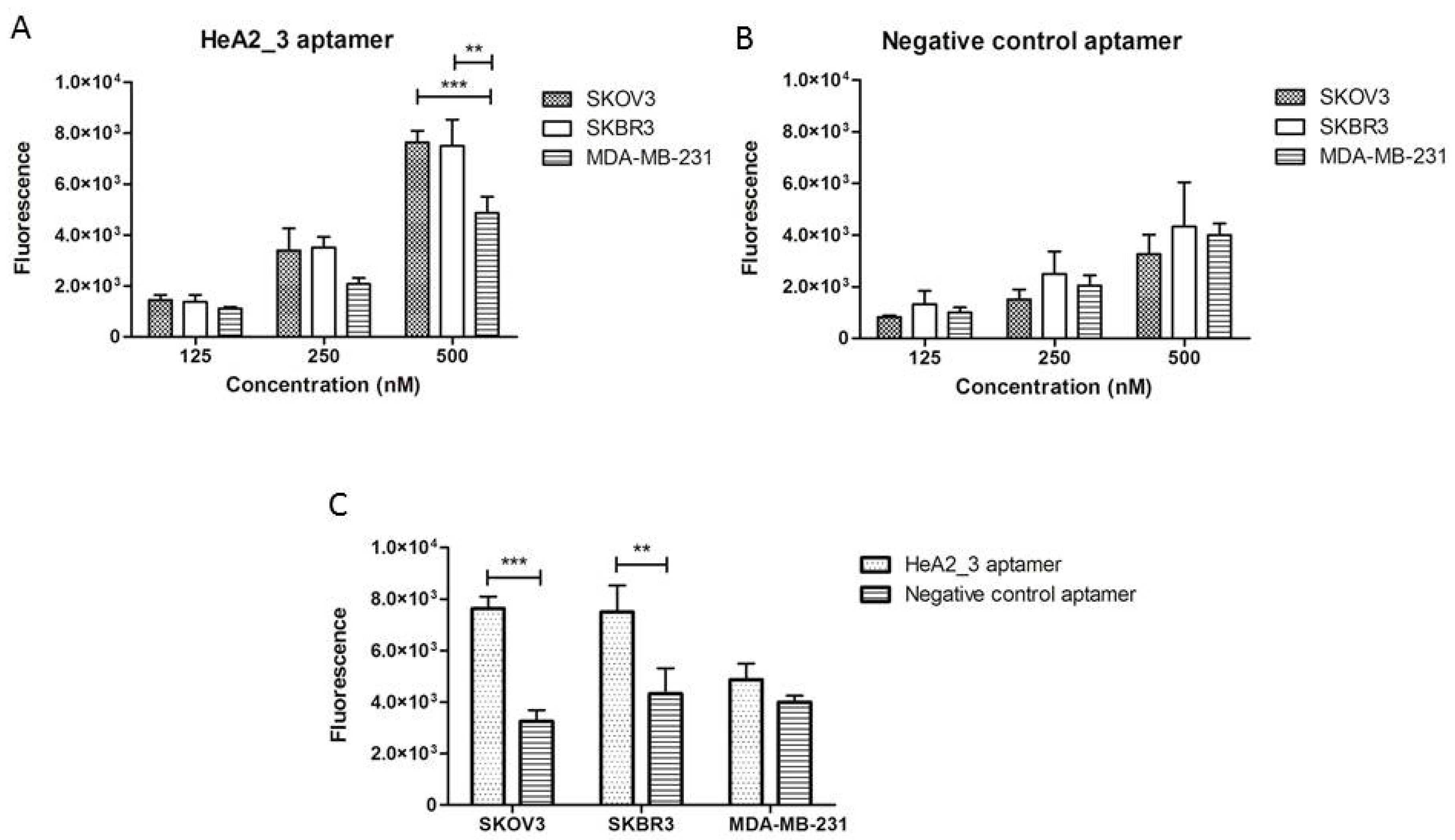
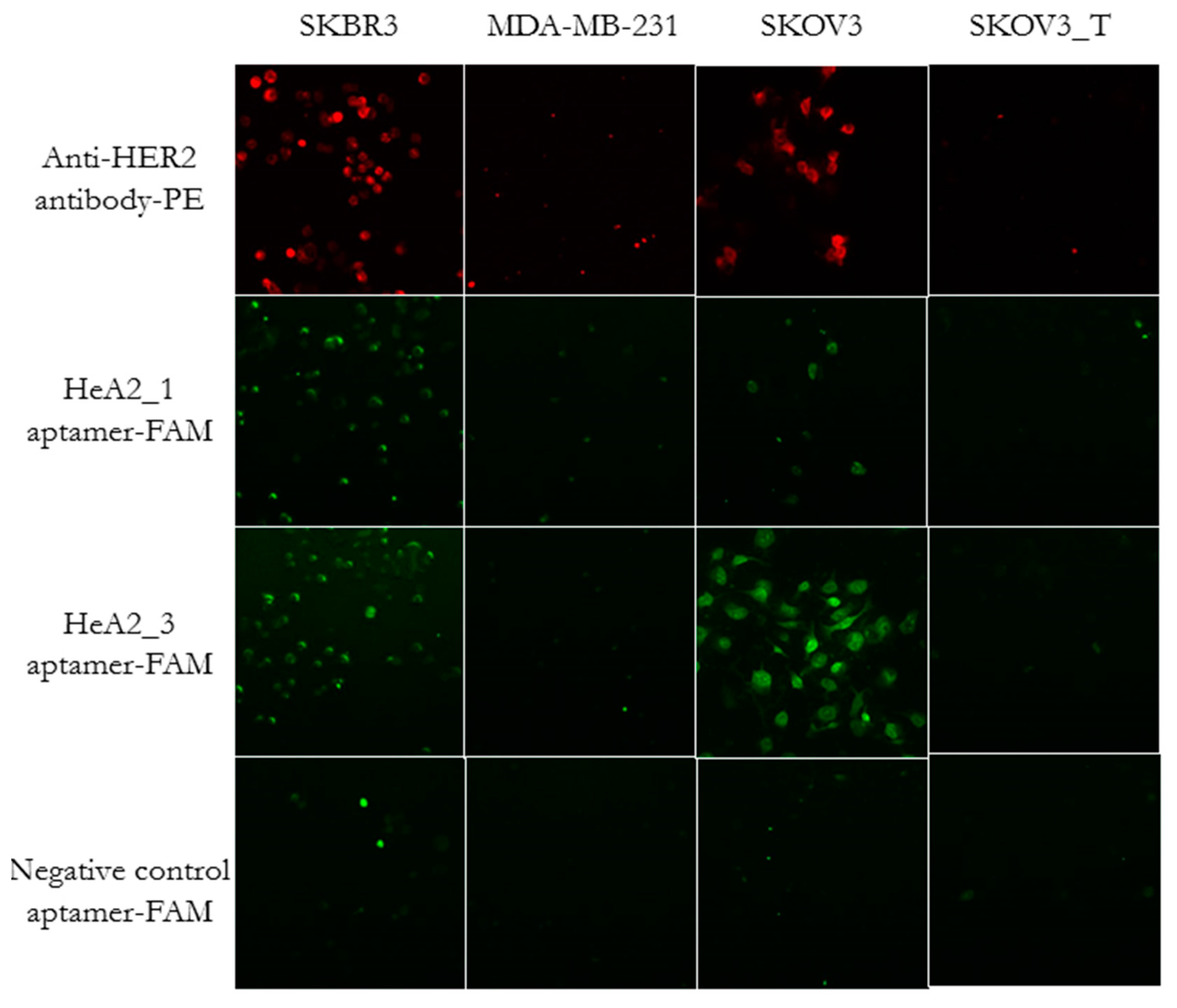
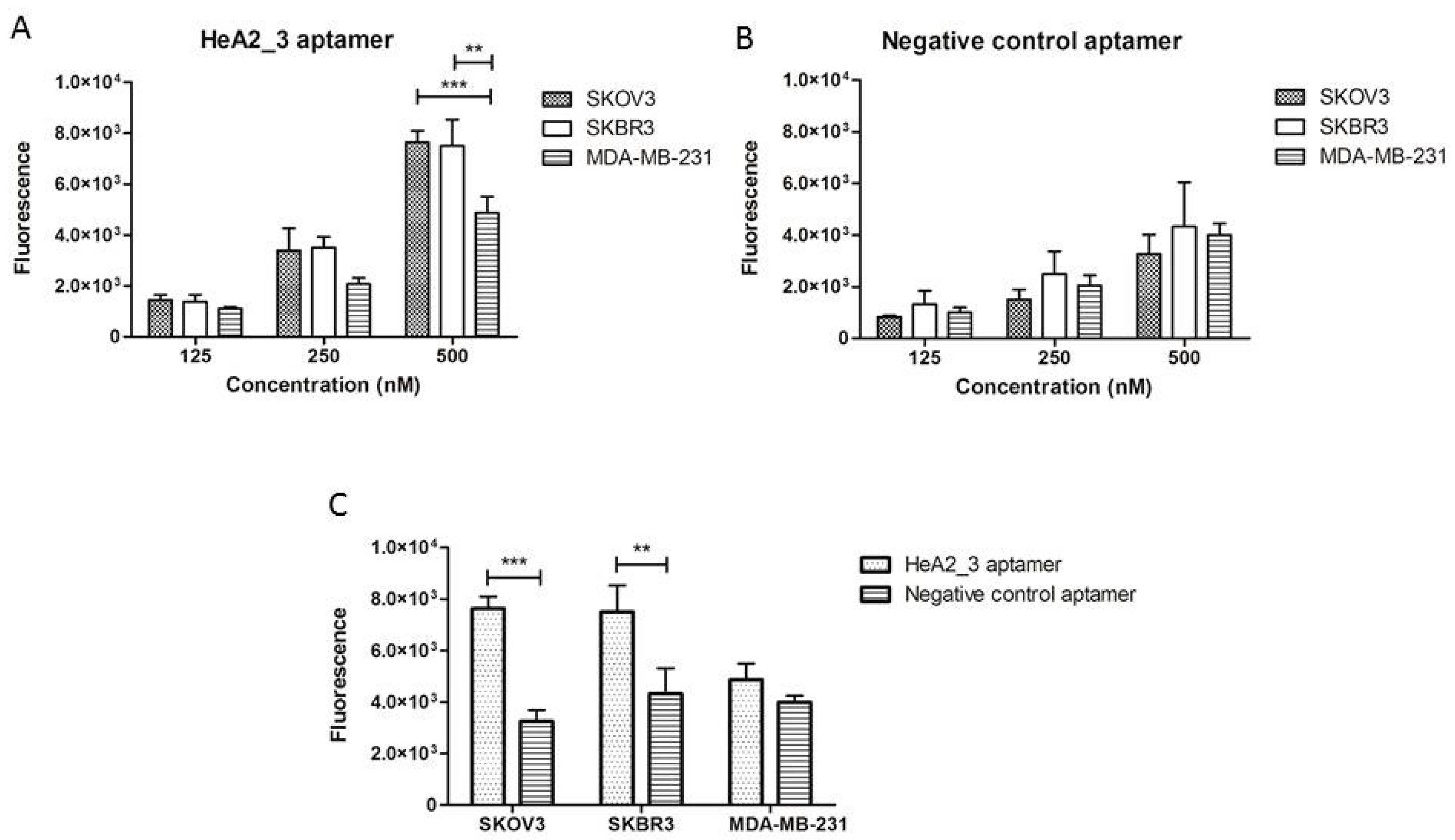
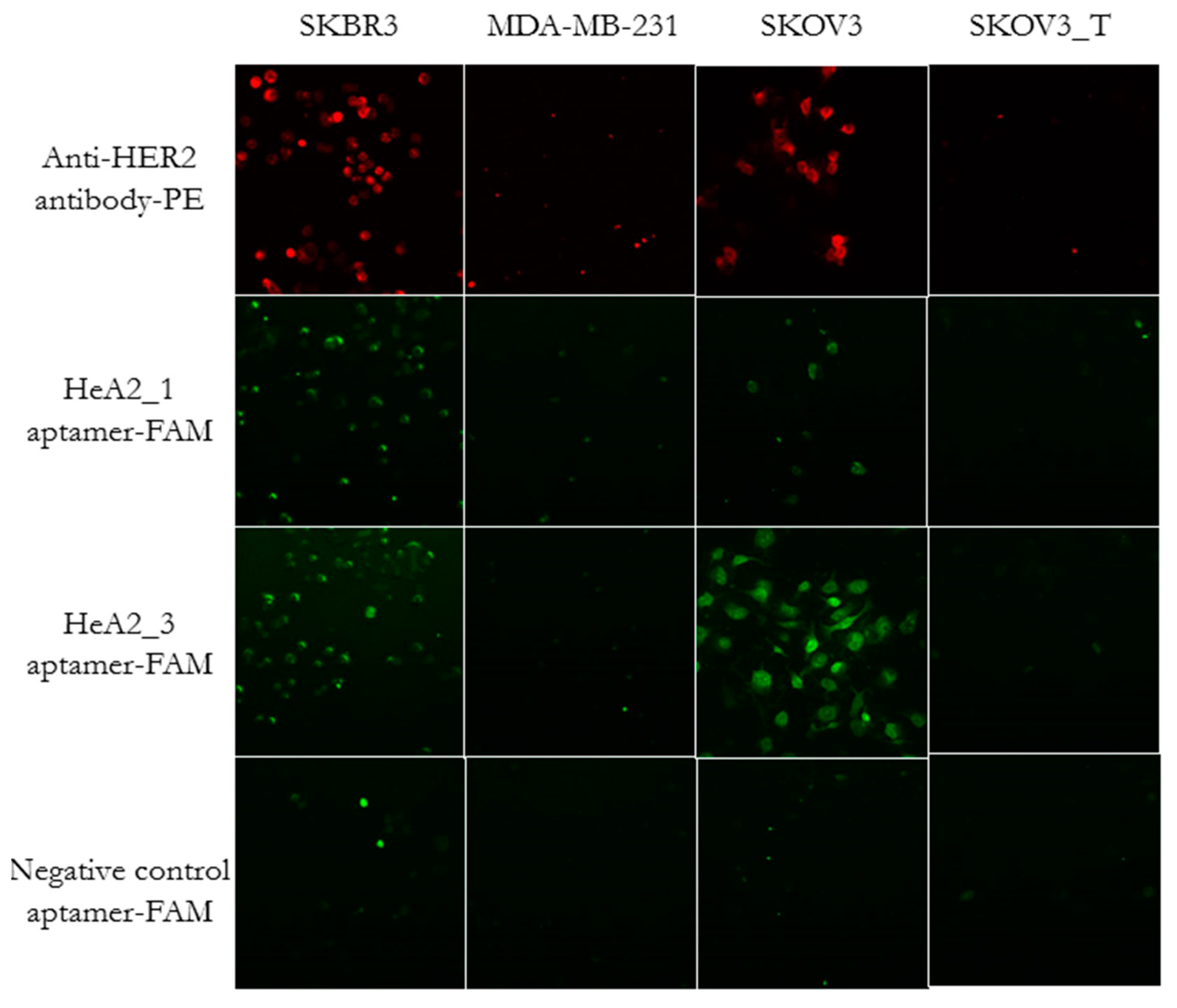
2.3. Aptamer Specificity for Different Cancer Cell Lines
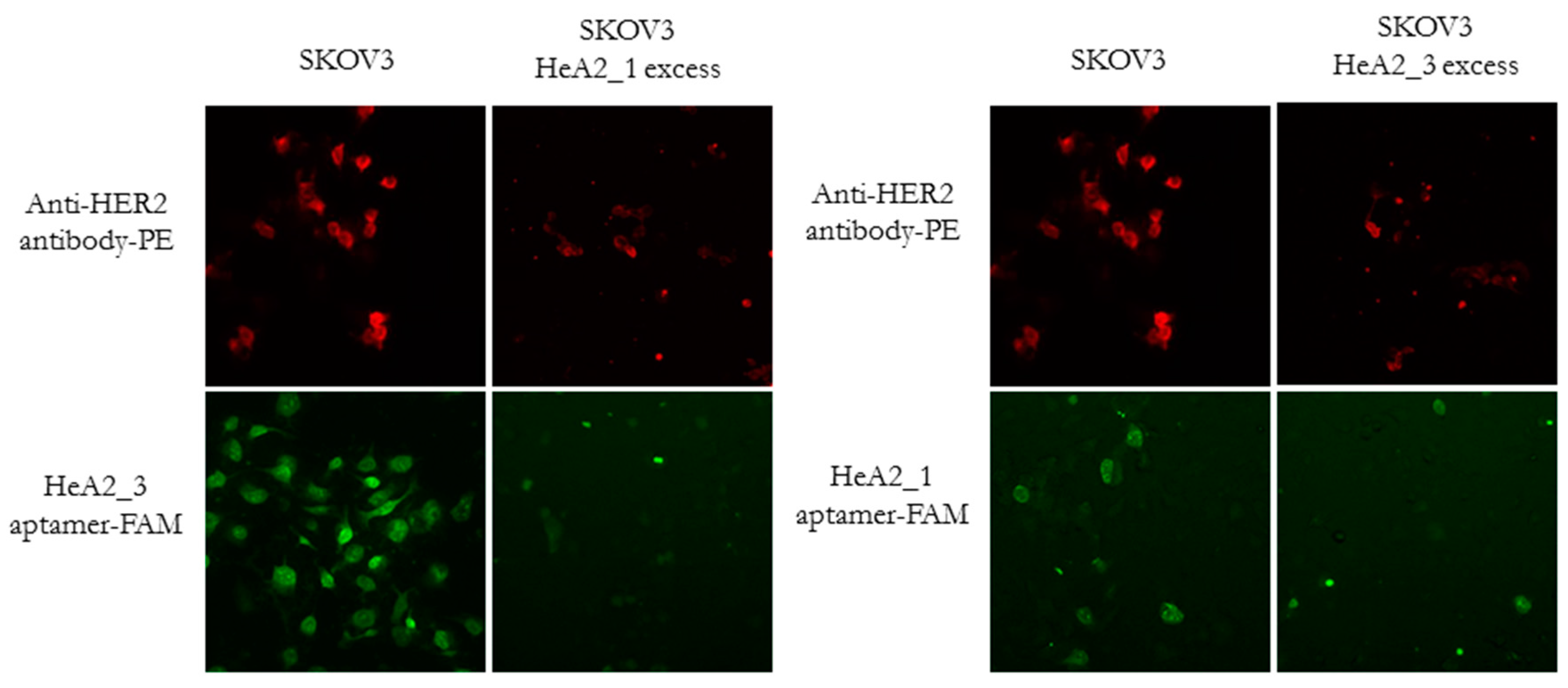
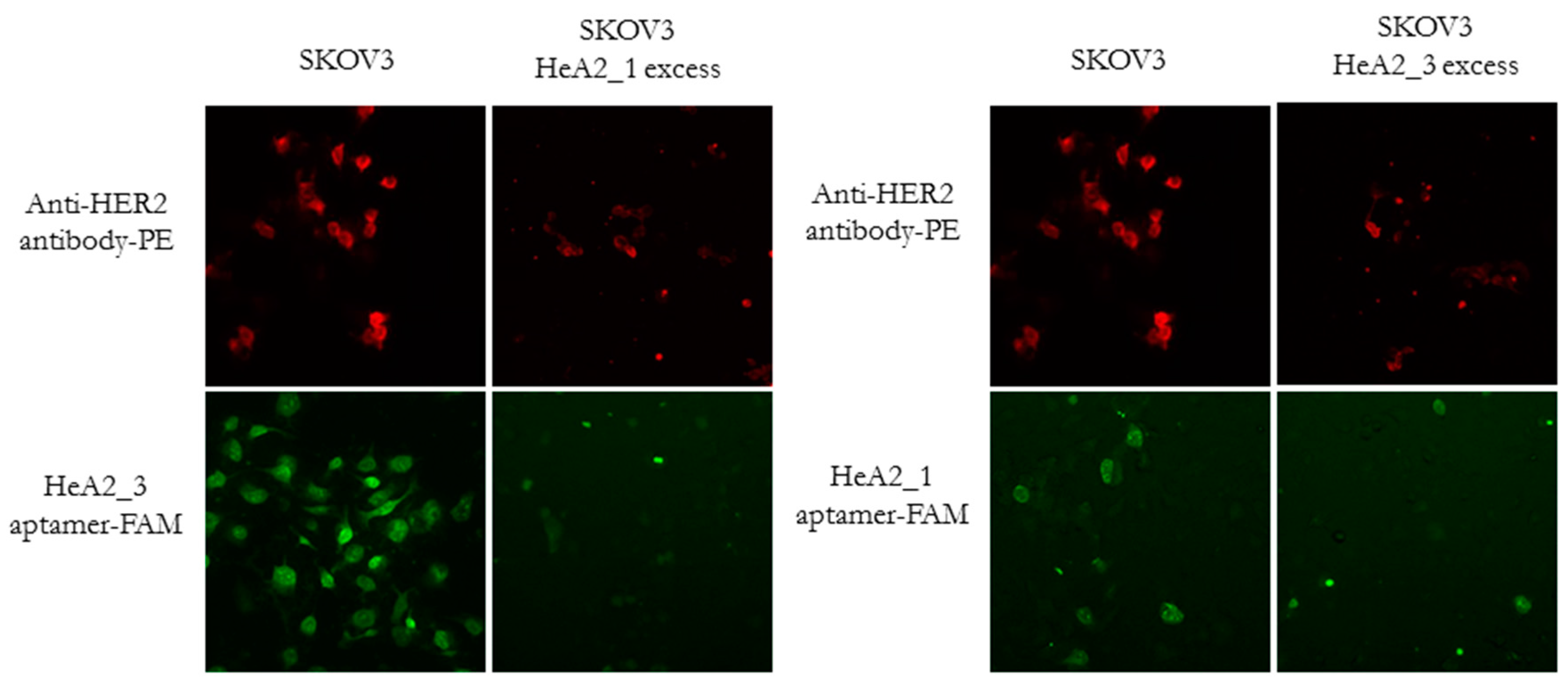
2.4. Competition for HER2 Binding
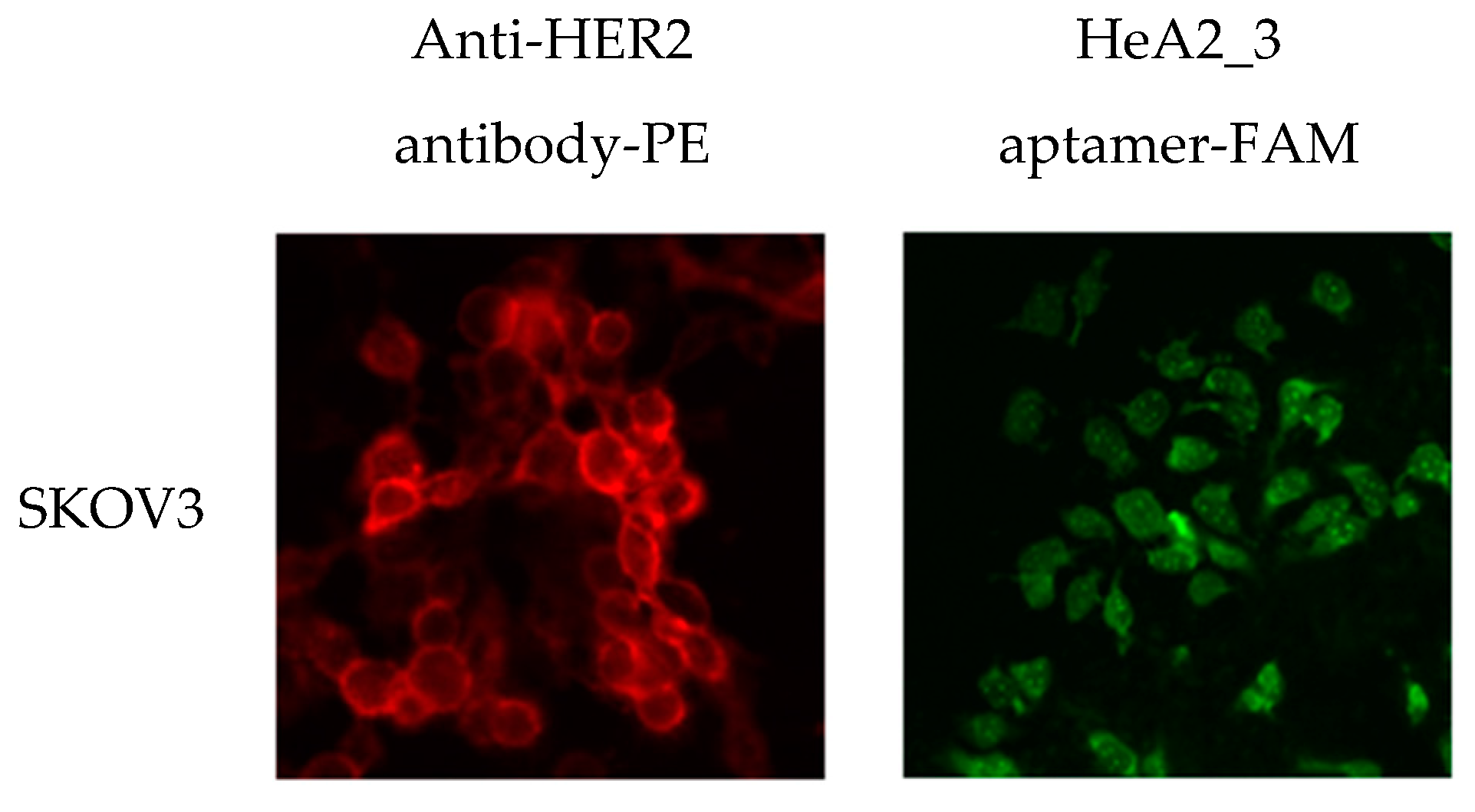
2.5. Internalization of Aptamer HeA2_3 into HER2 Overexpressing Cells
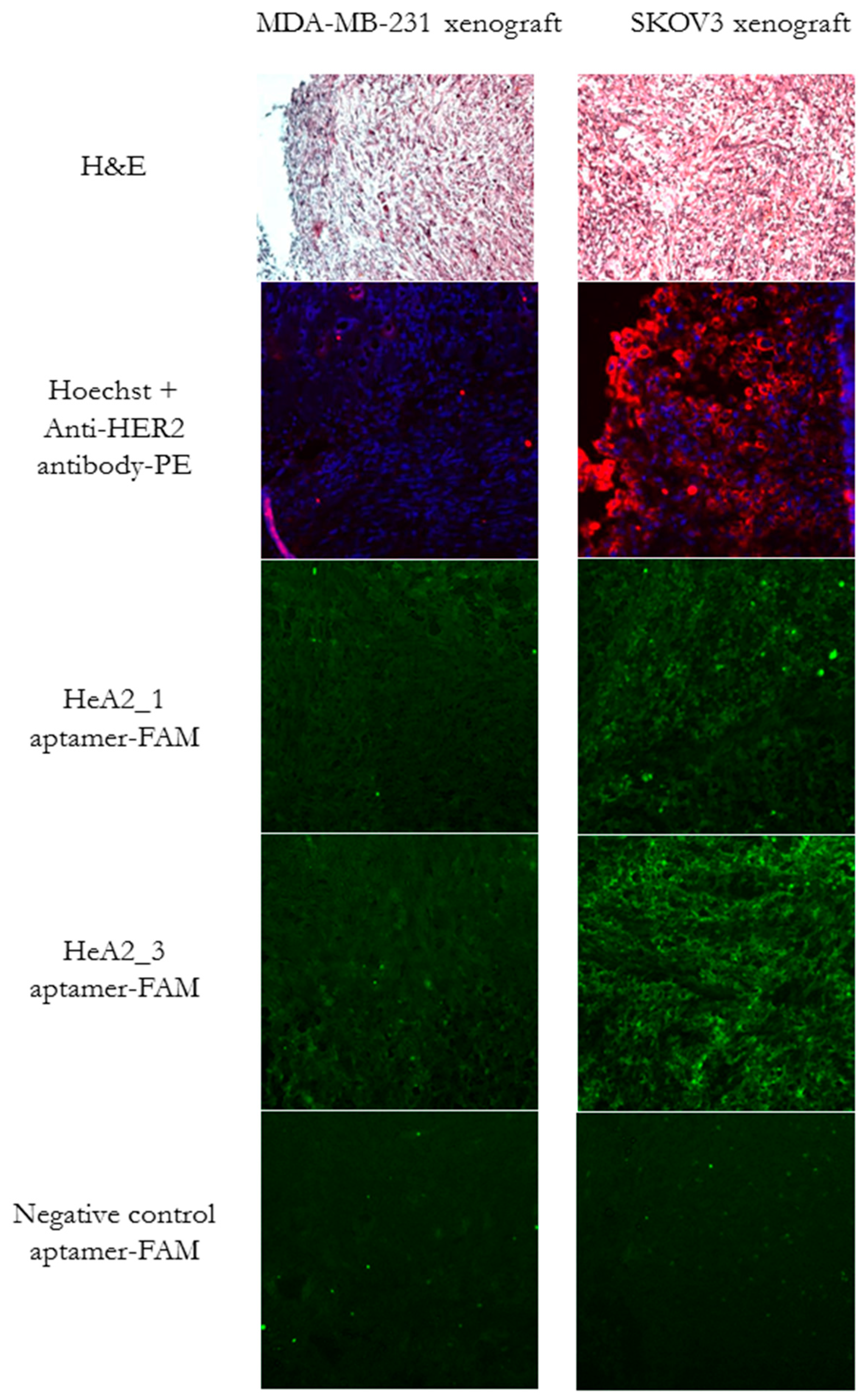
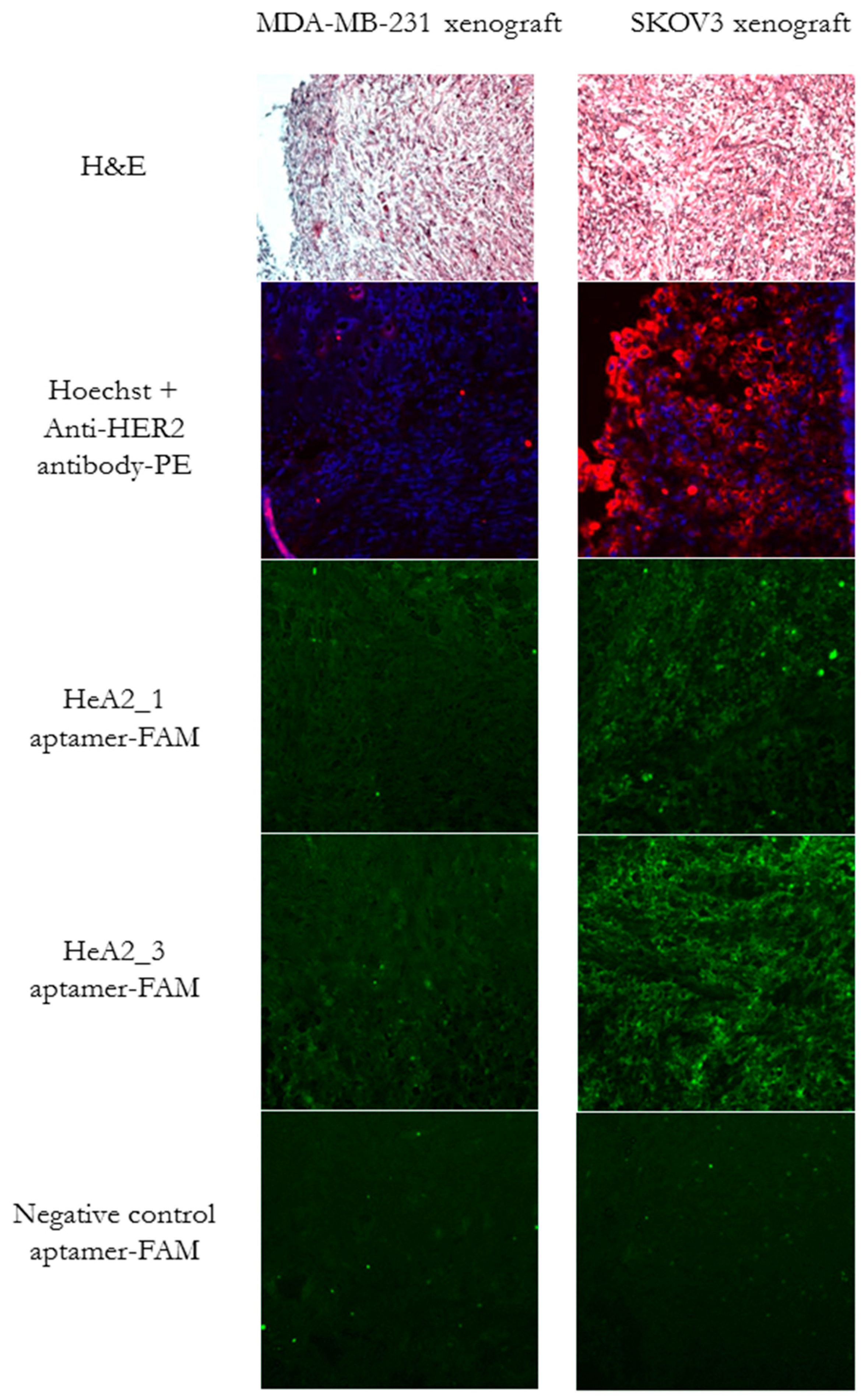
2.6. Ex Vivo Tumor Tissue Staining
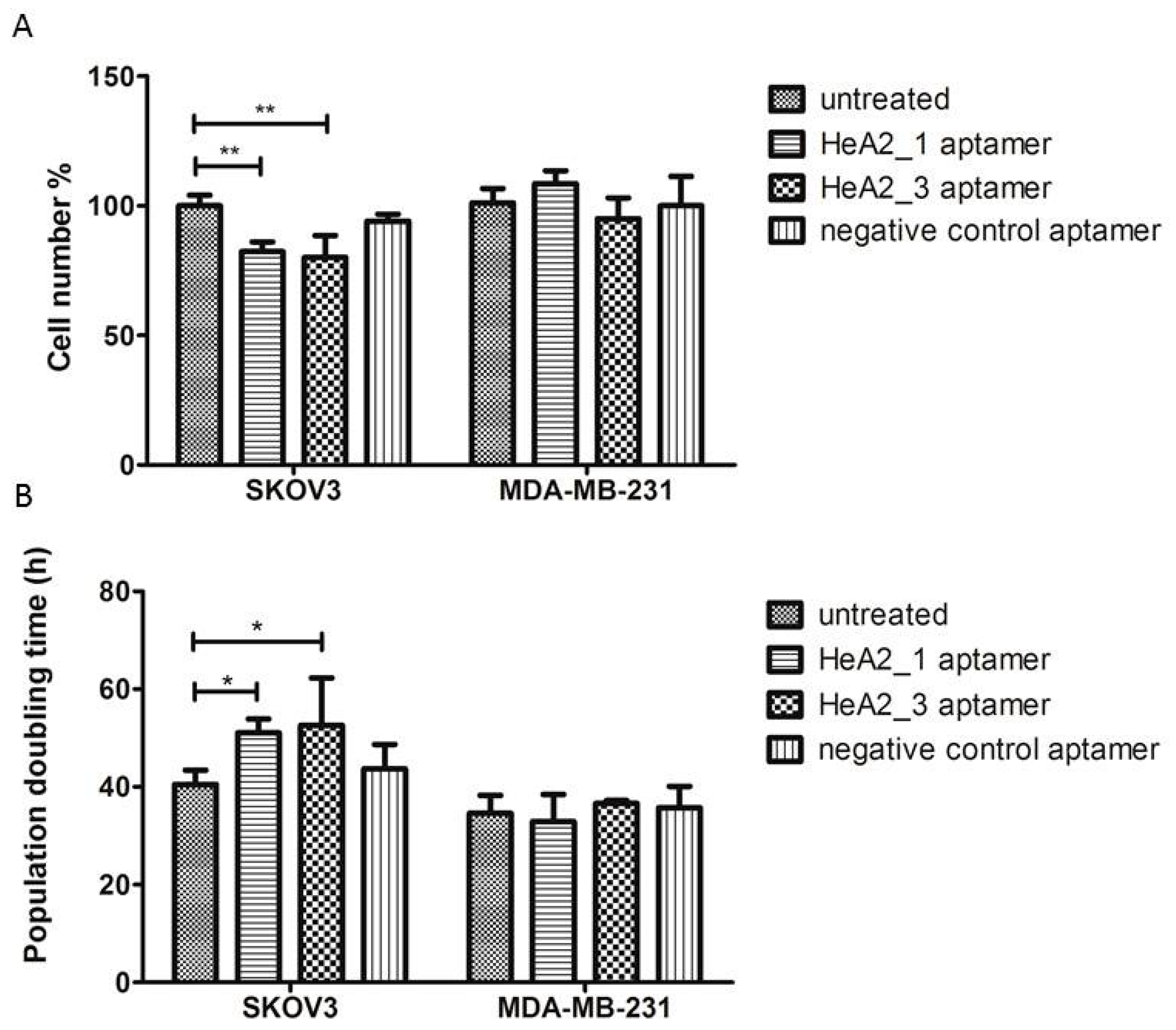
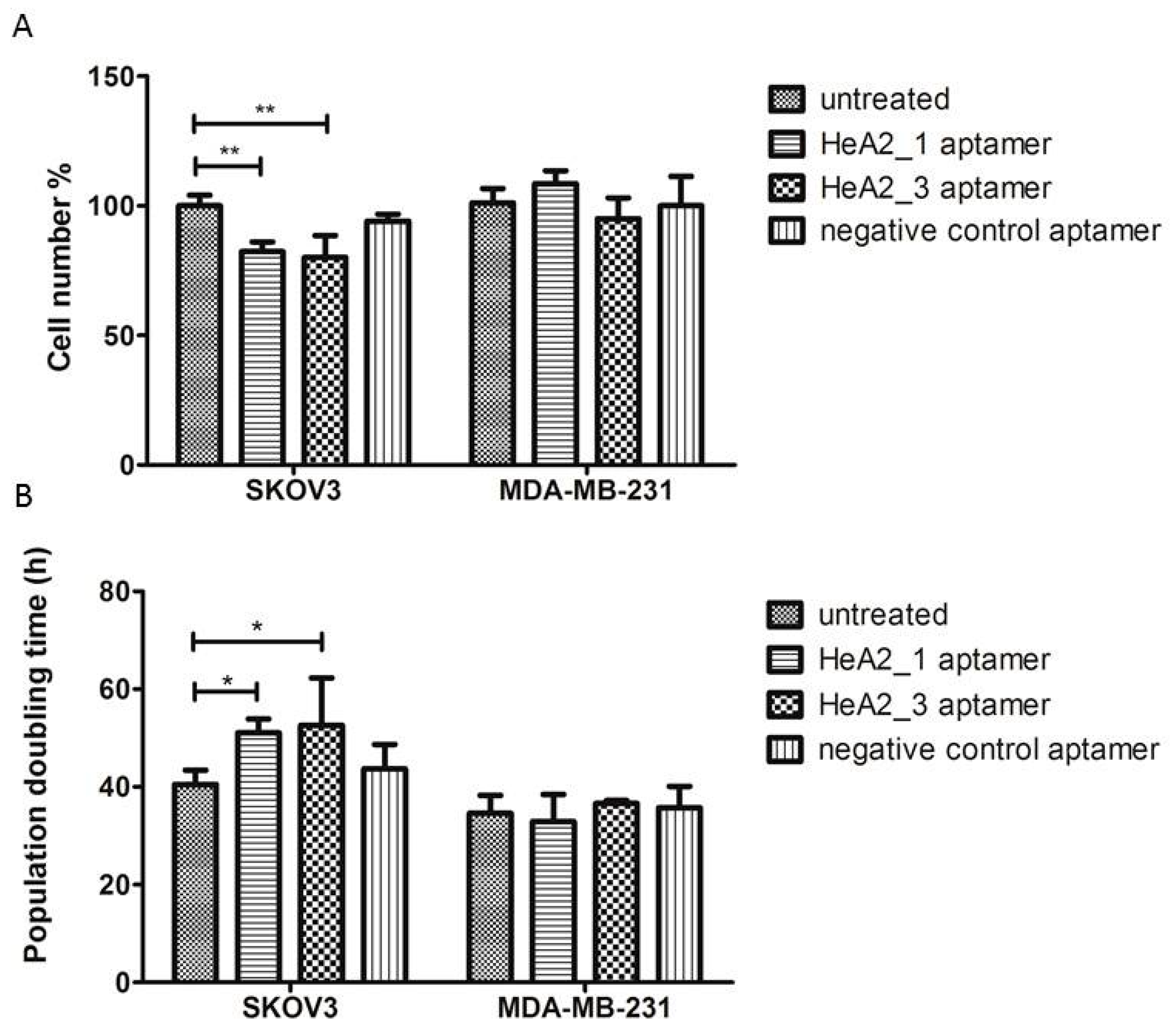
2.7. Targeted Inhibition of Cell Growth
3. Materials and Methods
3.1. Cell Lines and Cell Culture
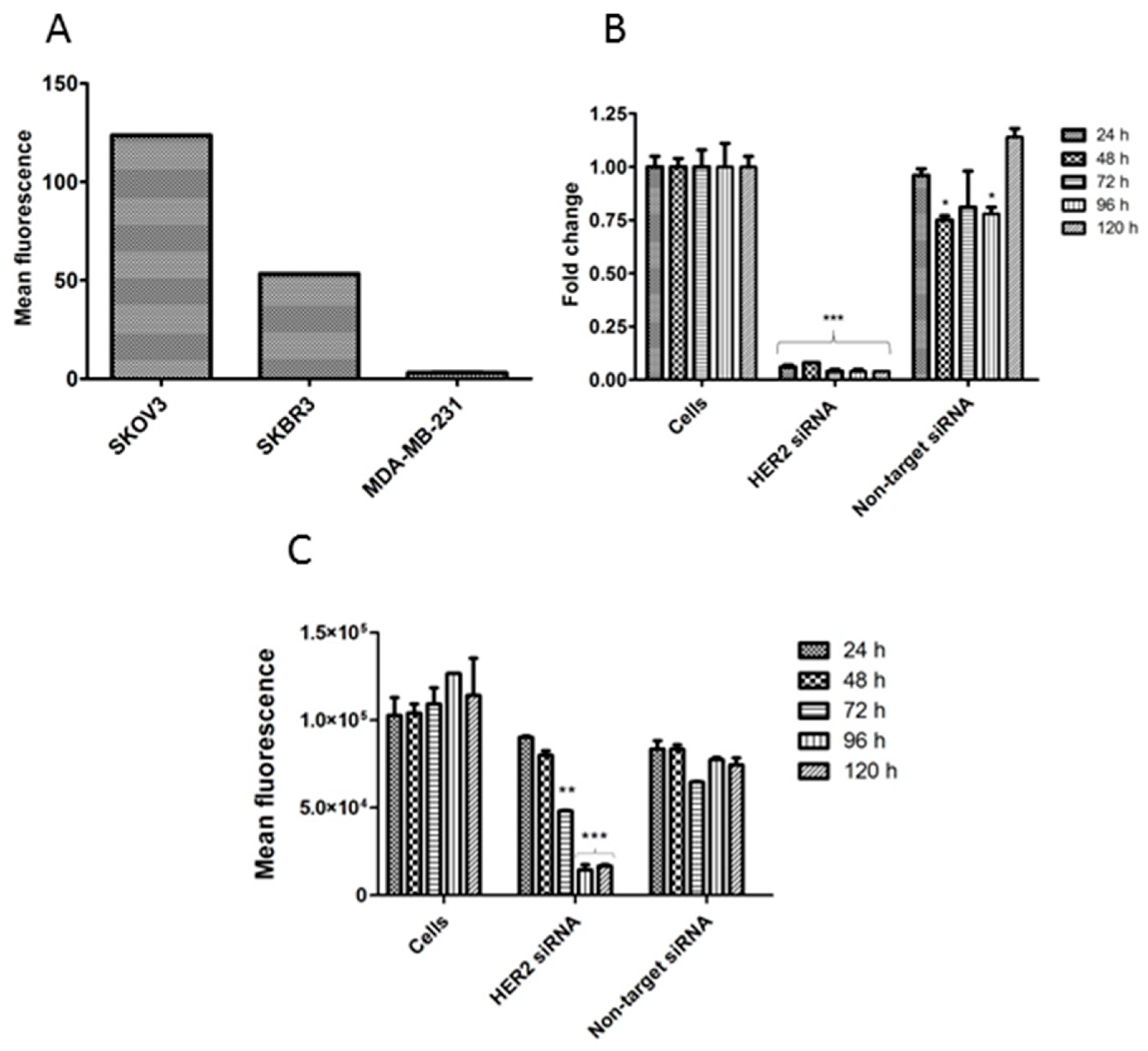
3.2. Transfection
