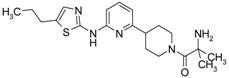
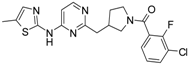
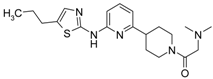
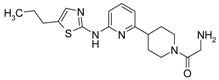
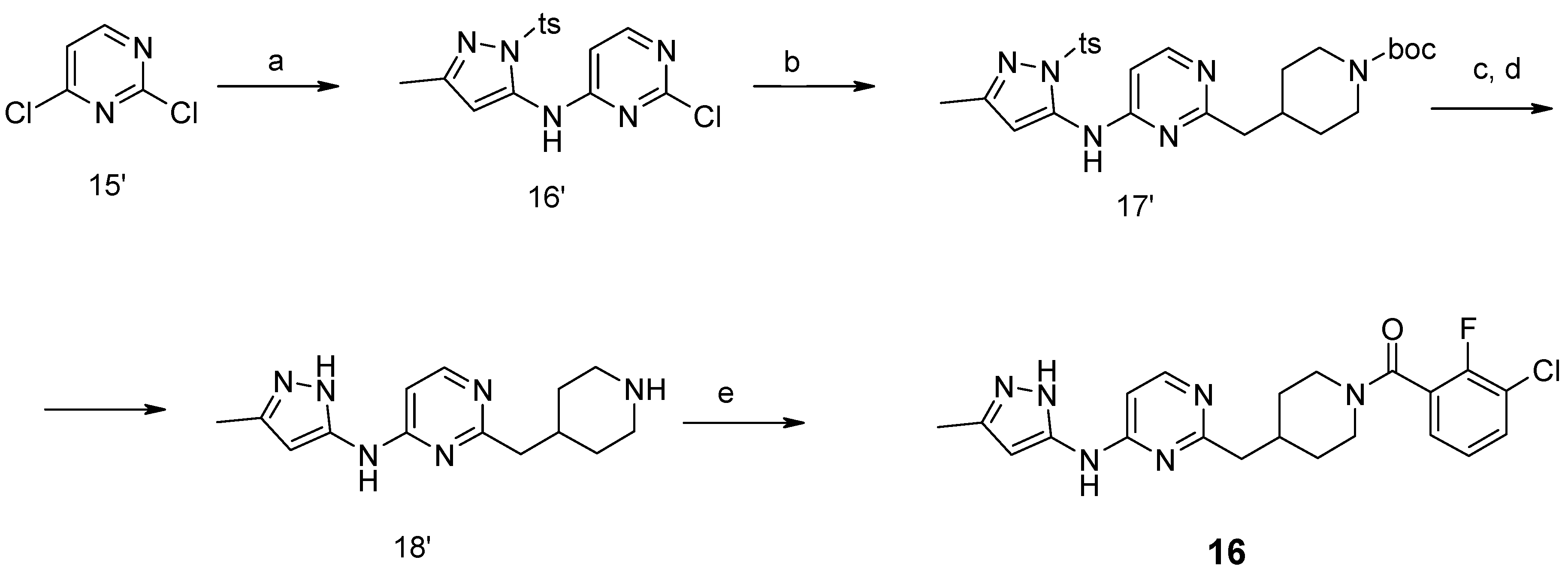
First, 3-Chloro-2-fluoro-benzoic acid (63 mg, 0.36 mmol), TBTU (116 mg, 0.36 mmol), amine dihydrochloride (0.3 mmol) and triethylamine (0.168 mL, 1.2 mmol) were dissolved in dry acetonitrile (5 mL). The mixture was stirred for 8 h at room temperature (TLC control). After the reaction was completed the mixture was concentrated to dryness in vacuo. Residue was treated with 10% potassium carbonate (10 mL). The resulting mixture was extracted with dichloromethane (2 × 20 mL), organic extract was concentrated and purified by column chromatography.
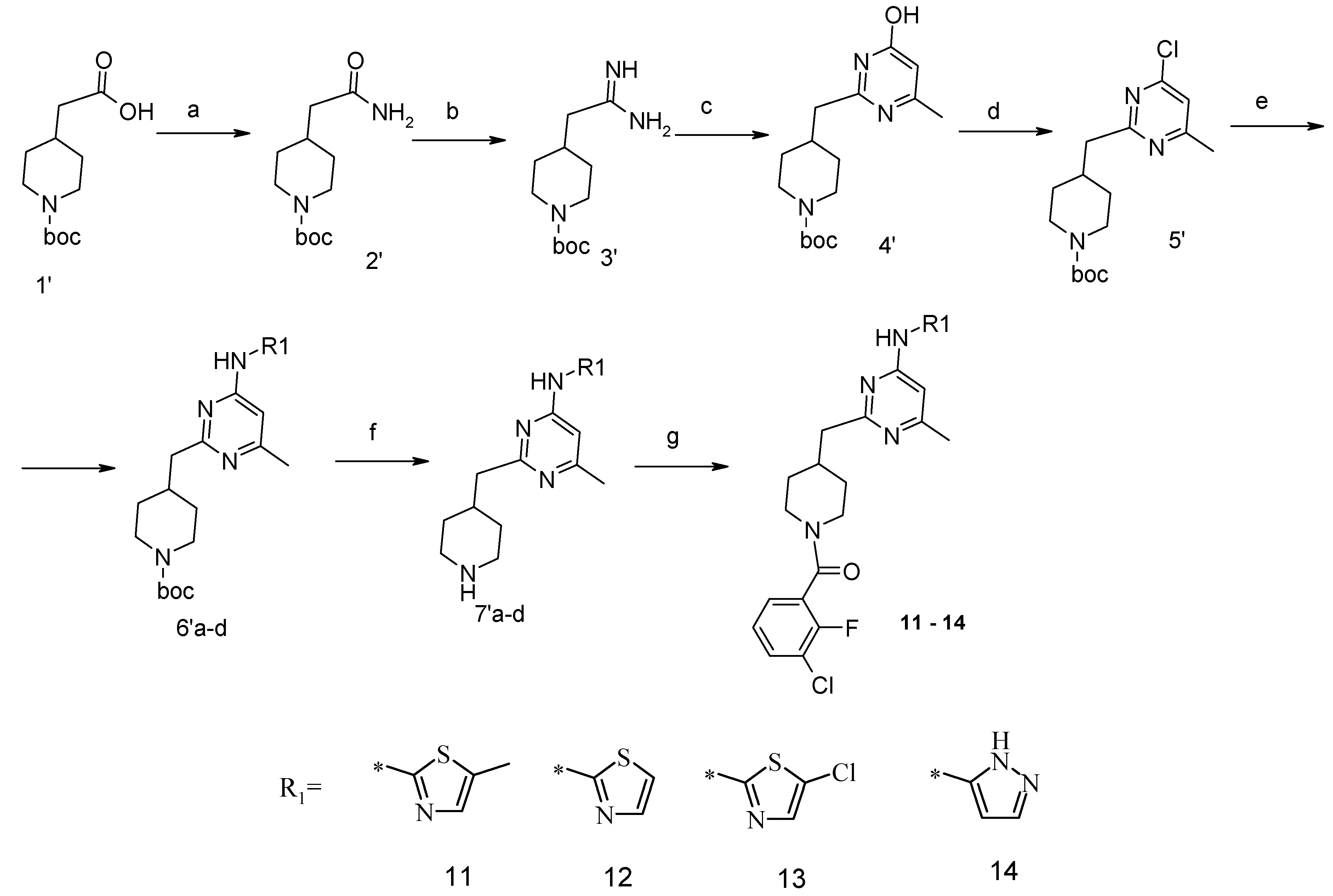
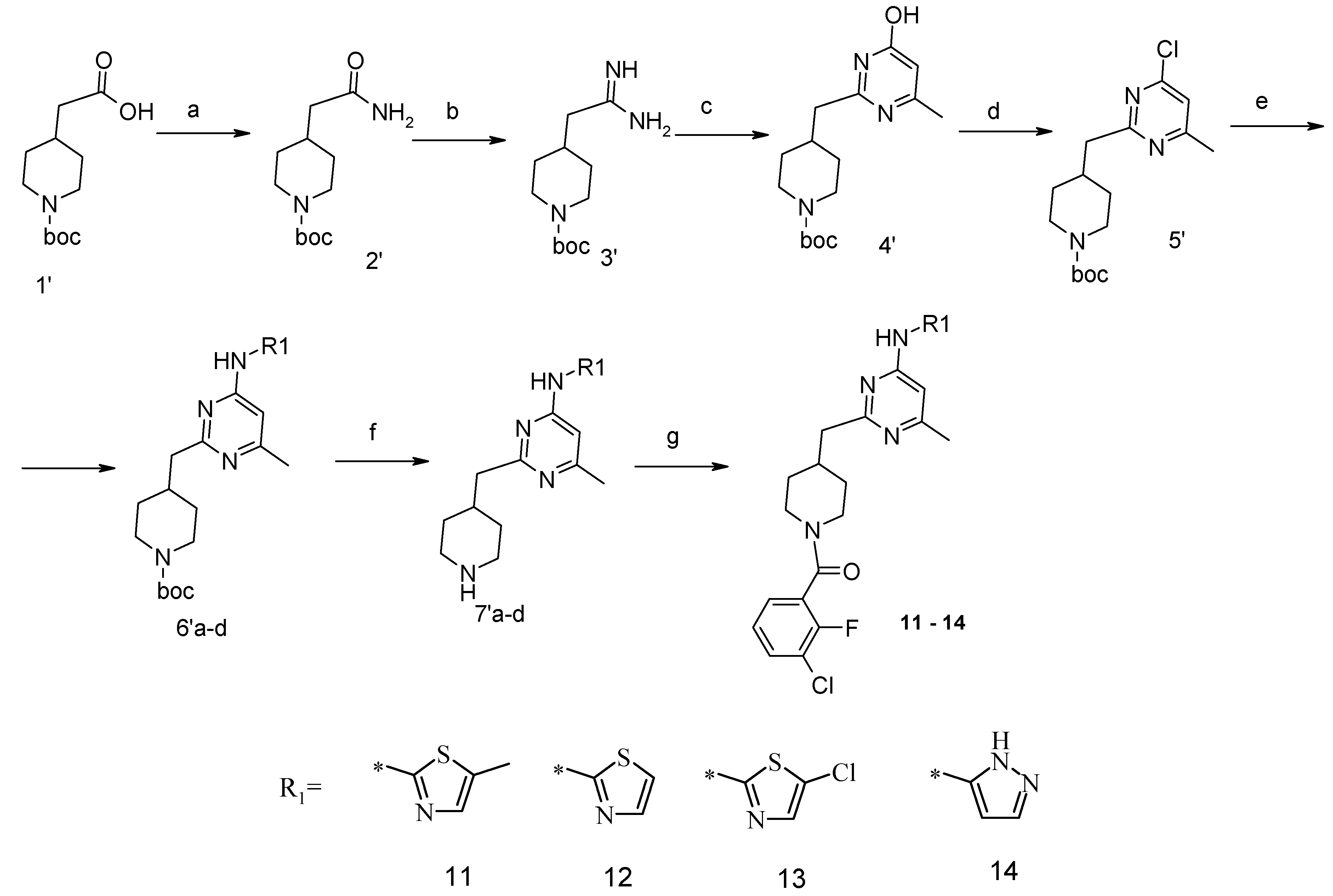
