Involvement of HDAC1 and HDAC3 in the Pathology of Polyglutamine Disorders: Therapeutic Implications for Selective HDAC1/HDAC3 Inhibitors

Abstract
:1. Introduction
2. Gene Expression Regulation: Chromatin, HATs and HDACs
3. HDAC Family of Proteins
3.1. HDAC1
3.2. HDAC3
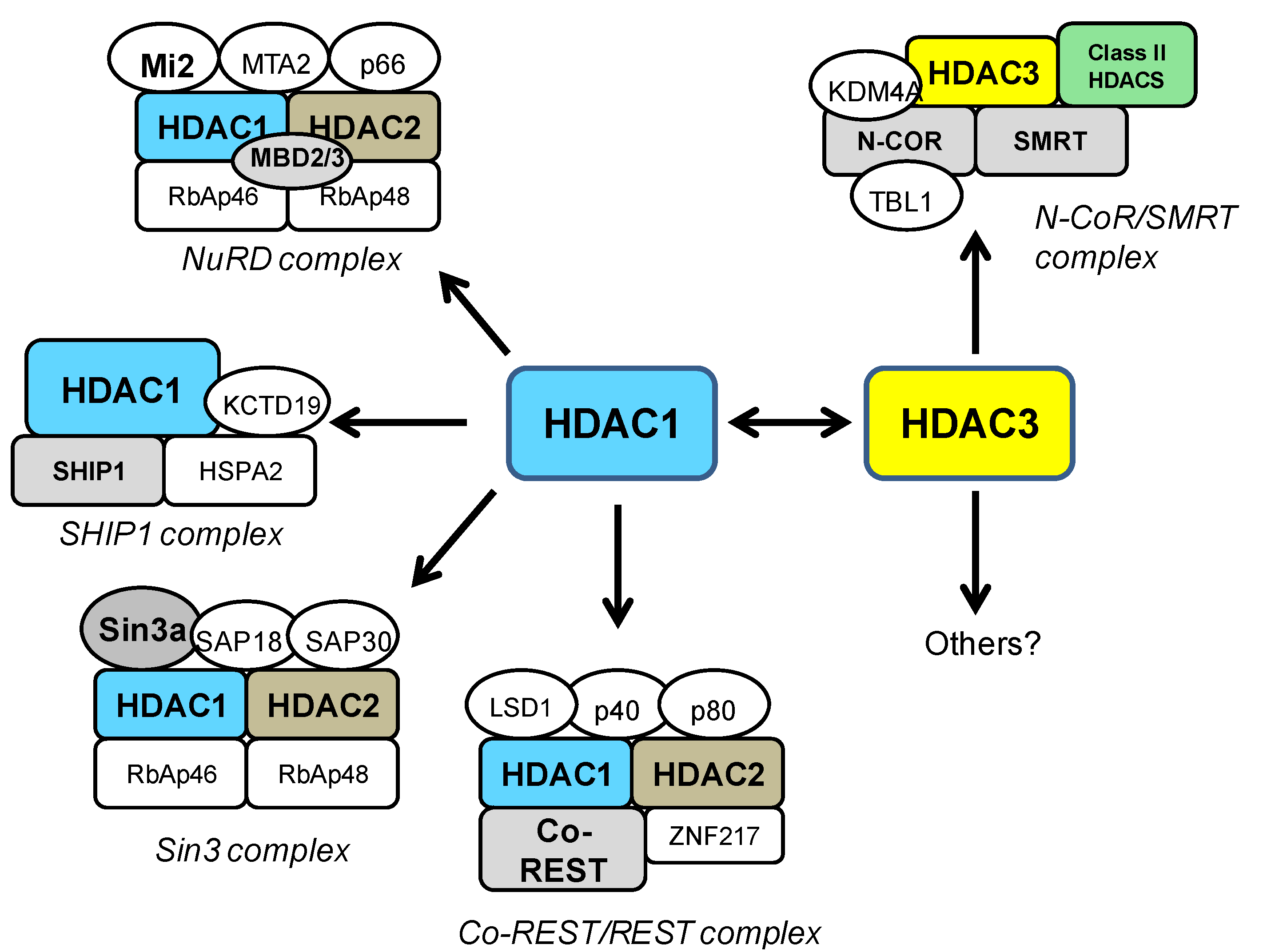
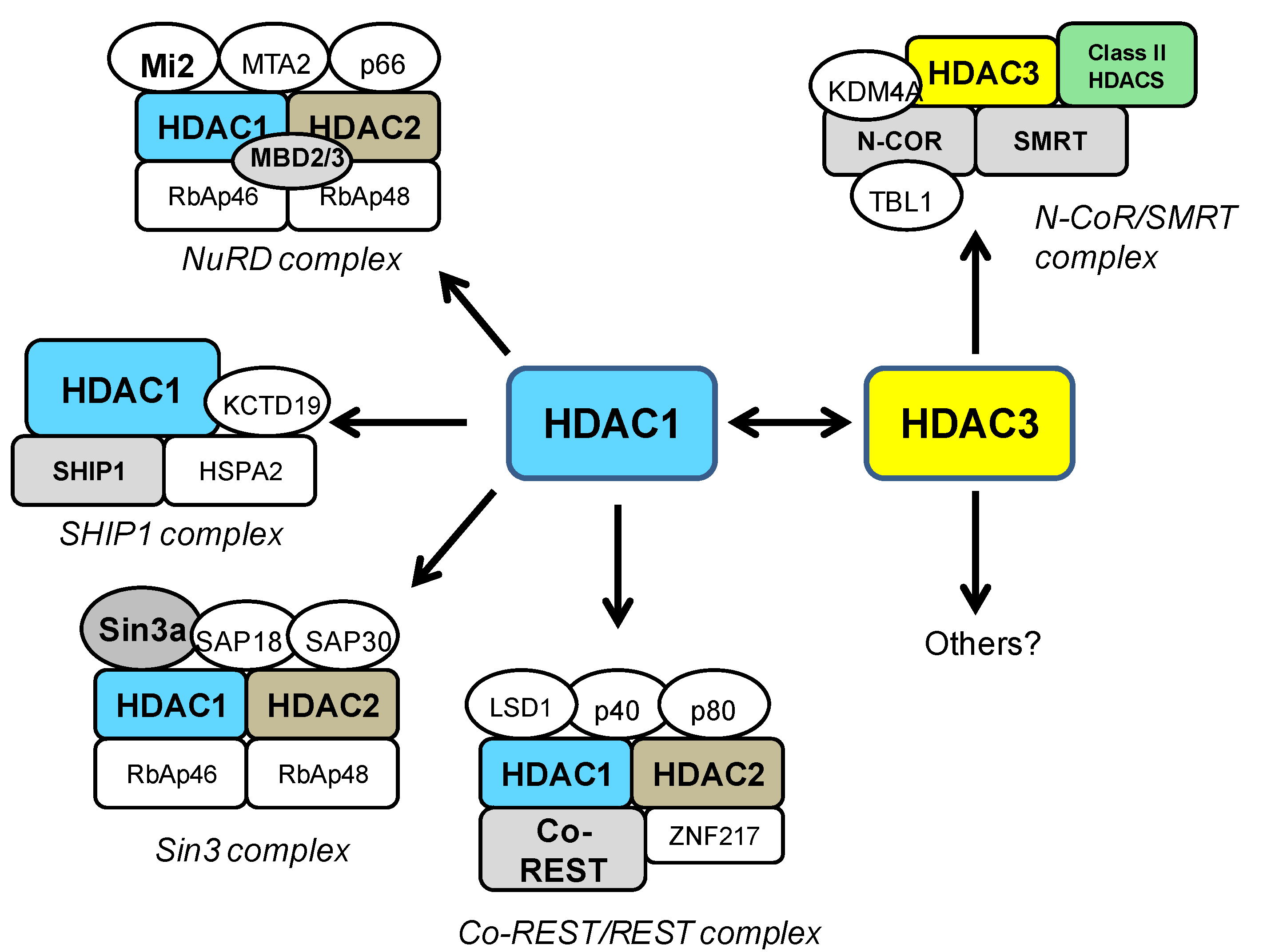
4. HDAC1 and HDAC3 in General Neurotoxicity
5. HDAC1 and HDAC3 in Polyglutamine Diseases
5.1. Huntington’s Disease
5.1.1. Expression Levels of HDAC1 and HDAC3 in HD Tissues
5.1.2. HDAC3 Binds Huntingtin Protein
5.1.3. HDAC1 and Regulation of Huntingtin Clearance
5.1.4. Genetic Knock-Down Studies in HD
5.2. Spinocerebellar Ataxias
5.2.1. Spinocerebellar Ataxia Type 1 (SCA1)
5.2.2. Spinocerebellar Ataxia type 3 (SCA3)
5.2.3. Spinocerebellar Ataxia Type 7 (SCA7)
6. Selective HDAC Inhibitors
7. Selective HDAC Inhibitors in Polyglutamine Disorders
7.1. Class I Specific HDAC Inhibitors
7.2. HDAC1/HDAC3-Targeting Inhibitors

{kind=link}
{kind=link}
| Class I-specific | HD Model and dose | Dose paradigm | Effects | Ref |
|---|---|---|---|---|
| Valproic Acid | N171-82Q transgenic mice | 100 mg/kg; i.p. | Ameliorated premature death and locomotor activity deficits. | [97] |
| N171-82Q transgenic mice | 25 g/kg in diet | Ameliorated premature death and depressive-like behavior. | [98] | |
