
UCP2 Modulates Cardioprotective Effects of Ru360 in Isolated Cardiomyocytes during Ischemia

Abstract
:1. Introduction
2. Results
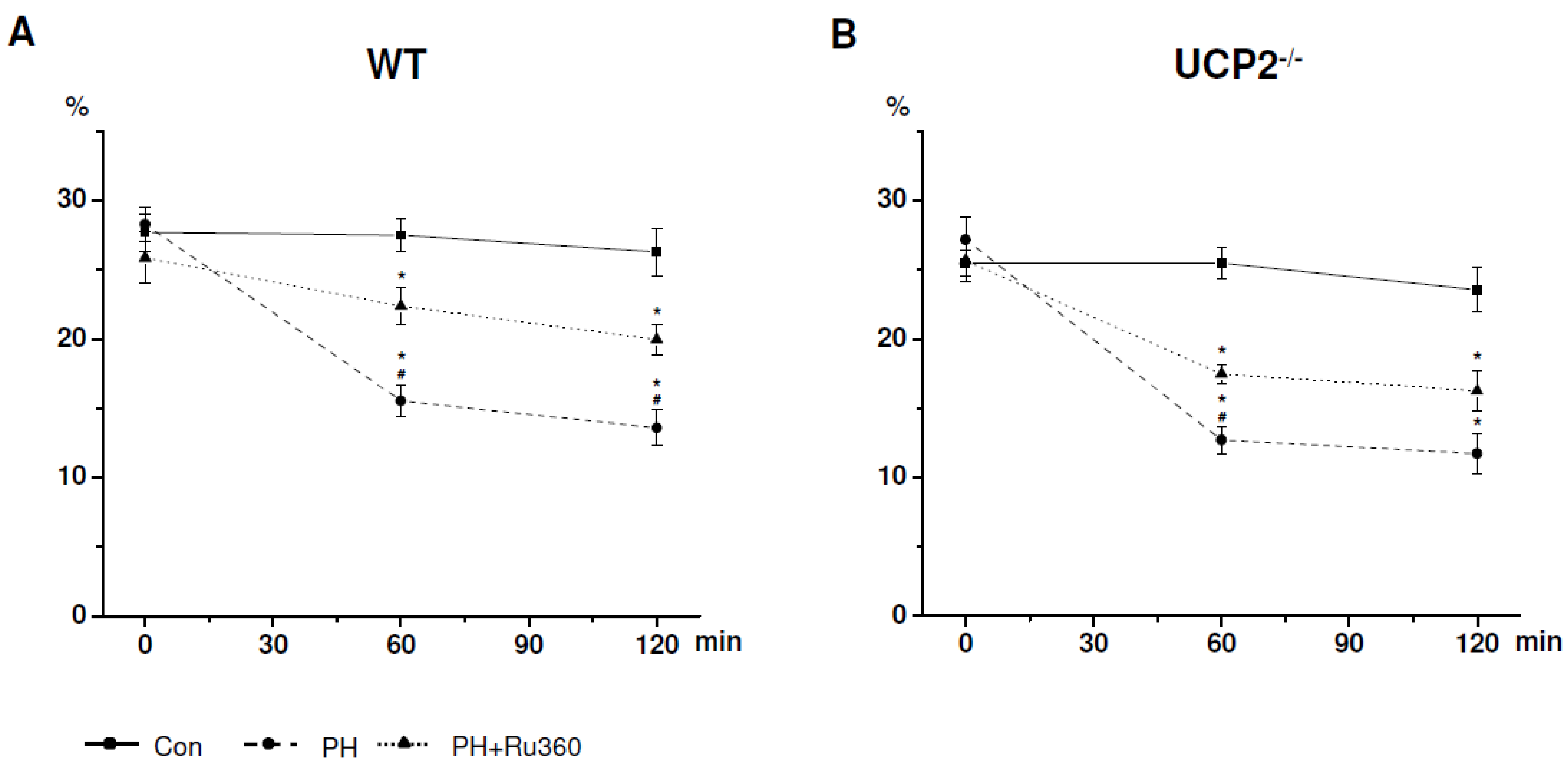

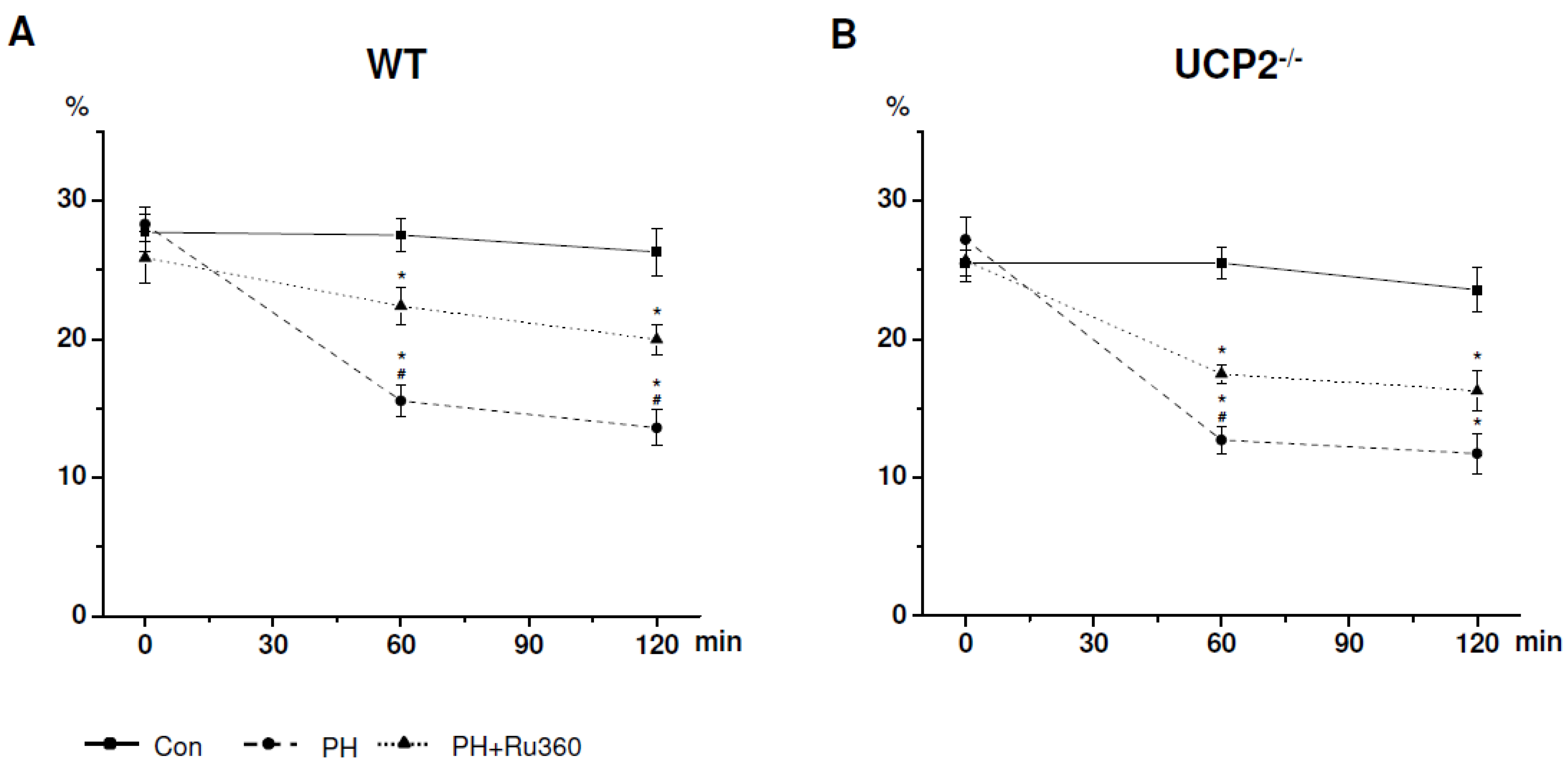{kind=link}
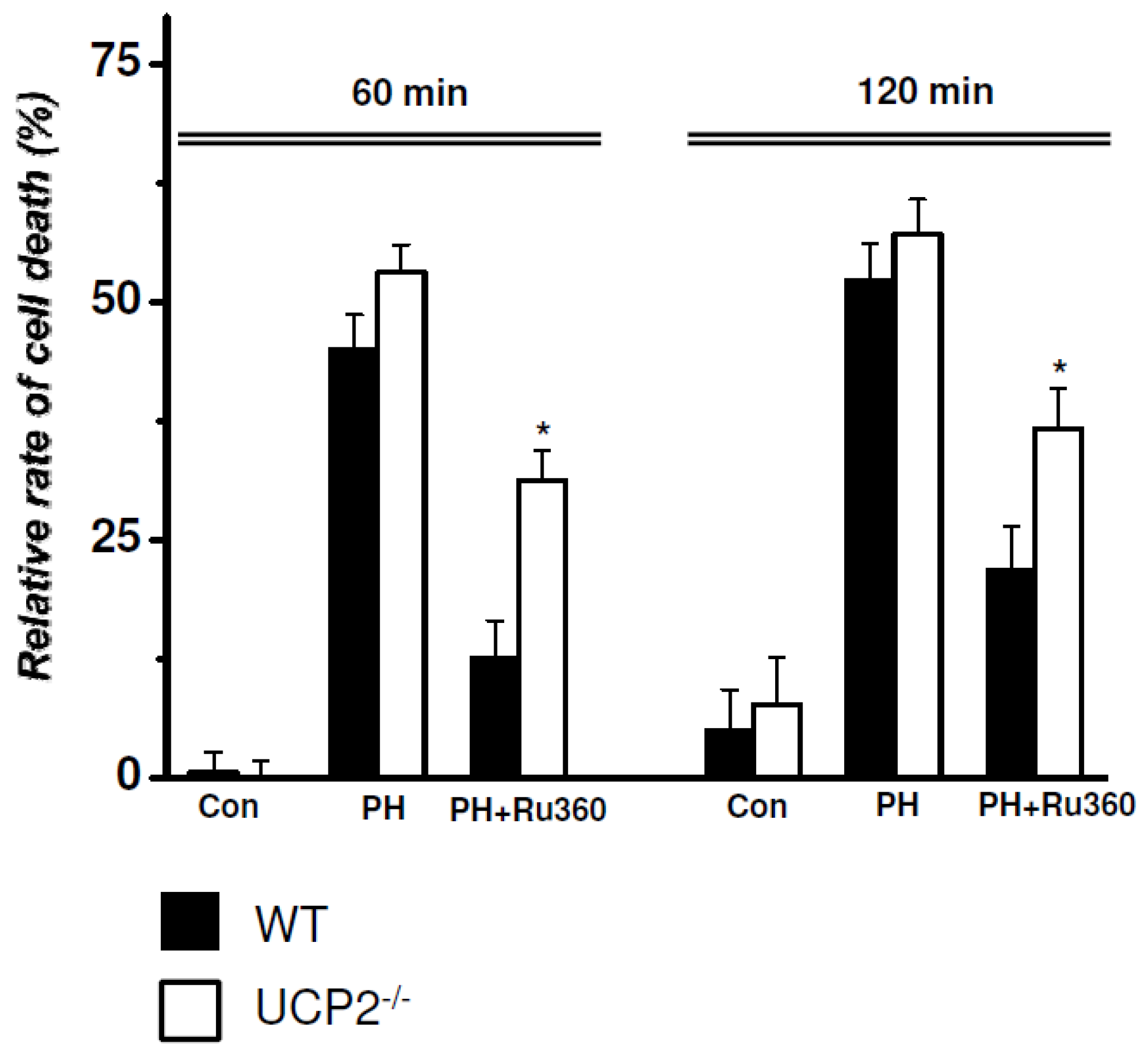{kind=link}
| WT (n = 6) | Cell Viability (%) | ||
|---|---|---|---|
| 0 min | 60 min | 120 min | |
| Control | 27.72 ± 1.25 | 27.51 ± 1.14 | 26.32 ± 1.58 |
| PH | 28.31 ± 1.13 | 15.55 ± 1.06 * | 13.60 ± 1.20 * |
| PH+Ru360 | 25.88 ± 1.72 | 22.39 ± 1.21 *# | 19.98 ± 0.98 *# |
2.1. CV after Induction of Ischemia in WT Cardiomyocytes
2.2. CV after Induction of Ischemia in UCP2−/− Cardiomyocytes
| UCP2−/− (n = 5) | Cell Viability (%) | ||
|---|---|---|---|
| 0 min | 0 min | 0 min | |
| Control | 25.50 ± 0.77 | 25.50 ± 0.97 | 23.56 ± 1.37 |
| PH | 27.19 ± 1.33 | 12.71 ± 0.83 * | 11.73 ± 1.23 * |
| PH+Ru360 | 25.67 ± 1.26 | 17.49 ± 0.58 *# | 16.26 ± 1.25 * |
2.3. RRCD in WT vs. UCP2−/− Cardiomyocytes
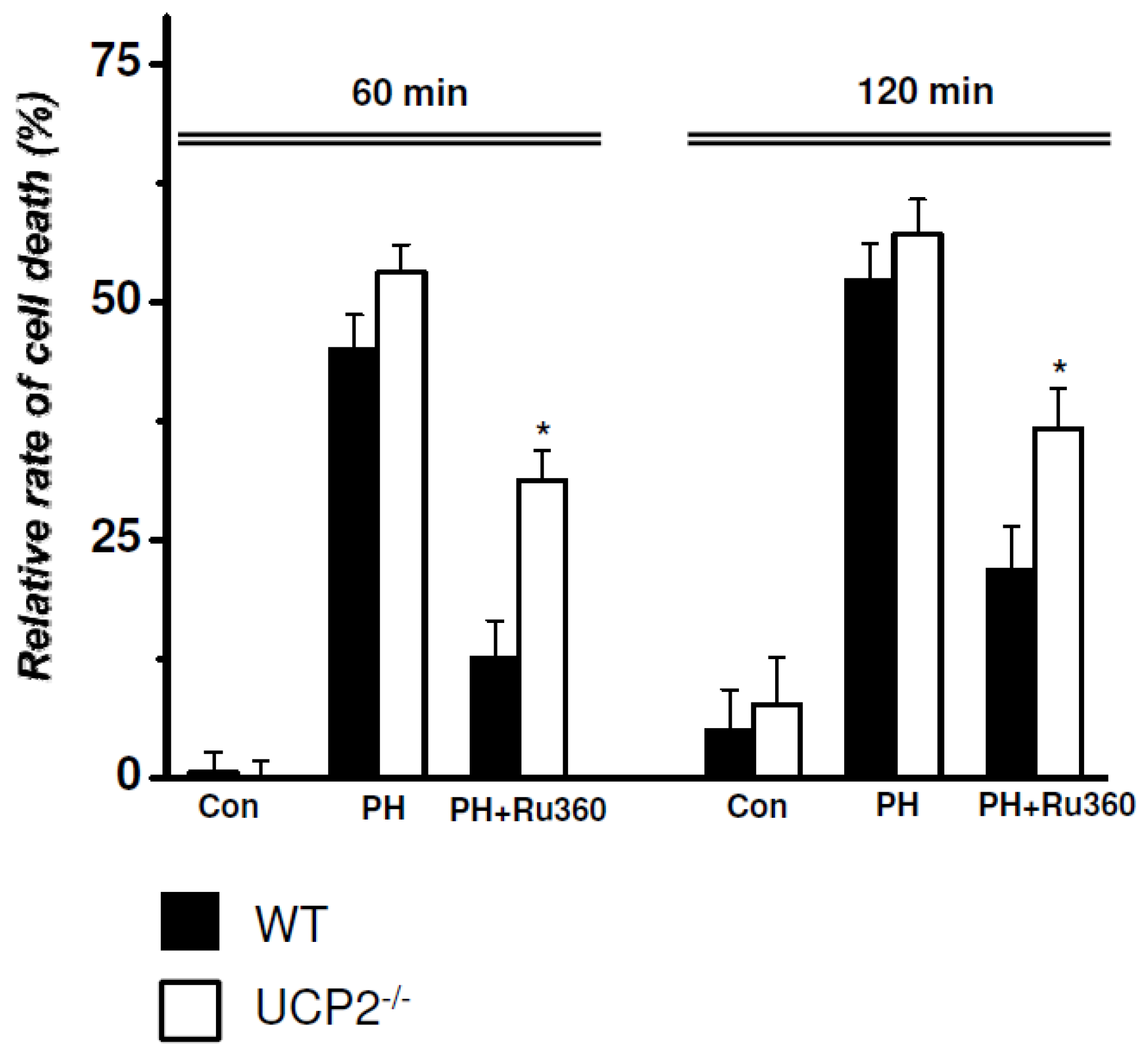
| WT (n = 6) vs. UCP2−/− (n = 5) | Relative Rate of Cell Death (%) | ||
|---|---|---|---|
| 60 min | 120 min | ||
| Control | WT | 0.54 ± 2.00 | 5.04 ± 3.95 |
| UCP2-/- | 0.12 ± 1.41 | 7.70 ± 4.27 | |
| PH | WT | 44.98 ± 3.45 | 52.25 ± 3.56 |
| UCP2-/- | 53.12 ± 2.43 | 57.10 ± 3.12 | |
| PH+Ru360 | WT | 12.64 ± 3.58 | 21.74 ± 4.26 |
| UCP2-/- | 31.30 ± 2.67 * | 36.68 ± 3.67 * | |
3. Discussion
4. Experimental Section
4.1. Animals
4.2. Preparation of Murine Cardiomyocytes
4.3. Pelleting Hypoxia