4-Carbamoylmethyl-piperidine-1-carboxylic acid tert-butyl Ester (2′). The suspension of 4-carboxymethyl-piperidine-1-carboxylic acid tert-butyl ester (1′) (3.04 g, 12.5 mmol), TBTU (4.49 g, 14 mmol), ammonium carbonate (2.4 g, 25 mmol) and triethylamine (5.32 mL, 38 mmol) in dry acetonitrile (80 mL) was stirred for 8 h at room temperature (TLC control). After the reaction was completed the mixture was evaporated to dryness in vacuo. Residue was treated with 10% aqueous solution of potassium carbonate (50 mL). The resulting mixture was extracted with dichloromethane (3 × 100 mL), organic extract was evaporated. Purification by column chromatography on silica gel (eluent: hexane/ethyl acetate—1/1) afforded (2′) (2.42 g, 83%). 1H-NMR δH (400 MHz, d6-DMSO): 0.98 (m, 2H, CH2), 1.37 (s, 9H, BOC), 1.61 (m, 2H, CH2), 1.79 (m, 1H, CH), 1.96 (m, 2H, CH2), 2.70 (m, 2H, CH2), 3.87 (d, 2H, CH2), 6.61 (br.s., 1H), 7.16 (br.s., 1H). APCI-MS (m/z (intensity)): 243.2 ([M + H]+, 100%).