| YAC128 transgenic mice | 25 g/kg in diet | Ameliorated body weight gain and anxiety-like behavior. | [98] | |
| Drosophila MJDtr-Q78 | 0.5–2 mM in diet | Prevented eye depigmentation, alleviated climbing disability, and extended the lifespan. | [99] | |
| MJDtr-Q68- expressing cells | 0.5–2 mM in culture | Reduced apoptosis. | [99] | |
| d-β-HB (d-β-H-butyrate) | 3-NP mouse model | 1.6 mmol/kg/day; minipump | Improved spontaneous locomotor activity. | [104] |
| R6/2 transgenic mice | 1.6 mmol/kg/day; minipump | Ameliorated premature death. | [104] | |
| HDAC1/HDAC3 | HD Model | Dose paradigm | Effects | Ref |
| HDACi 4b | R6/2 transgenic mice | 150 mg/kg/day; drinking water | Ameliorated body weight loss and locomotor deficits. | [60] |
| R6/2 transgenic mice | 150 mg/kg/day; s.c. | Prevented downregulation of a subset of HD-related genes. | [60] | |
| Drosophila Httex1p Q93 | 1–10 μM in diet | Ameliorated eye neurodegeneration. | [31] | |
| STHdhQ111 cells | 0.3–10 μM in culture | Improved metabolic deficit. | [31] | |
| N171-82Q transgenic mice | 50–100 mg/kg; s.c | Ameliorated body weight loss, locomotor deficits and cognitive decline. | [110] | |
| R6/2 transgenic mice | 0.85 mg/ml; drinking water | Ameliorated striatal atrophy and clasping phenotype. | [108] | |
| N171-82Q transgenic mice | 0.85 mg/ml; drinking water | No change in disease phenotypes. | [108] | |