4.4. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization (WHO). Facts and Figures: The World Health Report; WHO: Geneva, Switzerland, 2003; pp. 1–6. [Google Scholar]
- Buja, L.M.; Eigenbrodt, M.L.; Eigenbrodt, E.H. Apoptosis and necrosis. Basic types and mechanisms of cell death. Arch. Pathol. Lab. Med. 1993, 117, 1208–1214. [Google Scholar] [PubMed]
- Fliss, H.; Gattinger, D. Apoptosis in ischemic and reperfused rat myocardium. Circ. Res. 1996, 79, 949–956. [Google Scholar] [CrossRef] [PubMed]
- James, T.N. Normal and abnormal consequences of apoptosis in the human heart. From postnatal morphogenesis to paroxysmal arrhythmias. Circulation 1994, 90, 556–573. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B. Mitochondrial ion channels. Annu. Rev. Physiol. 2007, 69, 19–49. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial atp synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [PubMed]
- Territo, P.R.; Mootha, V.K.; French, S.A.; Balaban, R.S. Ca2+ activation of heart mitochondrial oxidative phosphorylation: Role of the f(0)/f(1)-atpase. Am. J. Physiol. Cell Physiol. 2000, 278, C423–C435. [Google Scholar] [PubMed]
- Kohlhaas, M.; Maack, C. Adverse bioenergetic consequences of Na+-Ca2+ exchanger-mediated Ca2+ influx in cardiac myocytes. Circulation 2010, 122, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Rasola, A. Calcium and cell death: The mitochondrial connection. Subcell. Biochem. 2007, 45, 481–506. [Google Scholar] [PubMed]
