
3,1-Benzothiazines, 1,4-Benzodioxines and 1,4-Benzoxazines as Inhibitors of Matriptase-2: Outcome of a Focused Screening Approach

 and
and 
Abstract
:
1. Introduction
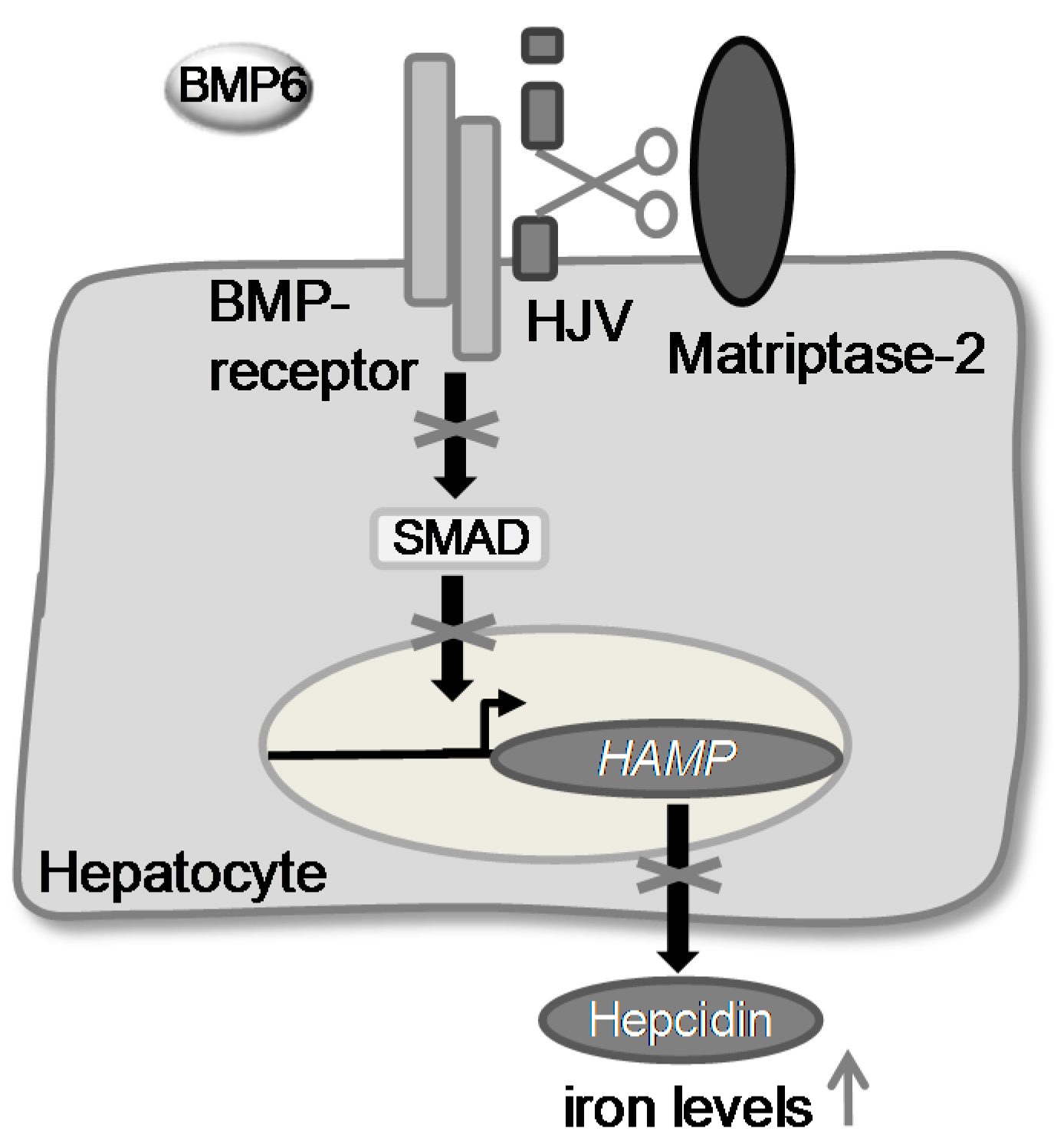
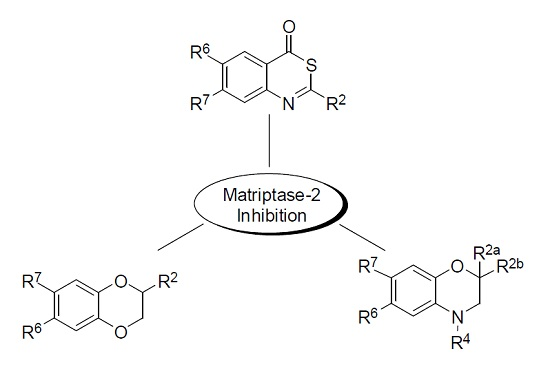
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| Compd | Structure | IC50 (µM) ± SEM a |
|---|---|---|
| 1 [34] | 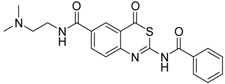 | 148 ± 28 |
| 2 [34] |  | >160 |
| 3 [32] |  | >160 |
| 4 [32] |  | >160 |
| 5 [32] |  | >160 |
| 6 [32] |  | >160 |
| 7 [32] |  | 119 ± 9 |
| 8 [32] |  | >160 |
| 9 [32] |  | >160 |
| Compd. | Structure | IC50 (µM) ± SEM a |
|---|---|---|
| (S)-10 [38] |  | 16.7 ± 1.7 |
| (R)-10 [38] | 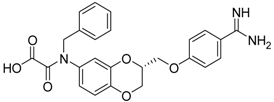 | 27.3 ± 2.0 |
| (S)-11 [38] |  | 29.8 ± 1.6 |
| (R)-11 [38] | 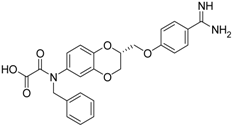 | 33.6 ± 1.1 |
| (S)-12 [38] |  | 8.47 ± 0.76 |
| 13 [39] |  | >40 |
| 14 [39] | 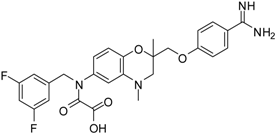 | 35.8 ± 1.1 |
| 15 [39] |  | 25.8 ± 4.0 |
| 16 [39] |  | 13.6 ± 2.31 |
| 17 [39] |  | 11.2 ± 1.61 |
| 18 [39,40] |  | 31.6 ± 2.69 |
| 19 [41] |  | 38.8 ± 3.14 |
| 20 [41] |  | 31.4 ± 3.6 |
| 21 [41] |  | 42.6 ± 4.7 |
| 22 [41] | 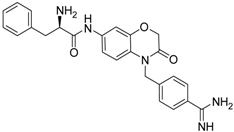 | 30.9 ± 3.4 |
| 23 [42] |  | 20.5 ± 2.0 |
| (R)-24 [43] |  | >40 |
| 25 b |  | >40 |
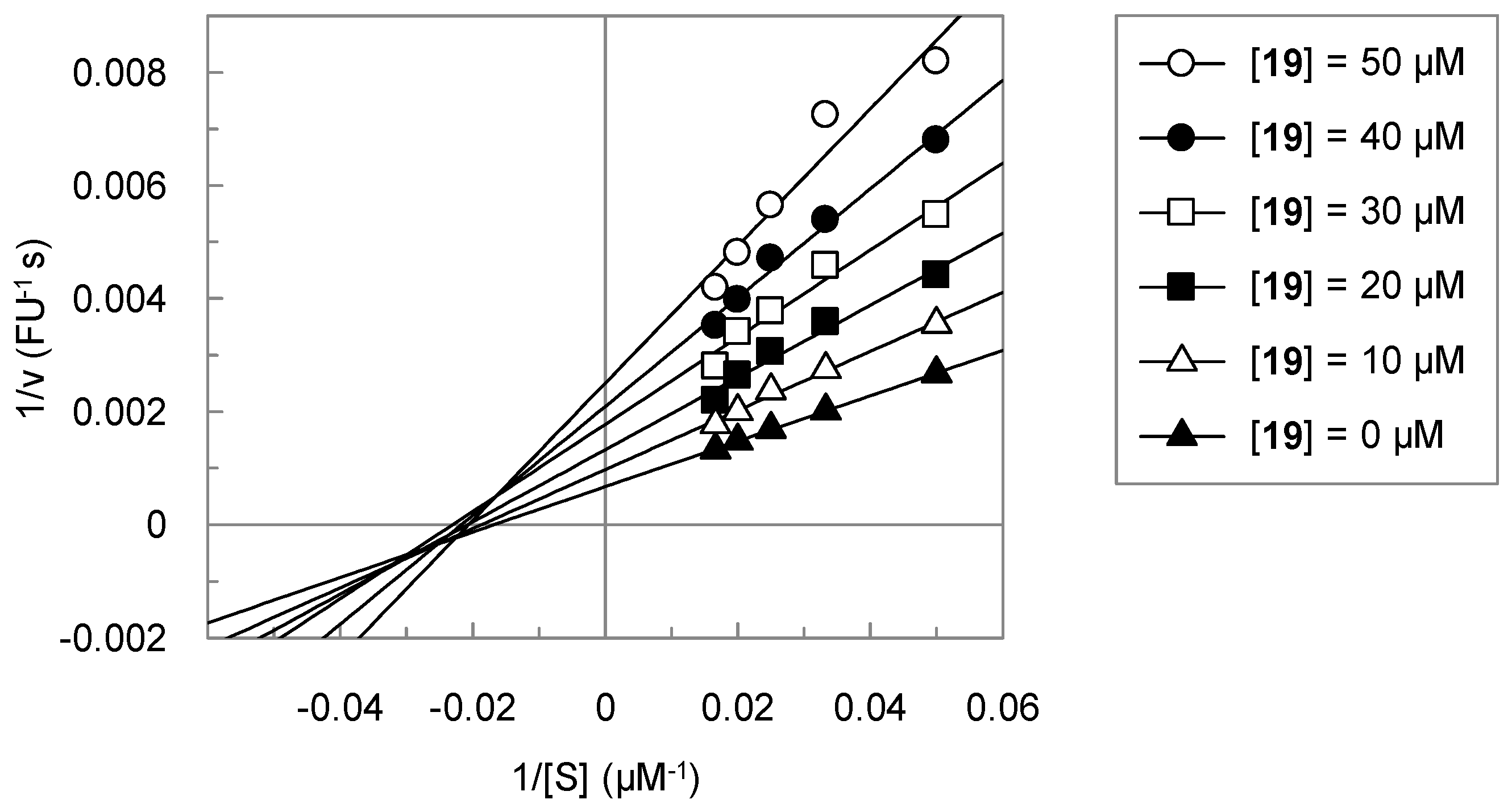
3. Experimental Section
3.1. Assays for Human Matriptase-2 Inhibition
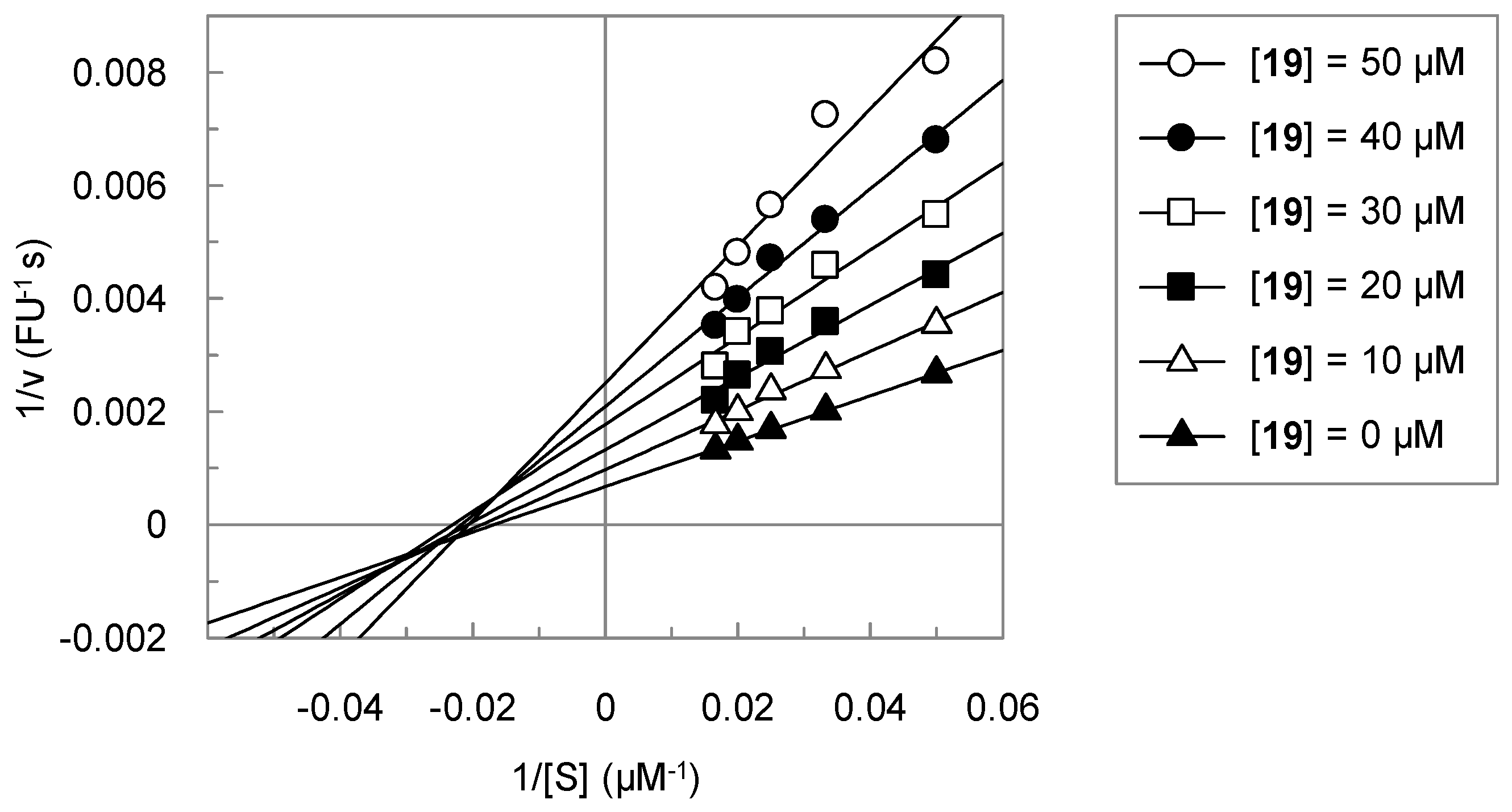
3.2. Analysis of the Kinetic Data
3.3. Purity of Tested Compounds
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ginzburg, Y.; Rivella, S. β-Thalassemia: A model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood 2011, 118, 4321–4330. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, G.; Tzilianos, M.; Christakis, J.I.; Bogdanos, D.; Tsimirika, K.; MacFarlane, J.; Goldberg, Y.P.; Sakellaropoulos, N.; Ganz, T.; Nemeth, E. Hepcidin in iron overload disorders. Blood 2005, 105, 4103–4105. [Google Scholar] [CrossRef] [PubMed]