| HDACi 874 | N171-82Q transgenic mice | 50 mg/kg; s.c. | Prevented mutant Htt aggregation. | [110] |
| HDACis 874, 968 and 974 | Drosophila Httex1p Q93 | 1–10 μM in diet | Ameliorated eye neurodegeneration. | [31] |
| STHdhQ111 cells | 0.3–10 μM in culture | Improved metabolic deficit. | [31] | |
| HDAC3-selective | HD Model | Dose paradigm | Effects | Ref |
| RGFP136 | R6/2 transgenic mice | 150 mg/kg; s.c. | Prevented downregulation of a subset of HD-related genes. | [31] |
| Drosophila Httex1p Q93 | 1–10 μM in diet | Ameliorated eye neurodegeneration. | [31] | |
| STHdhQ111 cells | 0.3–10 μM in culture | Improved metabolic deficit. | [31] | |
| RFGP966 | N171-82Q transgenic mice | 50 mg/kg; s.c. | Ameliorated body weight loss, locomotor deficits and cognitive decline. | [112] |
| Drosophila Httex1p Q93 | 1–10 μM in diet | Ameliorated eye neurodegeneration. | [31] | |
| STHdhQ111 cells | 0.3–10 μM in culture | Improved metabolic deficit. | [31] | |
| HDAC1-selective | HD Model | Dose paradigm | Effects | Ref |
| 228 | R6/2 transgenic mice | 150 mg/kg; s.c. | Prevented downregulation of a subset of HD-related genes. | [31] |
| 233, 941 and MS-275 | Drosophila Httex1p Q93 | 1–10 μM in diet | Ameliorated eye neurodegeneration. | [31] |
| STHdhQ111 cells | 0.3–10 μM in culture | Improved metabolic deficit. | [31] |
8. Mechanisms of Action of HDAC Inhibitors