4-(4-Hydroxy-6-methyl-pyrimidin-2-ylmethyl)-piperidine-1-carboxylic acid tert-butyl Ester (4′). To a solution of amide (2′) (2.42 g, 10 mmol) in dichloromethane (100 mL), triethyloxoniumtetrafluoroborate (1.78 g, 12 mmol) was added at constant stirring; the resulted mixture was stirred for 2 h at room temperature. Then the reaction mixture was concentrated at ~45 °C. The residue was treated with solution of NH3/CH3OH (pH should be 10), and left to stay overnight. The reaction mixture was evaporated to dryness, and the obtained product (3′) was used in the next step without purification. APCI-MS (m/z (intensity)): 242.1 ([M + H]+, 100%).
Crude compound (3′) was added to a solution of NaOtBu (1.15 g, 12 mmol) in anhydrous ethanol (100 mL). The mixture was stirred at room temperature for 10 min, and then methyl 3-oxobutanoate (1.95 g, 15 mmol) was added. The resulting mixture was refluxed for 10 h (TLC control). Then the reaction mixture was concentrated under reduced pressure, dissolved in water and acidified with 1 N HCl to pH = 5.0 and extracted with ethyl acetate (2 × 100 mL). An organic layer was dried over Na2SO4 and concentrated in vacuum. The residue was purified by flash-chromatography on silica gel (eluent: hexane/ethyl acetate—1/1). As a result, compound (4′) (1.99 g, 65%) was obtained. APCI-MS (m/z (intensity)): 308.2 ([M + H]+, 100%).
4-(4-Chloro-6-methyl-pyrimidin-2-ylmethyl)-piperidine-1-carboxylic acid tert-butyl Ester (5′). The compound (4′) (1.99 g, 6.5 mmol) and dimethylaniline (7.08 g, 58.5 mmol) were dissolved in toluene (100 mL). POCl3 (2.99 g, 19.5 mmol) was added dropwise. The reaction mixture was refluxed for 3 h, cooled down to room temperature and poured into water. Organic layer was separated, washed with 1 N HCl and water. Organic layer was concentrated under reduced pressure and purified with flash chromatography (eluent: hexane/ethylacetate 4/1). Yield 1.3 g (62%). 1H-NMR δH (400 MHz, CDCl3): 1.20–1.29 (m, 2H, CH2), 1.44 (s, 9H, BOC), 1.61 (m, 2H, CH2), 2.11 (m, 1H, CH), 2.49 (s, 3H, CH3), 2.71 (t, 2H, CH2), 2.83 (d, 2H, CH2), 4.07 (d, 2H, CH2), 7.03 (s, 1H, ArH). APCI-MS (m/z (intensity)): 325.4 ([M + H]+, 100%), 327.4 ([M + H]+, 35%).
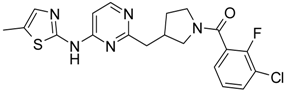
4-[4-Methyl-6-(thiazol-2-ylamino)-pyrimidin-2-ylmethyl]-piperidine-1-carboxylic acid tert-butyl Ester (6′a) was prepared using Buchwald reaction, yield 292 mg (75%). 1H-NMR δH (400 MHz, d6-DMSO): 1.10 (m, 2H, CH2), 1.32 (s, 9H, BOC), 1.60 (m, 2H, CH2), 2.21 (m, 1H, CH), 2.30 (s, 3H, CH3), 2.67 (m, 4H, CH2), 3.86 (d, 2H, CH2), 6.67 (s, 1H, ArH), 7.12 (m, 1H, ArH), 7.40 (m, 1H, ArH), 11.39 (br.s., 1H, NH). APCI-MS (m/z (intensity)): 390.12 ([M + H]+, 100%).