- Meynard, D.; Babitt, J.L.; Lin, H.Y. The liver: Conductor of systemic iron balance. Blood 2014, 123, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.S. Control of systemic iron homeostasis by the hemojuvelin-hepcidin axis. Adv. Nutr. 2010, 1, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Silvestri, L. Molecular mechanisms regulating hepcidin revealed by hepcidin disorders. Sci. World J. 2011, 11, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Fleming, M.D. Modulation of hepcidin as therapy for primary and secondary iron overload disorders. Preclinical models and approaches. Hematol. Oncol. Clin. North Am. 2014, 28, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Nai, A.; Pagani, A.; Mandelli, G.; Lidonnici, M.R.; Silvestri, L.; Ferrari, G.; Camaschella, C. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of β-thalassemia. Blood 2012, 119, 5021–5029. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Cal, S.; Quesada, V.; Sanchez, L.M.; Lopez-Otin, C. Matriptase-2, a membrane-bound mosaic serine proteinase predominantly expressed in human liver and showing degrading activity against extracellular matrix proteins. J. Biol. Chem. 2002, 277, 37637–37646. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, A.J.; Reid, J.C.; Velasco, G.; Quigley, J.P.; Hooper, J.D. The type II transmembrane serine protease matriptase-2—Identification, structural features, enzymology, expression pattern and potential roles. Front. Biosci. 2008, 13, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Stirnberg, M.; Gütschow, M. Matriptase-2, a regulatory protease of iron homeostasis: Possible substrates, cleavage sites and inhibitors. Curr. Pharm. Des. 2013, 19, 1052–1061. [Google Scholar] [PubMed]