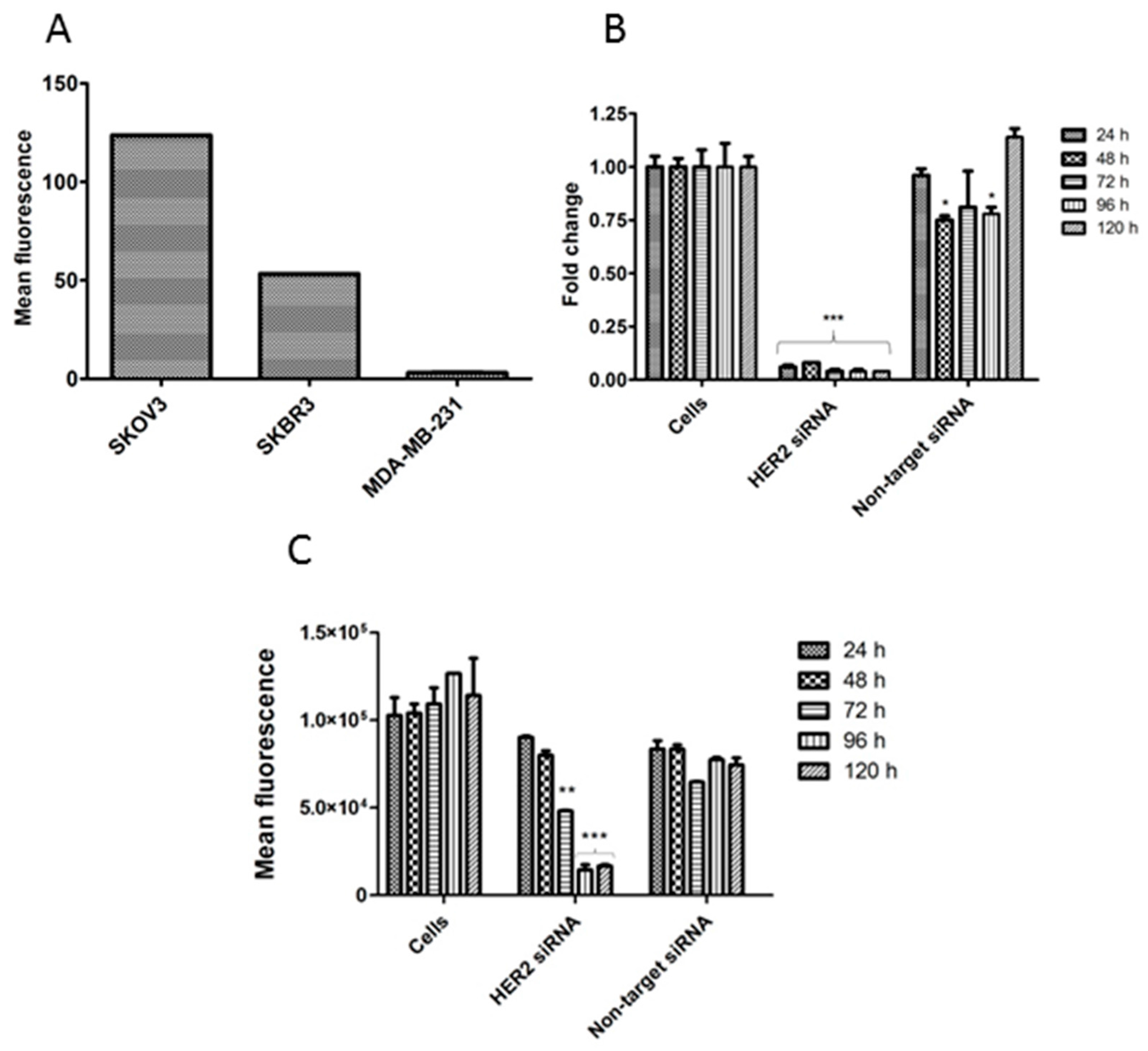
3.3. HER2 Expression Analysis by Q-PCR (mRNA Level)
3.4. HER2 Expression Analysis by Flow Cytometry (Protein Level)
3.5. Oligonucleotides
3.6. Aptamer Selection
3.7. Protein Binding by Surface Plasmon Resonance Imaging (SPRi)
3.8. Flow Cytometry
3.9. Fluorescent Microscopy—Cellular Staining
3.10. Fluorescent Microscopy—Tumor Tissue Staining
3.10.1. Animals
3.10.2. Ex Vivo Tumor Tissue Staining
3.11. Cell Proliferation Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Godolphin, W.; Jones, L.A.; Holt, J.A.; Wong, S.G.; Keith, D.E.; Levin, W.J.; Stuart, S.G.; Udove, J.; Ullrich, A.; et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989, 244, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y. HER2 over-expression and response to different chemotherapy regimens