2. Results
The results of our studies evaluating the overall effects of test drugs on exploratory/locomotor activity, fear response, and affective aggression in animals are shown in
Table 1 and
Table 2.
Our results (
Table 1) showed that administration of compound
L-17 resulted in decreased animal motor activity (3-fold and > 20-fold decreases at doses of 60 mg/kg and 120 mg/kg, respectively), and an increase in the threshold voltage (to 43 ± 1.5 V and 48 ± 1.5 V when administered at doses of 60 mg/kg and 120 mg/kg, respectively). Squeak intensity (the equivalent of an "emotional" fear response) changed significantly following administration of compound
L-17 at a dose of 120 mg/kg.
Administration of Eglonyl led to a significant decrease in overall exploratory/locomotor activity (1.6-fold at a dose of 40 mg/kg), an increase in the threshold voltage (to 35 ± 0.5 V and 38 ± 0.6 V when administered at doses of 20 mg/kg and 40 mg/kg, respectively), as well as changes in squeak intensity (at a dose of 20 mg/kg).
Thus, administration of compound L-17 led to the appearance of CNS-associated signs of depression similar to those induced by Eglonyl, which causes changes in spontaneous behavior (activity reduction), altered reactions to painful stimuli, and an increased threshold for a pain reaction. Compound L-17 produced an analgesic effect when given at a dose of 10 mg/kg, while a 120 mg/kg dose induced a cataleptic state.
Changes in apomorphine-induced stereotypy were evaluated in mice (
Table 2).
Animals that were administered L-17 displayed a significant increase in their latent period starting at a 30 mg/kg dose, and also a reduced duration of stereotypy starting at a 60 mg/kg dose. The estimated effective dose (ED50) of L-17 was 80 mg/kg. Animals which received the antipsychotic drug Eglonyl displayed significant increases in both their latent period and duration of stereotypy. The estimated effective dose (ED50) of Eglonyl was 19 mg/kg.
This test is specific to neuroleptic agents derived from phenothiazine and butyrophenone. An ability to suppress stereotypy indicates that the test substance inhibits dopaminergic neurotransmission in the nigrostriatal system of the brain. This trait was displayed by compound L-17.
Next, apomorphine-induced hypoactivity was evaluated in rats (
Table 3).
Our results showed that compound L-17 could potentiate the effects of presynaptic doses of apomorphine, and significantly reduce the motor activity of animals. A 40 mg/kg dose of L-17 administered in conjunction with apomorphine (0.15 mg/kg) reduced animal motor activity by 26.4 ± 3.8 movements when compared with a control group (217.75 ± 17.10 movements) and also when compared with a group of animals administered apomorphine alone at a dose of 0.15 mg/kg (75.88 ± 19.33 movements). In contrast, a 40 m/kg dose of Eglonyl induced an increase in locomotor activity (152.38 ± 12.42 movements) when compared locomotor activity displayed by animals treated with apomorphine alone (75.88 ± 19.33 movements).
The amphetamine-induced locomotion test was conducted with mice (
Table 4).
A 100 mg/kg dose of compound L-17 produced a significant increase in the duration of an animal’s latent period, and a 200 mg/kg dose produced a significant decrease in stereotypy. The estimated effective dose (ED50) of compound L-17 was 170 mg/kg. A 40 mg/kg dose of Eglonyl induced a significant decrease in the duration of stereotypy (40.3 ± 1.8 min) when compared with the duration in control animals (118.0 ± 1.2 min).
Liver enzyme activity was evaluated by using the hexenal test to examine the hypnotic effect of a barbiturate (hexenal) in mice (
Table 5). Our results showed that a 60 mg/kg dose of compound
L-17 significantly increased the duration of both latency (up to 254.0 ± 75.0 s) and sleep (65.7 ± 8.1 min), when compared with those parameters in a control group (126.3 ± 6.4 sec and 39.3 ± 6.4 min, respectively). The estimated effective dose (ED50) of
L-17 was 80 mg/kg. A 25 mg/kg dose of Eglonyl produced an increase in sleep duration (up to 39.8 ± 1.7 min) when compared with that in control animals (25.0 ± 0.8 min), and the estimated ED50 of Eglonil was 42 mg/kg.
As antipsychotic drugs typically potentiate the narcotic effect of barbiturates [
14], we also tested substances for their influence on the hypnogenetic effects of barbiturates (
Table 5).
The results shown in
Table 5 indicate that administration of a sub-threshold dose of hexenal caused 16.7% of the laboratory animals to fall asleep, while a 10 mg/kg dose of
L-17 administered in addition to hexenol caused a significantly greater percentage of the animals to fall asleep. This result suggests that compound
L-17 can decrease the ability of liver microsomes to metabolize barbiturates, which is a characteristic feature of many neuroleptics [
14].
The arecoline-evoked tremor was evaluated in mice. The results shown in
Table 6 indicate that a high dose (60 mg/kg) of compound
L-17 caused a significant reduction in the mean duration of tremor (14 ± 0.7 s), when compared with the mean duration in a control group; providing evidence for its anti-cholinergic effect [
15,
16].
The influence of compound
L-17 on the contractile activity of isolated smooth muscles (guinea pig ileum and rat seminal vesicles) was studied using acetylcholine and epinephrine in the standard method described by R. Blattner (
Table 7) [
17].