8.1. Chromatin/Transcription-Related Mechanisms of HDAC1/3 Inhibitors
8.1.1. Ubiquitination-Related Gene Expression
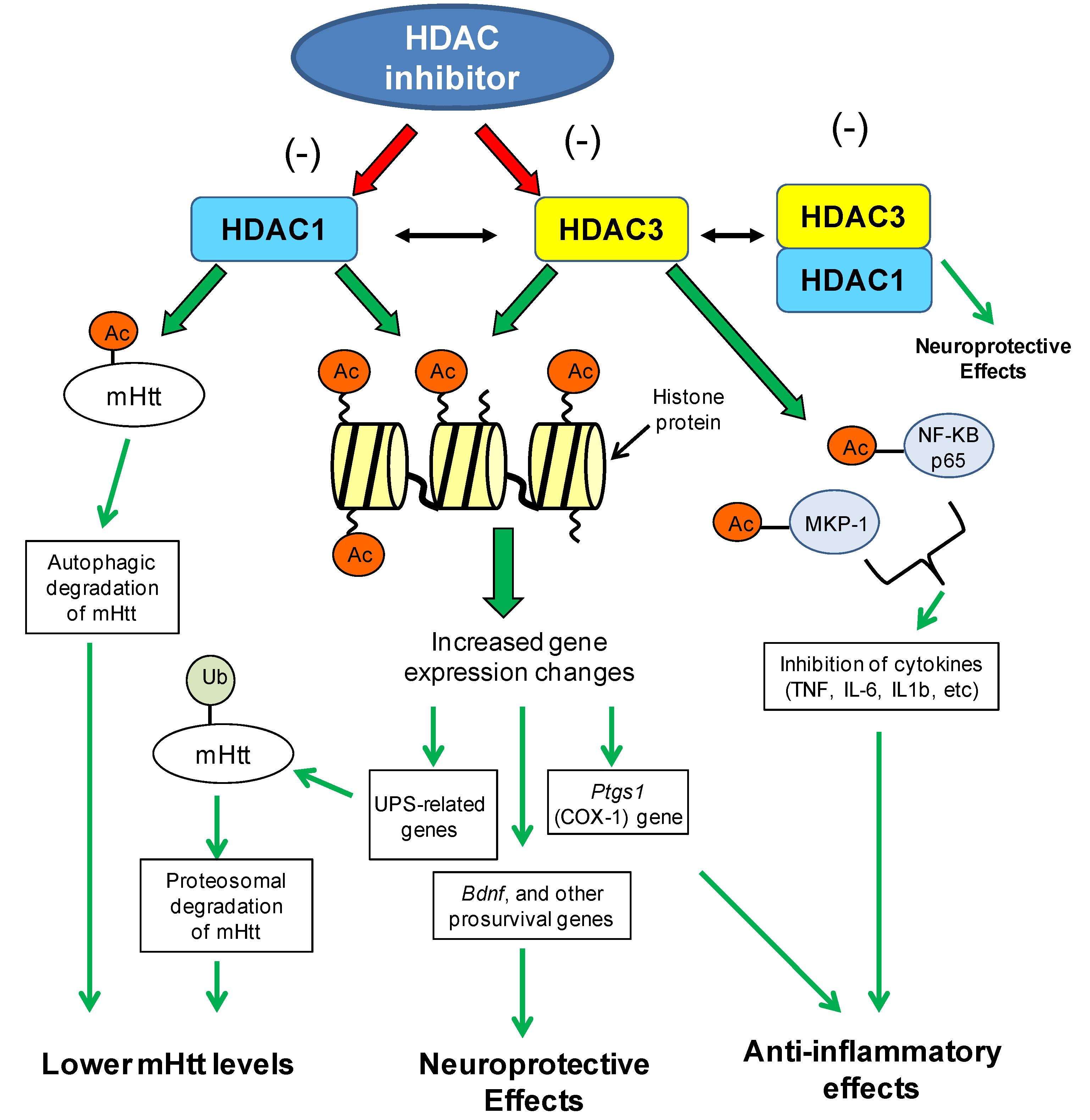
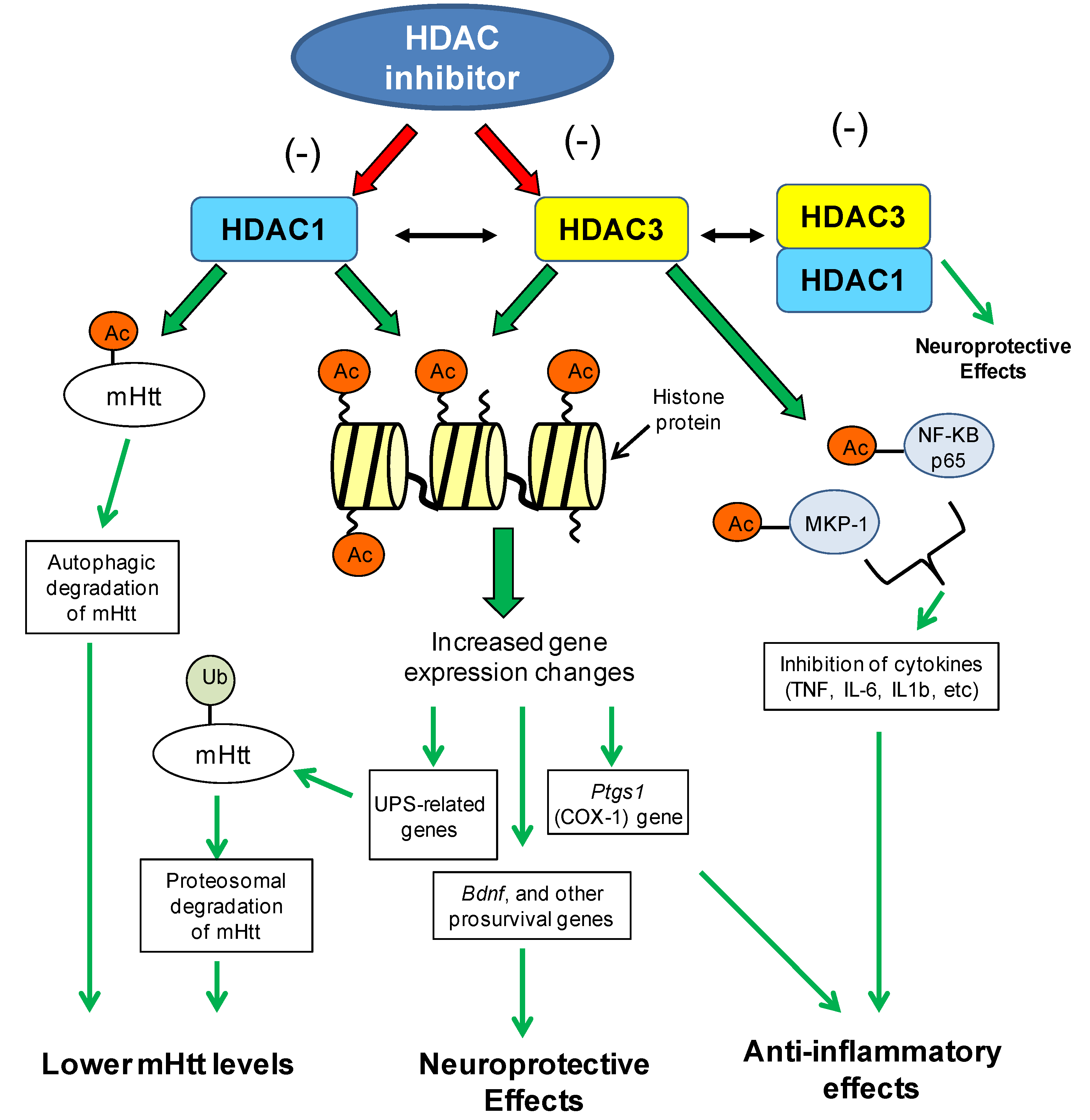
8.1.2. Brain-Derived Neurotrophic Factor Gene (BDNF) Expression Changes
8.2. Non-Chromatin Mechanisms Associated with HDAC1 and HDAC3
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Okazawa, H. Polyglutamine diseases: A transcription disorder? Cell Mol. Life Sci. 2003, 60, 1427–1439. [Google Scholar] [CrossRef]
- Cohen-Carmon, D.; Meshorer, E. Polyglutamine (polyQ) disorders: The chromatin connection. Nucleus 2012, 3, 433–441. [Google Scholar] [CrossRef]
- Butler, R.; Bates, G.P. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat. Rev. Neurosci. 2006, 7, 784–796. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Verner, E.; Buggy, J.J. Isoform-specific histone deacetylase inhibitors: The next step? Cancer Lett. 2009, 280, 211–221. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar]
- Quina, A.S.; Buschbeck, M.; di Croce, L. Chromatin structure and epigenetics. Biochem. Pharmacol. 2006, 72, 1563–1569. [Google Scholar] [CrossRef]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar] [CrossRef]