in breast cancer. J. Zhejiang Univ. Sci. B 2008, 9, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Musolino, A.; Ciccolallo, L.; Panebianco, M.; Fontana, E.; Zanoni, D.; Bozzetti, C.; Michiara, M.; Silini, E.M.; Ardizzoni, A. Multifactorial central nervous system recurrence susceptibility in patients with HER2-positive breast cancer: Epidemiological and clinical data from a population-based cancer registry study. Cancer 2011, 117, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Rouanet, P.; Roger, P.; Rousseau, E.; Thibault, S.; Romieu, G.; Mathieu, A.; Cretin, J.; Barneon, G.; Granier, M.; Maran-Gonzalez, A.; et al. HER2 overexpression a major risk factor for recurrence in pT1a-bN0M0 breast cancer: Results from a French regional cohort. Cancer Med. 2014, 3, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Fight HER2+ Breast Cancer with Herceptin. Available online: http://www.herceptin.com (accessed on 25 June 2015).
- Bartsch, R.; Wenzel, C.; Steger, G.G. Trastuzumab in the management of early and advanced stage breast cancer. Biologics 2007, 1, 19–31. [Google Scholar] [PubMed]
- Claret, F.X.; Vu, T.T. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef]
- Sengupta, P.P.; Northfelt, D.W.; Gentile, F.; Zamorano, J.L.; Khandheria, B.K. Trastuzumab-induced cardiotoxicity: Heart failure at the crossroads. Mayo Clin. Proc. 2008, 83, 197–203. [Google Scholar] [CrossRef]
- Ulrich, H. RNA aptamers: From basic science towards therapy. In RNA towards Medicine; Erdmann, V., Barciszewski, J., Brosius, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 173, pp. 305–326. [Google Scholar]