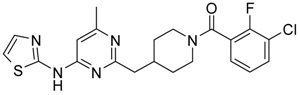
4-[4-Methyl-6-(5-methyl-thiazol-2-ylamino)-pyrimidin-2-ylmethyl]-piperidine-1-carboxylic acid tert-butyl Ester (6′b) was prepared using Buchwald reaction, yield 290 mg (72%). 1H-NMR δH (400 MHz, d6-DMSO): 1.09 (m, 2H, CH2), 1.36 (s, 9H, BOC), 1.61 (m, 2H, CH2), 2.14 (m, 1H, CH), 2.28 (s, 3H, CH3), 2.31 (s, 3H, CH3), 2.69(m, 4H, CH2), 3.88 (d, 2H, CH2), 6.62 (s, 1H, ArH), 7.04 (s, 1H, ArH), 11.13 (s, 1H, NH). APCI-MS (m/z (intensity)): 404.2 ([M + H]+, 100%).
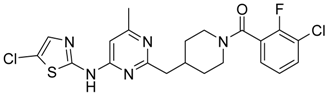
4-[4-Methyl-6-(5-chloro-thiazol-2-ylamino)-pyrimidin-2-ylmethyl]-piperidine-1-carboxylic acid tert-butyl Ester (6′c) was prepared from (6′a) (194 mg, 0.5 mmol) by reaction with N-chlorosuccinimide (67 mg, 0.5 mmol) in dichloromethane (20 mL) at room temperature for 4 h. Residue was treated with 10% aqueous solution of potassium carbonate (10 mL). The organic extract was evaporated and purified by column chromatography on silica gel (eluent: hexane/ethyl acetate—4/1) to afford (6′c) (148 mg, 70%). 1H-NMR δH (400 MHz, d6-DMSO): 1.12 (m, 2H, CH2), 1.36 (s, 9H, BOC), 1.61 (m, 2H, CH2), 2.13 (m, 1H, CH), 2.32 (s, 3H, CH3), 2.69 (m, 4H, CH2), 3.87 (d, 2H, CH2), 6.62 (s, 1H, ArH), 7.41 (s, 1H, ArH), 11.66 (s, 1H, NH). APCI-MS (m/z (intensity)): 423.4 ([M + H]+, 100%), 425.5 ([M + H]+, 30%).
4-[4-Methyl-6-(2H-pyrazol-3-ylamino)-pyrimidin-2-ylmethyl]-piperidine-1-carboxylic acid tert-butyl Ester (6′d). 2-Tosyl-2H-pyrazol-3-ylamine was obtained by reaction between 2H-pyrazol-3-ylamine and tosyl chloride (1 mmol) in presence of sodium bicarbonate (1.2 mmol) in acetonitrile at room temperature for 2 h (yield 60% after recrystallization from ethanol). Buchwald reaction led to tosyl-derivative of (6′d), yield 45%. The protective tosyl group was removed by treatment with 0.12 M NaOH in wet methanol at room temperature for 6 h. After reaction completion (TLC control) the mixture was poured into water (20 mL) and extracted with dichloromethane (3 × 20 mL), dried over Na2SO4 and concentrated in vacuum to give the titled compound (6′d). Yield 92%. 1H-NMR δH (400 MHz, d6-DMSO): 1.04 (m, 2H, CH2), 1.35 (s, 9H, BOC), 1.56 (m, 2H, CH2), 1.99 (m, 1H, CH), 2.21 (s, 3H, CH3), 2.51-2.65 (m, 4H, CH2), 3.86 (m, 2H, CH2), 6.33 (m, 1H, ArH), 6.85 (m, 1H, ArH), 7.58 (s, 1H, ArH), 9.63 (s, 1H, NH), 12.21 (s, 1H, NH). APCI-MS (m/z (intensity)): 373.2 ([M + H]+, 100%).
(6-Methyl-2-piperidin-4-ylmethyl-pyrimidin-4-yl)-thiazol-2-yl-amine Dihydrochloride (7′a) was prepared by Boc-deprotection. Yield 287 mg (99%). 1H-NMRδH (400 MHz, d6-DMSO): 1.57 (m, 2H, CH2), 1.86 (m, 2H, CH2), 2.33 (m, 1H, CH), 2.56 (s, 3H, CH3), 2.84 (m, 2H, CH2), 3.07 (m, 2H, CH2), 3.19 (m, 2H, CH2), 7.03 (br.s, 1H, ArH), 7.41 (m, 1H, ArH), 7.56 (s, 1H, ArH), 8.96 (m, 1H), 9.52 (m, 1H). APCI-MS (m/z (intensity)): 290.2 ([M + H]+, 100%).
(6-Methyl-2-piperidin-4-ylmethyl-pyrimidin-4-yl)-(5-methyl-thiazol-2-yl)-amine Dihydrochloride (7′b) was prepared by Boc-deprotection. Yield 265 mg (98%). 1H-NMR δH (400 MHz, d6-DMSO): 1.56 (m, 2H, CH2), 1.86 (m, 2H, CH2), 2.33 (m, 1H, CH), 2.45 (s, 3H, CH3), 2.52 (s, 3H, CH3), 2.84 (m, 2H, CH2), 2.98 (m, 2H, CH2), 3.23 (m, 2H, CH2), 6.92 (br.s, 1H, ArH), 7.26 (s, 1H, ArH), 8.95 (m, 1H), 9.20 (m, 1H). APCI-MS (m/z (intensity)): 304.2 ([M + H]+, 100%).
(5-Chloro-thiazol-2-yl)-(6-methyl-2-piperidin-4-ylmethyl-pyrimidin-4-yl)-amine Dihydrochloride (7′c) was prepared by Boc-deprotection. Yield 136 mg (98%). 1H-NMR δH (400 MHz, d6-DMSO): 1.55 (m, 2H, CH2), 1.84 (m, 2H, CH2), 2.28 (m, 1H, CH), 2.51 (s, 3H, CH3), 2.83 (m, 2H, CH2), 2.98 (m, 2H, CH2), 3.23 (m, 2H, CH2), 6.92 (s, 1H, ArH), 7.54 (s, 1H, ArH), 8.77 (m, 1H), 9.02 (m, 1H). APCI-MS (m/z (intensity)): 323.5 ([M + H]+, 100%), 325.5 ([M + H]+, 32%).