When injected at a concentration of 1 × 10
−5 mol/L, compound
L-17 demonstrated a moderate ability to block adrenoreceptors, which indicated its ability to interact with adrenergic receptors, and in particular, alpha-1-adrenoceptors. Eglonyl blocked adrenoceptors when injected at a concentration of 1 × 10
−6 mol/L. The ability of compound
L-17 to interact with 5-HT3 receptors was demonstrated in the model of serotonin-induced spasm of isolated guinea pig ileum muscle [
18]. Significant reductions in contraction amplitude were observed after injecting either compound
L-17 or Eglonyl at a concentration of 1 × 10
−5 mol/L.
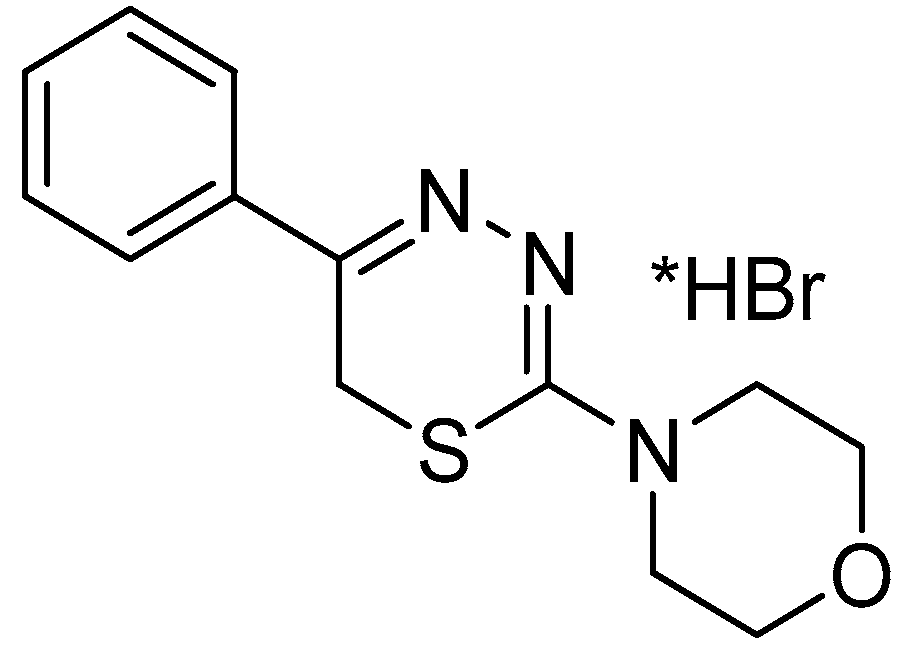
Thus, our data suggest that compound L-17, which was selected from a group of 5-phenyl substituted-6H-1,3,4-thiadiazine-2-amines, might exert its effects via one or more mechanisms of action. These include affecting cholinergic system agonists/antagonists (arecoline 15 mg/kg), dopaminergic neurotransmission (apomorphine 0.1 mg/kg and amphetamine 5 mg/kg), the adrenergic system (evaluation of smooth muscle contractile activity), and (or) 5-HT3 serotonin receptors (model of serotonin-induced muscle spasm).
3. Discussion
The results shown in
Table 1,
Table 2,
Table 3,
Table 4,
Table 5,
Table 6 and
Table 7 and by assays used to differentiate groups of drugs in initial screening tests [
11] indicated that compound
L-17, selected from a group of 5-phenyl substituted-6
H-1,3,4-thiadiazine-2-amines, possesses a combination of adrenergic properties, as well as certain characteristics of choline and serotonin blockers. Compound 17 demonstrated its effects when used in a concentration range similar to that employed when using the atypical antipsychotic agent Eglonyl (sulpiride), as well as antidepressants such as amitriptyline, and alpha-blockers such as pyrroxanum.
Practically all effective anti-psychotic medications are known to block a1-adrenoreceptors [
19]. While these receptors were initially thought to merely promote sedation, more recent data indicate that the ability to blockade a1-adrenoceptors contributes to the therapeutic effects anti-psychotic agents [
19]. A1-adrenoceptors are believed to play a regulatory role in the brain by enhancing certain excitatory afferent inputs to brain cells. The first evidence for tonic regulation of serotonin neurons (5-HT) by postsynaptic alpha-1-adrenergic receptors belonging to the noradrenergic system was reported by TH Svensson in 1975 [
20]. Those data were subsequently supported by several studies showing that alpha-1-blockers suppressed the activity of serotonergic neurons [
21,
22], and reduced serotonin levels in the hippocampus [
23]. These findings explain why medications that act on serotonin and noradrenaline systems demonstrate their most pronounced activity as antidepressants [
19]. Moreover, their synergistic effect may be due to the fact that alpha-1 and serotonin (5HT2A) receptors are involved in the phosphatidyl inositol/protein kinase C intracellular pathway via Gq proteins [
24].
At the same time, it should be noted that the affinity of norepinephrine for alpha-1 adrenoceptors is significantly less than its affinity for alpha-2-adrenoceptors. As a result, a substantial increase in norepinephrine levels (e.g., as induced by a stress reaction) is required for norepinephrine to interact with alpha-1-receptors [
19]. This explains why administration of an alpha-1-adrenergic receptor’s antagonist produces almost no effect under normal (non-stress) conditions, but has a protective effect during stressful conditions [
25].
The mechanisms described above may be responsible for the beneficial effects of thiadiazine compounds, as previously demonstrated in experimental models of acute stress [
13] and acute myocardial infarction [
10,
11].


 ,
, 

{kind=link}
{kind=link}