- Stirnberg, M.; Maurer, E.; Horstmeyer, A.; Kolp, S.; Frank, S.; Bald, T.; Arenz, K.; Janzer, A.; Prager, K.; Walter, J.; et al. Proteolytic processing of the serine protease matriptase-2: Identification of the cleavage sites required for its autocatalytic release from the cell surface. Biochem. J. 2010, 430, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; She, E.; Gelbart, T.; Truksa, J.; Lee, P.; Xia, Y.; Khovananth, K.; Mudd, S.; Mann, N.; Moresco, E.M.; et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science 2008, 320, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Finberg, K.E.; Heeney, M.M.; Campagna, D.R.; Aydinok, Y.; Pearson, H.A.; Hartman, K.R.; Mayo, M.M.; Samuel, S.M.; Strouse, J.J.; Markianos, K.; et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 2008, 40, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; de Lara, F.M.; Pendas, A.M.; Garabaya, C.; Rodriguez, F.; Astudillo, A.; Bernal, T.; Cabanillas, R.; Lopez-Otin, C.; Velasco, G. Membrane-bound serine protease matriptase-2 (TMPRSS6) is an essential regulator of iron homeostasis. Blood 2008, 112, 2539–2545. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Pagani, A.; Nai, A.; de Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metable 2008, 8, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Maxson, J.E.; Chen, J.; Enns, C.A.; Zhang, A.S. Matriptase-2- and proprotein convertase-cleaved forms of hemojuvelin have different roles in the down-regulation of hepcidin expression. J. Biol. Chem. 2010, 285, 39021–39028. [Google Scholar] [CrossRef] [PubMed]
- Rausa, M.; Ghitti, M.; Pagani, A.; Nai, A.; Campanella, A.; Musco, G.; Camaschella, C.; Silvestri, L. Identification of TMPRSS6 cleavage sites of hemojuvelin. J. Cell. Mol. Med. 2015, 19, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, M.; Gruba, N.; Miecznikowska, A.; Popow-Stellmaszyk, J.; Gütschow, M.; Stirnberg, M.; Furtmann, N.; Bajorath, J.; Lesner, A.; Rolka, K. Substrate specificity of human matriptase-2. Biochimie 2014, 97, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Dosa, S.; Stirnberg, M.; Lülsdorff, V.; Häuβler, D.; Maurer, E.; Gütschow, M. Active site mapping of trypsin, thrombin and matriptase-2 by sulfamoyl benzamidines. Bioorg. Med. Chem. 2012, 20, 6489–6505. [Google Scholar] [CrossRef] [PubMed]