- Hoppe, U.C. Mitochondrial calcium channels. FEBS Lett. 2010, 584, 1975–1981. [Google Scholar] [CrossRef] [PubMed]
- Piot, C.; Croisille, P.; Staat, P.; Thibault, H.; Rioufol, G.; Mewton, N.; Elbelghiti, R.; Cung, T.T.; Bonnefoy, E.; Angoulvant, D.; et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 2008, 359, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Kirichok, Y.; Krapivinsky, G.; Clapham, D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004, 427, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Michels, G.; Khan, I.F.; Endres-Becker, J.; Rottlaender, D.; Herzig, S.; Ruhparwar, A.; Wahlers, T.; Hoppe, U.C. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation 2009, 119, 2435–2443. [Google Scholar] [CrossRef] [PubMed]
- Matlib, M.A.; Zhou, Z.; Knight, S.; Ahmed, S.; Choi, K.M.; Krause-Bauer, J.; Phillips, R.; Altschuld, R.; Katsube, Y.; Sperelakis, N.; et al. Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J. Biol. Chem. 1998, 273, 10223–10231. [Google Scholar] [CrossRef] [PubMed]
- Zazueta, C.; Sosa-Torres, M.E.; Correa, F.; Garza-Ortiz, A. Inhibitory properties of ruthenium amine complexes on mitochondrial calcium uptake. J. Bioenerg. Biomembr. 1999, 31, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Benzi, R.H.; Lerch, R. Dissociation between contractile function and oxidative metabolism in postischemic myocardium. Attenuation by ruthenium red administered during reperfusion. Circ. Res. 1992, 71, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Figueredo, V.M.; Dresdner, K.P., Jr.; Wolney, A.C.; Keller, A.M. Postischaemic