- Bouchard, P.R.; Hutabarat, R.M.; Thompson, K.M. Discovery and development of therapeutic aptamers. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhang, K. The application of aptamers in cancer research: An up-to-date review. Future Oncol. 2013, 9, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Barbas, A.S.; Mi, J.; Clary, B.M.; White, R.R. Aptamer applications for targeted cancer therapy. Future Oncol. 2010, 6, 1117–1126. [Google Scholar] [CrossRef] [PubMed]
- Bruno, J.G. A review of therapeutic aptamer conjugates with emphasis on new approaches. Pharmaceuticals 2013, 6, 340–357. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Hahn, U.; Rentmeister, A. Cell-specific aptamers as emerging therapeutics. J. Nucleic Acids 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Thiel, K.W.; Giangrande, P.H. Therapeutic applications of DNA and RNA aptamers. Oligonucleotides 2009, 19, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, M.S. Therapeutic aptamers march on. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, P.; Kurniawan, H.; Byrne, M.E.; Wower, J. Therapeutic RNA aptamers in clinical trials. Eur. J. Pharm. Sci. 2013, 48, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Ellington, A.D.; Szostak, J.W. In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.C.; Zarbl, H. Use of cell-selex to generate DNA aptamers as molecular probes of HPV-associated cervical cancer cells. PLoS ONE 2012, 7, e36103. [Google Scholar] [CrossRef] [PubMed]
- Penner, G. Dubbles, an Alternative to Selex. Available online: https://www.youtube.com/watch?v=rmpvqCX1WqA (accessed on 25 June 2015).