- Furtmann, N.; Häußler, D.; Scheidt, T.; Stirnberg, M.; Steinmetzer, T.; Bajorath, J.; Gütschow, M. Limiting the number of potential binding modes by introducing symmetry into ligands: Structure-based design of inhibitors for trypsin-like serine proteases. Chem. Eur. J. 2016, 22, 610–625. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Casu, C.; Gardenghi, S.; Booten, S.; Aghajan, M.; Peralta, R.; Watt, A.; Freier, S.; Monia, B.P.; Rivella, S. Reducing TMPRSS6 ameliorates hemochromatosis and β-thalassemia in mice. J. Clin. Investig. 2013, 123, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Racie, T.; Westerman, M.; Fitzgerald, K.; Butler, J.S.; Fleming, M.D. Combination therapy with a Tmprss6 RNAi-therapeutic and the oral iron chelator deferiprone additively diminishes secondary iron overload in a mouse model of β-thalassemia intermedia. Am. J. Hematol. 2015, 90, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Toudjarska, I.; Sendamarai, A.K.; Racie, T.; Milstein, S.; Bettencourt, B.R.; Hettinger, J.; Bumcrot, D.; Fleming, M.D. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(−/−) mice and ameliorates anemia and iron overload in murine β-thalassemia intermedia. Blood 2013, 121, 1200–1208. [Google Scholar] [CrossRef] [PubMed]
- Casu, C.; Aghajan, M.; Oikonomidou, P.R.; Guo, S.; Monia, B.P.; Rivella, S. Combination of Tmprss6-ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of β-thalassemia intermedia. Haematologica 2016, 101, e8–e11. [Google Scholar] [CrossRef] [PubMed]
- Sisay, M.T.; Steinmetzer, T.; Stirnberg, M.; Maurer, E.; Hammami, M.; Bajorath, J.; Gütschow, M. Identification of the first low-molecular-weight inhibitors of matriptase-2. J. Med. Chem. 2010, 53, 5523–5535. [Google Scholar] [CrossRef] [PubMed]
- Hammami, M.; Rühmann, E.; Maurer, E.; Heine, A.; Gütschow, M.; Klebe, G.; Steinmetzer, T. New 3-amidinophenylalanine-derived inhibitors of matriptase. Med. Chem. Commun. 2012, 3, 807–813. [Google Scholar] [CrossRef]