reperfusion injury in the isolated rat heart: Effect of ruthenium red. Cardiovasc. Res. 1991, 25, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rivas Gde, J.; Carvajal, K.; Correa, F.; Zazueta, C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 2006, 149, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Grover, G.J.; Dzwonczyk, S.; Sleph, P.G. Ruthenium red improves postischemic contractile function in isolated rat hearts. J. Cardiovasc. Pharmacol. 1990, 16, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Z.; Gao, Q.; Cao, C.M.; Bruce, I.C.; Xia, Q. Involvement of the mitochondrial calcium uniporter in cardioprotection by ischemic preconditioning. Life Sci. 2006, 78, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Ricquier, D.; Bouillaud, F. The uncoupling protein homologues: Ucp1, ucp2, ucp3, stucp and atucp. Biochem. J. 2000, 345, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Teshima, Y.; Akao, M.; Jones, S.P.; Marban, E. Uncoupling protein-2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ. Res. 2003, 93, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Trenker, M.; Malli, R.; Fertschai, I.; Levak-Frank, S.; Graier, W.F. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 2007, 9, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Waldeck-Weiermair, M.; Jean-Quartier, C.; Rost, R.; Khan, M.J.; Vishnu, N.; Bondarenko, A.I.; Imamura, H.; Malli, R.; Graier, W.F. Leucine zipper ef hand-containing transmembrane protein 1 (letm1) and uncoupling proteins 2 and 3 (ucp2/3) contribute to two distinct mitochondrial Ca2+ uptake pathways. J. Biol. Chem. 2011, 286, 28444–28455. [Google Scholar] [CrossRef] [PubMed]