- Blind, M.; Blank, M. Aptamer selection technology and recent advances. Mol. Ther. Nucleic Acids 2015, 4. [Google Scholar] [CrossRef]
- Pan, W.; Xin, P.; Patrick, S.; Dean, S.; Keating, C.; Clawson, G. Primer-free aptamer selection using a random DNA library. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Clawson, G.A. The shorter the better: Reducing fixed primer regions of oligonucleotide libraries for aptamer selection. Molecules 2009, 14, 1353–1369. [Google Scholar] [CrossRef] [PubMed]
- Marimuthu, C.; Tang, T.H.; Tominaga, J.; Tan, S.C.; Gopinath, S.C. Single-stranded DNA (ssDNA) production in DNA aptamer generation. Analyst 2012, 137, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Le, L.C.; Cruz-Aguado, J.A.; Penner, G.A. DNA Ligands for Aflatoxin and Zearalenone. U.S. Patent 20,120,225,494, 6 September 2011. [Google Scholar]
- Penner, G. Detection Systems. Available online: http://neoventures.ca/products/mycotoxin-testing/ (accessed on 26 June 2015).
- Schütze, T.; Wilhelm, B.; Greiner, N.; Braun, H.; Peter, F.; Mörl, M.; Erdmann, V.A.; Lehrach, H.; Konthur, Z.; Menger, M.; et al. Probing the SELEX process with next-generation sequencing. PLoS ONE 2011, 6, e29604. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.; Xiao, Y.; Nie, J.; Stewart, R.; Csordas, A.T.; Oh, S.S.; Thomson, J.A.; Soh, H.T. Quantitative selection of DNA aptamers through microfluidic selection and high-throughput sequencing. Proc. Natl. Acad. Sci. USA 2010, 107, 15373–15378. [Google Scholar] [CrossRef] [PubMed]
- Dausse, E.; Taouji, S.; Evade, L.; di Primo, C.; Chevet, E.; Toulme, J.J. HAPIscreen, a method for high-throughput aptamer identification. J. Nanobiotechnol. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Bair, T.; Thiel, K.W.; Dassie, J.P.; Rockey, W.M.; Howell, C.A.; Liu, X.Y.; Dupuy, A.J.; Huang, L.; Owczarzy, R.; et al. Nucleotide bias observed with a short SELEX RNA aptamer library. Nucleic Acid Ther. 2011, 21, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.-S.; Huh, Y.-M.; Kim, S.; Lee, D.K. Isolation of RNA aptamers targeting HER2-overexpressing breast cancer cells using cell-SELEX. Bull. Korean Chem. Soc. 2009, 30, 1827–1831. [Google Scholar]
- Kim, M.Y.; Jeong, S. In vitro selection of RNA aptamer and specific targeting of ErbB2 in breast cancer cells. Nucleic Acid Ther. 2011, 21, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Thiel, K.W.; Hernandez, L.I.; Dassie, J.P.; Thiel, W.H.; Liu, X.; Stockdale, K.R.; Rothman, A.M.; Hernandez, F.J.; McNamara, J.O., 2nd; Giangrande, P.H. Delivery of chemo-sensitizing siRNAs to HER2+-breast cancer cells using RNA aptamers. Nucleic Acids Res. 2012, 40, 6319–6337. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Thirstrup, D.; Jarvis, T.C.; Schneider, D.J.; Wilcox, S.K.; Carter, J.; Zhang, C.; Gelinas, A.; Weiss, A.; Janjic, N.; et al. Rapid histochemistry using slow off-rate modified aptamers with anionic competition. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Duan, J.-H.; Song, Y.-M.; Ma, J.; Wang, F.-D.; Lu, X.; Yang, X.-D. Novel HER2 aptamer selectively delivers cytotoxic drug to HER2-positive breast cancer cells in vitro. J. Transl. Med. 2012, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Mahlknecht, G.; Maron, R.; Mancini, M.; Schechter, B.; Sela, M.; Yarden, Y. Aptamer to ErbB-2/HER2 enhances degradation of the target and inhibits tumorigenic growth. Proc. Natl. Acad. Sci. USA 2013, 110, 8170–8175. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Duan, J.; Cao, B.; Zhang, L.; Lu, X.; Wang, F.; Yao, F.; Zhu, Z.; Yuan, W.; Wang, C.; et al. Selection of a novel DNA thioaptamer against HER2 structure. Clin. Transl. Oncol. 2015, 17, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Ozer, A.; Pagano, J.M.; Lis, J.T. New technologies provide quantum changes in the scale, speed, and success of SELEX methods and aptamer characterization. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.S.; Guy-Caffey, J.K.; Ojwang, J.O.; Smith, S.R.; Hogan, M.E.; Cossum, P.A.; Rando, R.F.; Chaudhary, N. Intramolecular G-quartet motifs confer nuclease resistance to a potent anti-HIV oligonucleotide. J. Biol. Chem. 1996, 271, 5698–5703. [Google Scholar] [CrossRef] [PubMed]
- Casals, J.; Viladoms, J.; Pedroso, E.; Gonzalez, C. Structure and stability of a dimeric G-quadruplex formed by cyclic oligonucleotides. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Breaker, R.R. DNA aptamers and DNA enzymes. Curr. Opin. Chem. Biol. 1997, 1, 26–31. [Google Scholar] [CrossRef]
- Tucker, W.O.; Shum, K.T.; Tanner, J.A. G-quadruplex DNA aptamers and their ligands: Structure, function and application. Curr. Pharm. Des. 2012, 18, 2014–2026. [Google Scholar] [CrossRef] [PubMed]
- Kikin, O.; D’Antonio, L.; Bagga, P.S. QGRS mapper: A web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res. 2006, 34, W676–W682. [Google Scholar] [CrossRef] [PubMed]
- Cload, S.T.; McCauley, T.G.; Keefe, A.D.; Healy, J.M.; Wilson, C. Properties of therapeutic aptamers. In The Aptamer Handbook; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006; pp. 363–416. [Google Scholar]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- The Unafold Web Server. Available online: http://mfold.rna.albany.edu/ (accessed on 25 June 2015).
- DeFazio-Eli, L.; Strommen, K.; Dao-Pick, T.; Parry, G.; Goodman, L.; Winslow, J. Quantitative assays for the measurement of HER1-HER2 heterodimerization and phosphorylation in cell lines and breast tumors: Applications for diagnostics and targeted drug mechanism of action. Breast Cancer Res. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Tolmachev, V. Imaging of HER2 overexpression in tumors for guiding therapy. Curr. Pharm. Des. 2008, 14, 2999–3019. [Google Scholar] [CrossRef] [PubMed]
- Subik, K.; Lee, J.F.; Baxter, L.; Strzepek, T.; Costello, D.; Crowley, P.; Xing, L.; Hung, M.C.; Bonfiglio, T.; Hicks, D.G.; et al. The expression patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by immunohistochemical analysis in breast cancer cell lines. Breast Cancer 2010, 4, 35–41. [Google Scholar] [PubMed]
- Savinainen, K.J.; Saramaki, O.R.; Linja, M.J.; Bratt, O.; Tammela, T.L.; Isola, J.J.; Visakorpi, T. Expression and gene copy number analysis of ErbB2 oncogene in prostate cancer. Am. J. Pathol. 2002, 160, 339–345. [Google Scholar] [CrossRef]
- Hermanto, U.; Zong, C.S.; Wang, L.H. ErbB2-overexpressing human mammary carcinoma cells display an increased requirement for the phosphatidylinositol 3-kinase signaling pathway in anchorage-independent growth. Oncogene 2001, 20, 7551–7562. [Google Scholar] [CrossRef] [PubMed]
- Ginestier, C.; Adelaide, J.; Goncalves, A.; Repellini, L.; Sircoulomb, F.; Letessier, A.; Finetti, P.; Geneix, J.; Charafe-Jauffret, E.; Bertucci, F.; et al. ErbB2 phosphorylation and trastuzumab sensitivity of breast cancer cell lines. Oncogene 2007, 26, 7163–7169. [Google Scholar] [CrossRef] [PubMed]
- Magnifico, A.; Albano, L.; Campaner, S.; Delia, D.; Castiglioni, F.; Gasparini, P.; Sozzi, G.; Fontanella, E.; Menard, S.; Tagliabue, E. Tumor-initiating cells of HER2-positive carcinoma cell lines express the highest oncoprotein levels and are sensitive to trastuzumab. Clin. Cancer Res. 2009, 15, 2010–2021. [Google Scholar] [CrossRef] [PubMed]
- Cuello, M.; Ettenberg, S.A.; Clark, A.S.; Keane, M.M.; Posner, R.H.; Nau, M.M.; Dennis, P.A.; Lipkowitz, S. Down-regulation of the ErbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress ErbB-2. Cancer Res. 2001, 61, 4892–4900. [Google Scholar] [PubMed]