- Duchêne, D.; Colombo, E.; Désilets, A.; Boudreault, P.L.; Leduc, R.; Marsault, E.; Najmanovich, R. Analysis of subpocket selectivity and identification of potent selective inhibitors for matriptase and matriptase-2. J. Med. Chem. 2014, 57, 10198–10204. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, A.; Dębowski, D.; Karna, N.; Łęgowska, A.; Stirnberg, M.; Gütschow, M.; Rolka, K. Inhibitors of matriptase-2 based on the trypsin inhibitor SFTI-1. ChemBioChem 2015, 16, 1601–1607. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Bacchi, C.J.; Kode, N.R.; Zhang, Q.; Wang, G.; Yartlet, N.; Rattendi, D.; Londono, I.; Mazumder, L.; Vanden Eynde, J.J.; et al. Trypanocidal activity of piperazine-linked bisbenzamidines and bisbenzamidoxime, an orally active prodrug. Int. J. Antimicrob. Agents 2007, 30, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Vanden Eynde, J.J.; Mayence, A.; Collins, M.S.; Cushion, M.T.; Rattendi, D.; Londono, I.; Mazumder, L.; Bacchi, C.J.; Yarlett, N. Synthesis and SAR of alkanediamide-linked bisbenzamidines with anti-trypanosomal and anti-pneumocystis activity. Bioorg. Med. Chem. Lett. 2009, 19, 5884–5886. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Mayence, A.; Vanden Eynde, J.J. Some non-conventional biomolecular targets for diamidines. A short survey. Bioorg. Med. Chem. 2014, 22, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Gütschow, M.; Schlenk, M.; Gäb, J.; Paskaleva, M.; Alnouri, M.W.; Scolari, S.; Iqbal, J.; Müller, C.E. Benzothiazinones: A novel class of adenosine receptor antagonists structurally unrelated to xanthine and adenine derivatives. J. Med. Chem. 2012, 55, 3331–3341. [Google Scholar] [CrossRef] [PubMed]
- Stössel, A.; Schlenk, M.; Hinz, S.; Küppers, P.; Heer, J.; Gütschow, M.; Müller, C.E. Dual targeting of adenosine A(2A) receptors and monoamine oxidase B by 4H-3,1-benzothiazin-4-ones. J. Med. Chem. 2013, 56, 4580–4596. [Google Scholar] [CrossRef] [PubMed]