- Waldeck-Weiermair, M.; Malli, R.; Naghdi, S.; Trenker, M.; Kahn, M.J.; Graier, W.F. The contribution of ucp2 and ucp3 to mitochondrial Ca2+ uptake is differentially determined by the source of supplied Ca2+. Cell Calcium 2010, 47, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Motloch, L.J.; Larbig, R.; Gebing, T.; Reda, S.; Weichselbaumer, S.; Kokoschinegg, D.; Schwaiger, A.; Wolny, M.; Hoppe, U.C. Ucp2 modulates mitochondrial calcium uniporter. Biophys. J. 2014, 106, 593a. [Google Scholar] [CrossRef]
- Larbig, R.K.; Motloch, L.J.; Gebing, T.; Reda, S.; Deininger, E.; Schwaiger, A.; Wolny, M.; Hoppe, U.C. Ucp2 modulates cellular excitation contraction coupling via mitochondrial calcium uptake. Biophys. J. 2014, 106, 593a. [Google Scholar] [CrossRef]
- Diaz, R.J.; Wilson, G.J. Studying ischemic preconditioning in isolated cardiomyocyte models. Cardiovasc. Res. 2006, 70, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Daniel, K.W.; Liu, D.; Lyght, O.; Edelstein, D.; Brownlee, M.; Corkey, B.E.; Collins, S. Persistent oxidative stress due to absence of uncoupling protein 2 associated with impaired pancreatic beta-cell function. Endocrinology 2009, 150, 3040–3048. [Google Scholar] [CrossRef] [PubMed]