(6-Methyl-2-piperidin-4-ylmethyl-pyrimidin-4-yl)-(2H-pyrazol-3-yl)-amine Dihydrochloride (7′d) was prepared by Boc-deprotection. Yield 140 mg (99%). APCI-MS (m/z (intensity)): 273.1 ([M + H]+, 100%).
(3-Chloro-2-fluoro-phenyl)-{4-[4-methyl-6-(thiazol-2-ylamino)-pyrimidin-2-ylmethyl]-piperidin-1-yl}-methanone (12) was prepared by Boc-deprotection. Yield 113 mg (85%). M.p. 207–209 °C. 1H-NMR δH (400 MHz, d6-DMSO): 1.09–1.26 (m, 2H), 1.64 (m, 1H), 1.80 (m, 1H), 2.23 (s, 3H), 2.35 (m, 1H), 2.65 (m, 2H), 2.84 (m, 1H), 3.04 (m, 1H), 3.33 (m, 1H), 4.43 (m, 1H), 6.45 (s, 1H), 6.89 (d, 1H), 7.28 (m, 3H), 7.62 (m, 2H). 13C-NMR δ (400 MHz, d6-DMSO): 23.45 (CH3), 31.30 (CH), 32.06 (CH2), 34.43 (CH2), 41.31 (CH2), 44.82 (CH2), 46.59 (CH2), 103.22 (CH), 112.25 (CH), 119.9 (d, J = 20 Hz, C), 125.80 (d, J = 4 Hz, CH), 125.91 (d, J = 20 Hz, C), 126.19 (CH), 127.09 (d, J = 4 Hz, CH), 131.08 (CH), 137.28 (CH), 152.77 (d, J = 247 Hz, C), 157.63 (C), 159.91 (C), 162.40 (C), 164.50 (C), 167.01 (C). APCI-MS (m/z (intensity)): 445.9 ([M + H]+, 100%), 447.7([M + H]+, 40%). HRMS (ESI): Calcd for C21H21ClFN5OS ([M + H]+), 446.1212; found, 446.1219.
(3-Chloro-2-fluoro-phenyl)-{4-[4-methyl-6-(5-methyl-thiazol-2-ylamino)-pyrimidin-2-ylmethyl]-piperidin-1-yl}-methanone (11) was prepared by Boc-deprotection. Yield 120 mg (87%). M. p. 211–213 °C. 1H-NMR δH (400 MHz, d6-DMSO): 1.11–1.27 (m, 2H), 1.62 (m, 1H), 1.78 (m, 1H), 2.27 (s, 3H), 2.34 (s, 3H), 2.72 (m, 2H), 2.83 (m, 1H), 3.05 (m, 1H), 3.34 (m, 1H), 3.45 (m, 1H), 6.61 (s, 1H), 7.05 (s, 1H), 7.25–7.36 (m, 2H), 7.62 (m, 1H), 11.18 (br.s., 1H). 13C-NMR δ (400 MHz, d6-DMSO): 10.87 (CH3), 23.46 (CH3), 31.25 (CH), 31.98 (CH2), 34.43 (CH2), 41.29 (CH2), 44.71 (CH2), 46.56 (CH2), 102.41 (CH), 119.9 (d, J = 19 Hz, C), 125.48 (C), 125.76 (d, J = 4 Hz, CH), 126.09 (d, J = 19 Hz, C), 127.08 (d, J = 4 Hz, CH), 131.06 (CH), 134.43 (CH), 152.77 (d, J = 247 Hz, C), 156.81 (C), 156.91 (C), 162.38 (C), 162.03 (C), 167.16 (C). APCI-MS (m/z (intensity)): 459.8 ([M + H]+, 100%), 461.7 ([M + H]+, 37%). HRMS (ESI): Calcd for C22H23ClFN5OS ([M + H]+), 460.1369; found, 460.1367.
(3-Chloro-2-fluoro-phenyl)-{4-[4-(5-chloro-thiazol-2-ylamino)-6-methyl-pyrimidin-2-ylmethyl]-piperidin-1-yl}-methanone (13) was prepared by Boc-deprotection. Yield 130 mg (90%). M. p. 221–223 °C. 1H-NMR δH (400 MHz, d6-DMSO): 1.12–1.27 (m, 2H), 1.63 (m, 1H), 1.79 (m, 1H), 2.29 (s, 3H), 2.73 (m, 2H), 2.83 (m, 1H), 3.06 (m, 1H), 3.35 (m, 1H), 4.45 (m, 1H), 6.62 (s, 1H), 7.39 (s, 1H), 7.27–7.37 (m, 2H), 7.63 (m, 1H), 11.65 (s, 1H). APCI-MS (m/z (intensity)): 480.09 ([M + H]+, 100%), 482.09 ([M + H]+, 68%). HRMS (ESI): Calcd for C21H20Cl2FN5OS ([M + H]+), 480.0822; found, 480.0825.