- Blättermann, S.; Peters, L.; Ottersbach, P.A.; Bock, A.; Konya, V.; Weaver, C.D.; Gonzalez, A.; Schröder, R.; Tyagi, R.; Luschnig, P.; et al. A biased ligand for OXE-R uncouples Gα and Gβγ signaling within a heterotrimer. Nat. Chem. Biol. 2012, 8, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Krantz, A.; Spencer, R.W.; Tam, T.F.; Liak, T.J.; Copp, L.J.; Thomas, E.M.; Rafferty, S.P. Design and synthesis of 4H-3,1-benzoxazin-4-ones as potent alternate substrate inhibitors of human leukocyte elastase. J. Med. Chem. 1990, 33, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Neumann, U.; Schechter, N.M.; Gütschow, M. Inhibition of human chymase by 2-amino-3,1-benzoxazin-4-ones. Bioorg. Med. Chem. 2001, 9, 947–954. [Google Scholar] [CrossRef]
- Neumann, U.; Gütschow, M. 3,1-Benzothiazin-4-ones and 3,1-benzoxazin-4-ones: Highly different activities in chymotrypsin inactivation. Bioorg. Chem. 1995, 23, 72–88. [Google Scholar] [CrossRef]
- Ilić, M.; Dunkel, P.; Ilaš, J.; Chabielska, E.; Zakrzeska, A.; Mátyus, P.; Kikelj, D. Towards dual antithrombotic compounds—Balancing thrombin inhibitory and fibrinogen GPIIb/IIIa binding inhibitory activities of 2,3-dihydro-1,4-benzodioxine derivatives through regio- and stereoisomerism. Eur. J. Med. Chem. 2013, 62, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Ilić, M.; Kikelj, D.; Ilaš, J. Fluorinated dual antithrombotic compounds based on 1,4-benzoxazine scaffold. Eur. J. Med. Chem. 2012, 50, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Ilić, M.; Kontogiorgis, C.; Hadjipavlou-Litina, D.; Ilaš, J.; Kikelj, D. Thrombin inhibitors with lipid peroxidation and lipoxygenase inhibitory activities. Bioorg. Med. Chem. Lett. 2011, 21, 4705–4709. [Google Scholar] [CrossRef] [PubMed]
- Ilaš, J.; Jakopin, Z.; Borštnar, T.; Stegnar, M.; Kikelj, D. 3,4-Dihydro-2H-1,4-benzoxazine derivatives combining thrombin inhibitory and glycoprotein IIb/IIIa receptor antagonistic activity as a novel class of antithrombotic compounds with dual function. J. Med. Chem. 2008, 51, 5617–5629. [Google Scholar] [CrossRef] [PubMed]