- An, W. Histone acetylation and methylation: Combinatorial players for transcriptional regulation. Subcell Biochem. 2007, 41, 351–369. [Google Scholar]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Leipe, D.D.; Landsman, D. Histone deacetylases, acetoin utilization proteins and acetylpolyamine amidohydrolases are members of an ancient protein superfamily. Nucleic Acids Res. 1997, 25, 3693–3697. [Google Scholar] [CrossRef]
- Blander, G.; Guarente, L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004, 73, 417–435. [Google Scholar] [CrossRef]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Hildmann, C.; Riester, D.; Schwienhorst, A. Histone deacetylases—An important class of cellular regulators with a variety of functions. Appl. Microbiol. Biotechnol. 2007, 75, 487–497. [Google Scholar] [CrossRef]
- Adcock, I.M.; Ford, P.; Ito, K.; Barnes, P.J. Epigenetics and airways disease. Respir. Res. 2006, 7, 21–39. [Google Scholar] [CrossRef]
- Reichert, N.; Choukrallah, M.A.; Matthias, P. Multiple roles of class I HDACs in proliferation, differentiation, and development. Cell Mol. Life Sci. 2012, 69, 2173–2187. [Google Scholar] [CrossRef]
- Simoes-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.A.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7–23. [Google Scholar] [CrossRef]
- Hoshino, M.; Tagawa, K.; Okuda, T.; Murata, M.; Oyanagi, K.; Arai, N.; Mizutani, T.; Kanazawa, I.; Wanker, E.E.; Okazawa, H. Histone deacetylase activity is retained in primary neurons expressing mutant huntingtin protein. J. Neurochem. 2003, 87, 257–267. [Google Scholar] [CrossRef]