- Lange, P.S.; Er, F.; Gassanov, N.; Hoppe, U.C. Andersen mutations of kcnj2 suppress the native inward rectifier current ik1 in a dominant-negative fashion. Cardiovasc. Res. 2003, 59, 321–327. [Google Scholar] [CrossRef]
- Hoppe, U.C.; Beuckelmann, D.J. Characterization of the hyperpolarization-activated inward current in isolated human atrial myocytes. Cardiovasc. Res. 1998, 38, 788–801. [Google Scholar] [CrossRef]
- Li, X.; Heinzel, F.R.; Boengler, K.; Schulz, R.; Heusch, G. Role of connexin 43 in ischemic preconditioning does not involve intercellular communication through gap junctions. J. Mol. Cell. Cardiol. 2004, 36, 161–163. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motloch, L.J.; Reda, S.; Wolny, M.; Hoppe, U.C. UCP2 Modulates Cardioprotective Effects of Ru360 in Isolated Cardiomyocytes during Ischemia. Pharmaceuticals 2015, 8, 474-482. https://doi.org/10.3390/ph8030474
Motloch LJ, Reda S, Wolny M, Hoppe UC. UCP2 Modulates Cardioprotective Effects of Ru360 in Isolated Cardiomyocytes during Ischemia. Pharmaceuticals. 2015; 8(3):474-482. https://doi.org/10.3390/ph8030474
Chicago/Turabian StyleMotloch, Lukas J., Sara Reda, Martin Wolny, and Uta C. Hoppe. 2015. "UCP2 Modulates Cardioprotective Effects of Ru360 in Isolated Cardiomyocytes during Ischemia" Pharmaceuticals 8, no. 3: 474-482. https://doi.org/10.3390/ph8030474
APA StyleMotloch, L. J., Reda, S., Wolny, M., & Hoppe, U. C. (2015). UCP2 Modulates Cardioprotective Effects of Ru360 in Isolated Cardiomyocytes during Ischemia. Pharmaceuticals, 8(3), 474-482. https://doi.org/10.3390/ph8030474