(3-Chloro-2-fluoro-phenyl)-{4-[4-methyl-6-(2H-pyrazol-3-ylamino)-pyrimidin-2-ylmethyl]-piperidin-1-yl}-methanone (14) was prepared by Boc-deprotection. Yield 114 mg (89%). M. p. 125–128 °C. 1H-NMR δH (400 MHz, d6-DMSO): 1.11–1.34 (m, 2H), 1.62 (m, 1H), 1.83 (m, 1H), 2.16 (m, 1H), 2.25 (s, 3H), 2.60 (m, 2H), 2.81 (m, 1H), 3.06 (m, 1H), 3.29 (m, 1H), 4.43 (m, 1H), 6.35 (s, 1H), 6.89 (s, 1H), 7.26–7.36 (m, 2H), 7.56-7.64 (m, 2H), 9.55 (s, 1H), 12.25 (s, 1H). APCI-MS (m/z (intensity)): 428.8 ([M + H]+, 100%), 430.8 ([M + H]+, 41%). HRMS (ESI): Calcd for C21H21ClFN6O ([M + H]+), 429.1600; found, 429.1608.
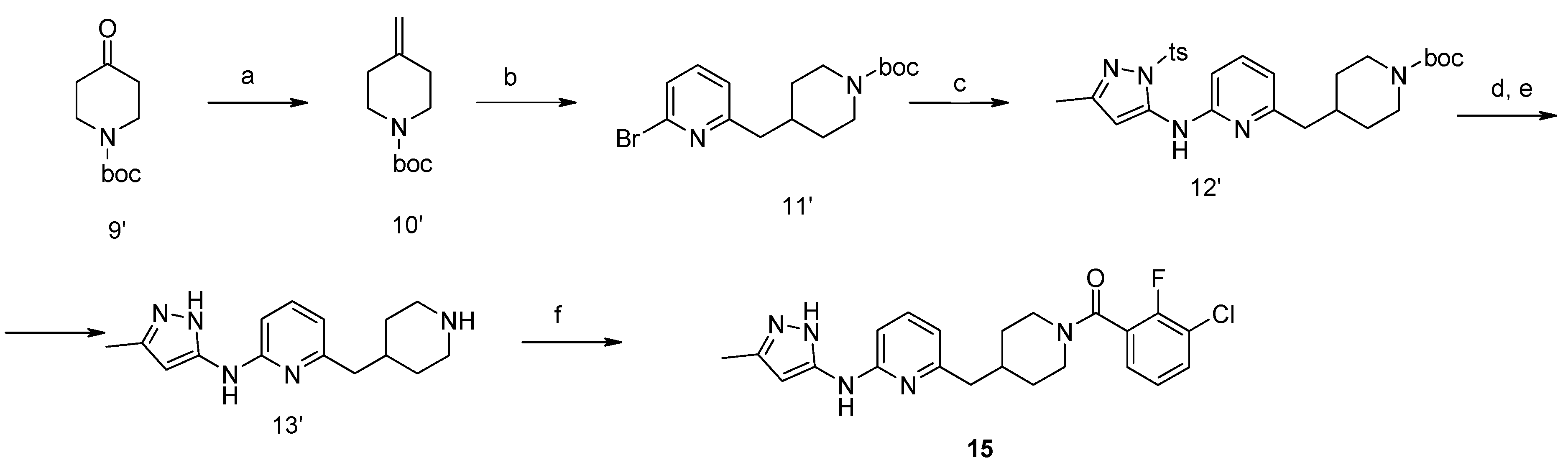
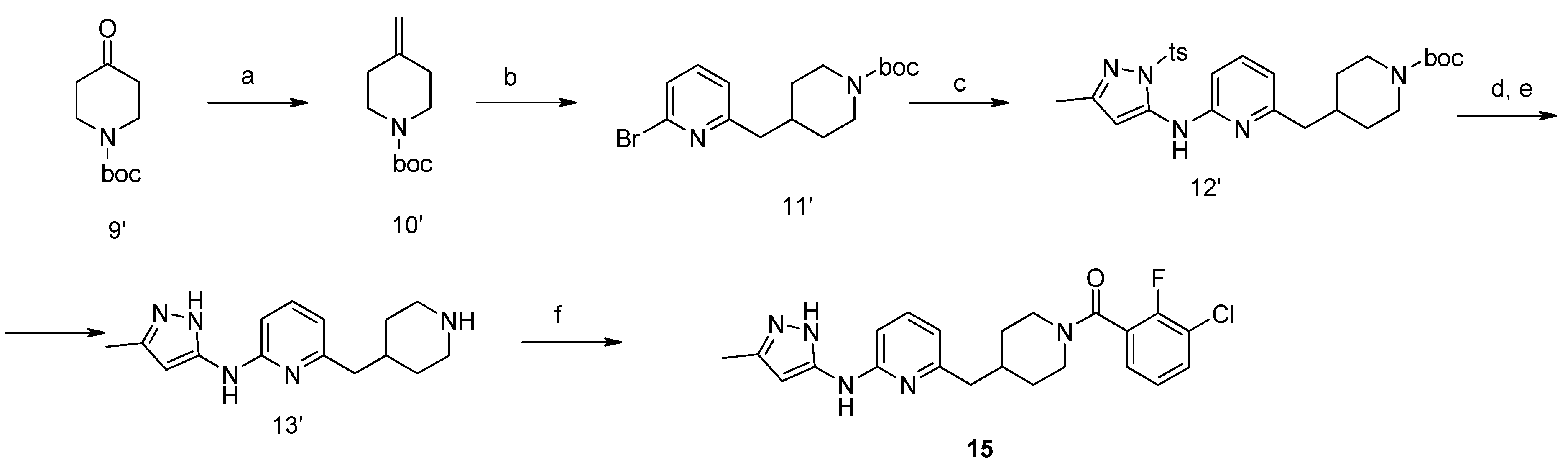
4-Methylene-piperidine-1-carboxylic acid tert-butyl Ester (10′). NaH in mineral oil (2.28 g, 57 mmol, 60%) was added portion-wise under argon atmosphere to DMSO (35 mL). The reaction mixture was stirred on a water bath up to 75 °C for about 20 min until completion of hydrogen formation. The reaction mixture was cooled down to 20 °C and treated with methyl triphenylsulfonium iodide (23.1 g, 57 mmol). After stirring for about 15 min, ketone (9′) (9.96 g, 50 mmol) in THF (15 mL) was added. The mixture was stirred at 25 °C for about 2 h and diluted with water, ethyl acetate and hexane. The organic layer was washed with water (2 × 25 mL), diluted with dichloromethane and washed with brine. The combined aqueous layers were extracted with ethyl acetate (50 mL) and hexane (50 mL). The organic layer was filtered through SiO2. The combined filtrate was concentrated and resulted in the title compound. Yield 7.4 g (75%).