- Anderluh, M.; Cesar, C.; Štefanič, P.; Kikelj, D.; Janeš, D.; Murn, J.; Nadrah, K.; Tominc, M.; Addicks, E.; Giannis, A.; et al. Design and synthesis of novel platelet fibrinogen receptor antagonists with 2H-1,4-benzoxazine-3(4H)-one scaffold. A systematic study. Eur. J. Med. Chem. 2005, 40, 25–49. [Google Scholar] [CrossRef] [PubMed]
- Trstenjak, U.; Ilaš, J.; Kikelj, D. Low molecular weight dual inhibitors of factor Xa and fibrinogen binding to GPIIb/IIIa with highly overlapped pharmacophores. Eur. J. Med. Chem. 2013, 64, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Zega, A.; Mlinsek, G.; Solmajer, T.; Trampus-Bakija, A.; Stegnar, M.; Urleb, U. Thrombin inhibitors built on an azaphenylalanine scaffold. Bioorg. Med. Chem. Lett. 2004, 14, 1563–1567. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roydeva, P.G.; Beckmann, A.-M.; Stirnberg, M.; Cesar, J.; Kikelj, D.; Ilaš, J.; Gütschow, M. 3,1-Benzothiazines, 1,4-Benzodioxines and 1,4-Benzoxazines as Inhibitors of Matriptase-2: Outcome of a Focused Screening Approach. Pharmaceuticals 2016, 9, 2. https://doi.org/10.3390/ph9010002
Roydeva PG, Beckmann A-M, Stirnberg M, Cesar J, Kikelj D, Ilaš J, Gütschow M. 3,1-Benzothiazines, 1,4-Benzodioxines and 1,4-Benzoxazines as Inhibitors of Matriptase-2: Outcome of a Focused Screening Approach. Pharmaceuticals. 2016; 9(1):2. https://doi.org/10.3390/ph9010002
Chicago/Turabian StyleRoydeva, Polya G., Anna-Madeleine Beckmann, Marit Stirnberg, Jožko Cesar, Danijel Kikelj, Janez Ilaš, and Michael Gütschow. 2016. "3,1-Benzothiazines, 1,4-Benzodioxines and 1,4-Benzoxazines as Inhibitors of Matriptase-2: Outcome of a Focused Screening Approach" Pharmaceuticals 9, no. 1: 2. https://doi.org/10.3390/ph9010002
APA StyleRoydeva, P. G., Beckmann, A.-M., Stirnberg, M., Cesar, J., Kikelj, D., Ilaš, J., & Gütschow, M. (2016). 3,1-Benzothiazines, 1,4-Benzodioxines and 1,4-Benzoxazines as Inhibitors of Matriptase-2: Outcome of a Focused Screening Approach. Pharmaceuticals, 9(1), 2. https://doi.org/10.3390/ph9010002