- Govindarajan, S.; Sivakumar, J.; Garimidi, P.; Rangaraj, N.; Kumar, J.; Rao, N.; Gopal, V. Targeting human epidermal growth factor receptor 2 by a cell-penetrating peptide-affibody bioconjugate. Biomaterials 2011, 33, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.; Figari, I.; Fendly, B.; Wong, W.; Carter, P.; Gorman, C.; Shepard, H. Differential responses of human tumor cell lines to anti-p185HER2 monoclonal antibodies. Cancer Immunol. Immunother. 1993, 37, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Schnell, U.; Dijk, F.; Sjollema, K.A.; Giepmans, B.N. Immunolabeling artifacts and the need for live-cell imaging. Nat. Methods 2012, 9, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.D.; de Maziere, A.M.; Pisacane, P.I.; van Dijk, S.M.; Eigenbrot, C.; Sliwkowski, M.X.; Klumperman, J.; Scheller, R.H. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol. Biol. Cell 2004, 15, 5268–5282. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- De Goeij, B.E.; Peipp, M.; de Haij, S.; van den Brink, E.N.; Kellner, C.; Riedl, T.; de Jong, R.; Vink, T.; Strumane, K.; Bleeker, W.K.; et al. HER2 monoclonal antibodies that do not interfere with receptor heterodimerization-mediated signaling induce effective internalization and represent valuable components for rational antibody-drug conjugate design. MAbs 2014, 6, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Pruszynski, M.; Koumarianou, E.; Vaidyanathan, G.; Revets, H.; Devoogdt, N.; Lahoutte, T.; Lyerly, H.K.; Zalutsky, M.R. Improved tumor targeting of anti-HER2 nanobody through N-succinimidyl 4-guanidinomethyl-3-iodobenzoate radiolabeling. J. Nucl. Med. 2014, 55, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Wallberg, H.; Orlova, A. Slow internalization of anti-HER2 synthetic affibody monomer 111In-DOTA-ZHER2:342-pep2: Implications for development of labeled tracers. Cancer Biother. Radiopharm. 2008, 23, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, S.; Orlova, A.; Wallberg, H.; Hansson, M.; Sandstrom, M.; Lewsley, R.; Wennborg, A.; Abrahmsen, L.; Tolmachev, V.; Feldwisch, J. Targeting of HER2-expressing tumors using 111In-ABY-025, a second-generation affibody molecule with a fundamentally reengineered scaffold. J. Nucl. Med. 2010, 51, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Levy-Nissenbaum, E.; Alexis, F.; Lupták, A.; Teply, B.A.; Chan, J.M.; Shi, J.; Digga, E.; Cheng, J.; Langer, R.; et al. Engineering of targeted nanoparticles for cancer therapy using internalizing aptamers isolated by cell-uptake selection. ACS Nano 2012, 6, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Dellamaggiore, K.R.; Ouellette, C.P.; Sedano, C.D.; Lizadjohry, M.; Chernis, G.A.; Gonzales, M.; Baltasar, F.E.; Fan, A.L.; Myerowitz, R.; et al. Aptamer-based endocytosis of a lysosomal enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 15908–15913. [Google Scholar] [CrossRef] [PubMed]
- Porciani, D.; Signore, G.; Marchetti, L.; Mereghetti, P.; Nifosi, R.; Beltram, F. Two interconvertible folds modulate the activity of a DNA aptamer against transferrin receptor. Mol. Ther. Nucleic Acids 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, S.; Laufer, S.D.; Restle, T. Recent developments in peptide-based nucleic acid delivery. Int. J. Mol. Sci. 2008, 9, 1276–1320. [Google Scholar] [CrossRef] [PubMed]
- Gourronc, F.A.; Rockey, W.M.; Thiel, W.H.; Giangrande, P.H.; Klingelhutz, A.J. Identification of RNA aptamers that internalize into HPV-16 E6/E7 transformed tonsillar epithelial cells. Virology 2013, 446, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.; Berk, A.; Kaiser, C.A.; Krieger, M.; Scott, M.P.; Bretscher, A.; Ploegh, H.; Matsudaira, P. Molecular Cell Biology; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Brockhoff, G.; Heckel, B.; Schmidt-Bruecken, E.; Plander, M.; Hofstaedter, F.; Vollmann, A.; Diermeier, S. Differential impact of Cetuximab, Pertuzumab and Trastuzumab on BT474 and SK-BR-3 breast cancer cell proliferation. Cell Prolif. 2007, 40, 488–507. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, H.P.; Kim, Y.J.; Oh do, Y.; Im, S.A.; Lee, D.; Jong, H.S.; Kim, T.Y.; Bang, Y.J. Trastuzumab inhibits the growth of human gastric cancer cell lines with HER2 amplification synergistically with cisplatin. Int. J. Oncol. 2008, 32, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Famulok, M.; Hartig, J.; Mayer, G. Functional aptamers and aptazymes in biotechnology, diagnostics, and therapy. Chem. Rev. 2007, 107, 3715–3743. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Roy, I. Therapeutic applications of aptamers. Expert Opin. Investig. Drugs 2008, 17, 43–60. [Google Scholar] [CrossRef] [PubMed]
- QGRS Mapper. Available online: http://bioinformatics.ramapo.edu/QGRS/index.php (accessed on 25 June 2015).
- Ohuchi, S. Cell-SELEX technology. BioRes. Open Access 2012, 1, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Han, D.; Ocsoy, I.; Tan, W. Aptamers selected by cell-SELEX for application in cancer studies. Bioanalysis 2010, 2, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Sefah, K.; Shangguan, D.; Xiong, X.; O’Donoghue, M.B.; Tan, W. Development of DNA aptamers using cell-SELEX. Nat. Protoc. 2010, 5, 1169–1185. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Hu, J.; Peng, M.; Liu, J.; Liu, J.; Liu, H.; Zhao, X.; Tan, W. Generating aptamers by cell-SELEX for applications in molecular medicine. Int. J. Mol. Sci. 2012, 13, 3341–3353. [Google Scholar] [CrossRef] [PubMed]
- Thiel, W.H.; Bair, T.; Peek, A.S.; Liu, X.; Dassie, J.; Stockdale, K.R.; Behlke, M.A.; Miller, F.J., Jr.; Giangrande, P.H. Rapid identification of cell-specific, internalizing rna aptamers with bioinformatics analyses of a cell-based aptamer selection. PLoS ONE 2012, 7, e43836. [Google Scholar] [CrossRef] [PubMed]
- Juweid, M.; Neumann, R.; Paik, C.; Perez-Bacete, M.J.; Sato, J.; van Osdol, W.; Weinstein, J.N. Micropharmacology of monoclonal antibodies in solid tumors: Direct experimental evidence for a binding site barrier. Cancer Res. 1992, 52, 5144–5153. [Google Scholar] [PubMed]
- Fujimori, K.; Covell, D.G.; Fletcher, J.E.; Weinstein, J.N. A modeling analysis of monoclonal antibody percolation through tumors: A binding-site barrier. J. Nucl. Med. 1990, 31, 1191–1198. [Google Scholar] [PubMed]
- Van Osdol, W.; Fujimori, K.; Weinstein, J.N. An analysis of monoclonal antibody distribution in microscopic tumor nodules: Consequences of a “binding site barrier”. Cancer Res. 1991, 51, 4776–4784. [Google Scholar] [PubMed]
- Adams, G.P.; Schier, R.; McCall, A.M.; Simmons, H.H.; Horak, E.M.; Alpaugh, R.K.; Marks, J.D.; Weiner, L.M. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001, 61, 4750–4755. [Google Scholar] [PubMed]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for growth factors in cancer progression. Physiology 2010, 25, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.Y.; Chipuk, J.E. Survival of HER2-positive breast cancer cells: Receptor signaling to apoptotic control centers. Genes Cancer 2013, 4, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Human epidermal growth factor receptor 2 (HER2) in cancers: Overexpression and therapeutic implications. Mol. Biol. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Neve, R.M.; Lane, H.A.; Hynes, N.E. The role of overexpressed HER2 in transformation. Ann. Oncol. 2001, 12, S9–S13. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gijs, M.; Penner, G.; Blackler, G.B.; Impens, N.R.E.N.; Baatout, S.; Luxen, A.; Aerts, A.M. Improved Aptamers for the Diagnosis and Potential Treatment of HER2-Positive Cancer. Pharmaceuticals 2016, 9, 29. https://doi.org/10.3390/ph9020029
Gijs M, Penner G, Blackler GB, Impens NREN, Baatout S, Luxen A, Aerts AM. Improved Aptamers for the Diagnosis and Potential Treatment of HER2-Positive Cancer. Pharmaceuticals. 2016; 9(2):29. https://doi.org/10.3390/ph9020029
Chicago/Turabian StyleGijs, Marlies, Gregory Penner, Garth B. Blackler, Nathalie R.E.N. Impens, Sarah Baatout, André Luxen, and An M. Aerts. 2016. "Improved Aptamers for the Diagnosis and Potential Treatment of HER2-Positive Cancer" Pharmaceuticals 9, no. 2: 29. https://doi.org/10.3390/ph9020029
APA StyleGijs, M., Penner, G., Blackler, G. B., Impens, N. R. E. N., Baatout, S., Luxen, A., & Aerts, A. M. (2016). Improved Aptamers for the Diagnosis and Potential Treatment of HER2-Positive Cancer. Pharmaceuticals, 9(2), 29. https://doi.org/10.3390/ph9020029