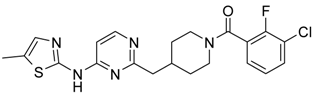
4-(6-Bromo-pyridin-2-ylmethyl)-piperidine-1-carboxylic acid tert-butyl Ester (11′). To a solution of alkene (10′) (7.4 g, 37.5 mmol) in 50 mL of anhydrous THF 9-borabicyclo[3.3.1]nonane (90 mL, 0.5 M solution in THF, 45 mmol) was added under argon atmosphere. A resulted solution was stirred at room temperature for 24 h, then 2,6-dibromopyridine (8.9 g, 37.5 mmol), potassium carbonate (7.65 g, 56 mmol), water (15 mL), and catalyst Pd(PPh3)4 (2.16 g, 1.87 mmol) were added therein. The reaction mixture was refluxed for 4 h, cooled to room temperature, poured into water (100 mL) and extracted with ethyl acetate (2 × 100 mL). The combined organic extracts were evaporated to give a crude oil, which was purified by a silica gel column chromatography to give the titled compound. Yield 10.3 g (78%). 1H-NMR δH (400 MHz, d6-DMSO): 1.06 (m, 2H, CH2), 1.35 (s, 9H, BOC), 1.47 (m, 2H, CH2), 1.69–1.87 (m, 2H, CH2), 2.65 (m, 3H, CH, CH2), 3.84 (d, 2H, CH2), 7.26 (m, 1H, ArH), 7.46 (m, 1H, ArH), 7.64 (m, 1H, ArH). APCI-MS (m/z (intensity)): 355.1 ([M + H]+, 100%), 357.0 ([M + H]+, 80%).
4-{6-[5-Methyl-2-(toluene-4-sulfonyl)-2H-pyrazol-3-ylamino]-pyridin-2-ylmethyl}-piperidine-1-carboxylic acid tert-butyl Ester (12′). Compound (12′) was prepared by Buchwald reaction, yield 367 mg (70%). 5-Methyl-2-(toluene-4-sulfonyl)-2H-pyrazol-3-ylamine was obtained similar to compound (6′d) by reaction of 5-methyl-2H-pyrazol-3-ylamine and tosyl chloride (65%). APCI-MS (m/z (intensity)): 526.4 ([M + H]+, 100%).
(5-Methyl-2H-pyrazol-3-yl)-(6-piperidin-4-ylmethyl-pyridin-2-yl)-amine Dihydrochloride (13′). Tosyl- and Boc-protective groups of the compound (12′) were removed by subsequent treatment with NaOH in methanol and HCl in 1,4-dioxane). Yield 170 mg (90%). 1H-NMR δH (400 MHz, d6-DMSO): 1.11 (m, 2H, CH2), 1.38 (s, 9H, BOC), 1.61 (m, 2H, CH2), 1.93 (m, 1H, CH), 2.27 (s, 3H, CH3), 2.63–2.79 (m, 4H, CH2), 3.78 (d, 2H, CH2), 5.96 (s, 1H, ArH), 6.93 (d, 1H, ArH), 7.26 (d, 1H, ArH), 7.94 (m, 2H, ArH, NH), 11.4 (br.s., 1H, NH). APCI-MS (m/z (intensity)): 372.2 ([M + H]+, 100%).
(3-Chloro-2-fluoro-phenyl)-{4-[6-(5-methyl-2H-pyrazol-3-ylamino)-pyridin-2-ylmethyl]-piperidin-1-yl}-Methanone (15) was prepared by acylation general method. Yield 103 mg (80%). M. p. 120–122 °C. 1H-NMR δH (400 MHz, d6-DMSO): 1.10–1.25 (m, 2H), 1.58 (m, 1H), 1.74 (m, 1H), 2.02 (m, 1H), 2.18 (s, 3H), 2.53 (m, 2H), 2.79 (m, 1H), 3.00 (m, 1H), 3.32 (m, 1H), 4.43 (m, 1H), 5.90 (s, 1H), 6.50 (d, 1H), 7.05 (s, 1H), 7.23–7.41 (m, 3H), 7.62 (m, 1H), 8.76 (s, 1H), 11.61 (s, 1H). APCI-MS (m/z (intensity)): 427.8 ([M + H]+, 100%), 429.7 ([M + H]+, 38%). HRMS (ESI): Calcd for C22H23ClFN5O ([M + H]+), 428.1648; found, 428.1642.