- Quinti, L.; Chopra, V.; Rotili, D.; Valente, S.; Amore, A.; Franci, G.; Meade, S.; Valenza, M.; Altucci, L.; Maxwell, M.M.; et al. Evaluation of histone deacetylases as drug targets in Huntington’s disease models. Study of HDACs in brain tissues from R6/2 and CAG140 knock-in HD mouse models and human patients and in a neuronal HD cell model. PLoS Curr. 2010, 1, 1–16. [Google Scholar]
- Mielcarek, M.; Benn, C.L.; Franklin, S.A.; Smith, D.L.; Woodman, B.; Marks, P.A.; Bates, G.P. SAHA decreases HDAC 2 and 4 levels in vivo and improves molecular phenotypes in the R6/2 mouse model of Huntington’s disease. PLoS One 2011, 6, e27746. [Google Scholar]
- Yeh, H.H.; Young, D.; Gelovani, J.G.; Robinson, A.; Davidson, Y.; Herholz, K.; Mann, D.M. Histone deacetylase class II and acetylated core histone immunohistochemistry in human brains with Huntington’s disease. Brain Res. 2013, 1504, 16–24. [Google Scholar]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef]
- Mielcarek, M.; Landles, C.; Weiss, A.; Bradaia, A.; Seredenina, T.; Inuabasi, L.; Osborne, G.F.; Wadel, K.; Touller, C.; Butler, R.; et al. HDAC4 reduction: A novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 2013, 11, e1001717. [Google Scholar] [CrossRef]
- Tsai, S.C.; Seto, E. Regulation of histone deacetylase 2 by protein kinase CK2. J. Biol. Chem. 2002, 277, 31826–31833. [Google Scholar] [CrossRef]