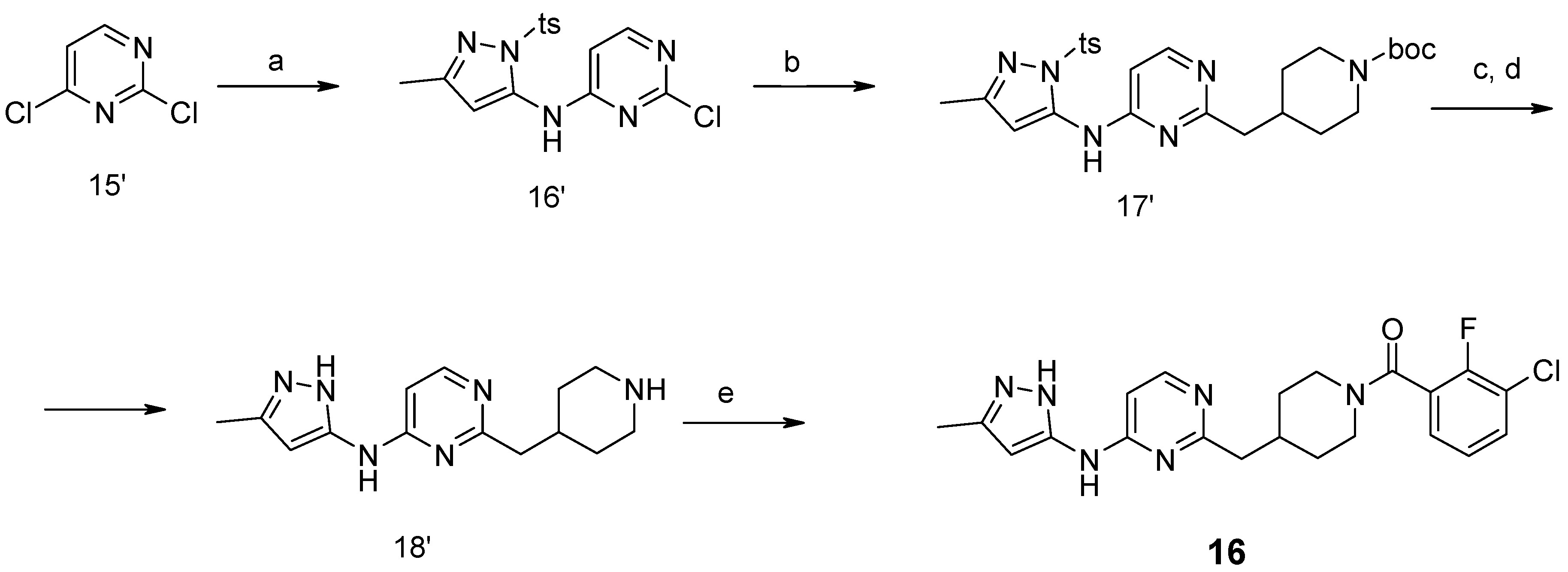

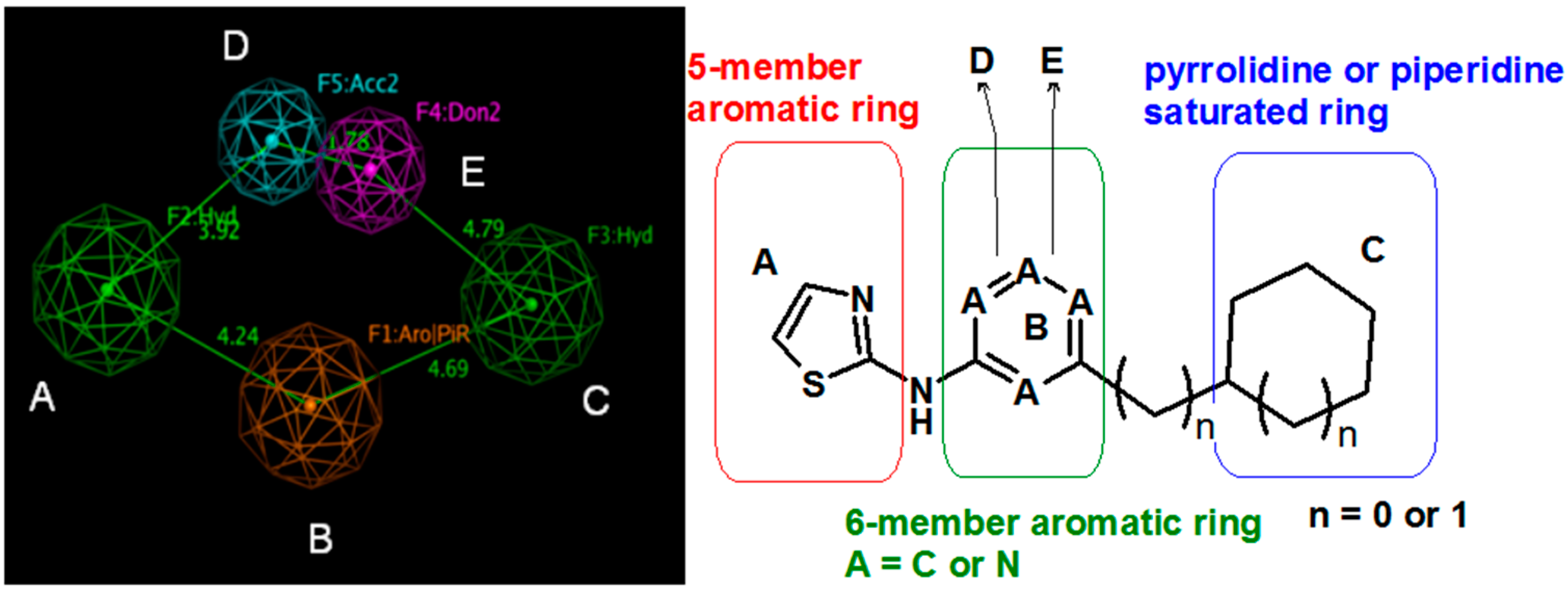{kind=link}
{kind=link}
{kind=link}
{kind=link}