- Brunmeir, R.; Lagger, S.; Seiser, C. Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. Int. J. Dev. Biol. 2009, 53, 275–289. [Google Scholar] [CrossRef]
- Lagger, G.; O’Carroll, D.; Rembold, M.; Khier, H.; Tischler, J.; Weitzer, G.; Schuettengruber, B.; Hauser, C.; Brunmeir, R.; Jenuwein, T.; et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002, 21, 2672–2681. [Google Scholar] [CrossRef]
- Trivedi, C.M.; Luo, Y.; Yin, Z.; Zhang, M.; Zhu, W.; Wang, T.; Floss, T.; Goettlicher, M.; Noppinger, P.R.; Wurst, W.; et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat. Med. 2007, 13, 324–331. [Google Scholar] [CrossRef]
- Zimmermann, S.; Kiefer, F.; Prudenziati, M.; Spiller, C.; Hansen, J.; Floss, T.; Wurst, W.; Minucci, S.; Gottlicher, M. Reduced body size and decreased intestinal tumor rates in HDAC2-mutant mice. Cancer Res. 2007, 67, 9047–9054. [Google Scholar] [CrossRef]
- Jia, H.; Pallos, J.; Jacques, V.; Lau, A.; Tang, B.; Cooper, A.; Syed, A.; Purcell, J.; Chen, Y.; Sharma, S.; et al. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol. Dis. 2012, 46, 351–361. [Google Scholar] [CrossRef]
- Broide, R.S.; Redwine, J.M.; Aftahi, N.; Young, W.; Bloom, F.E.; Winrow, C.J. Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 2007, 31, 47–58. [Google Scholar] [CrossRef]
- Thomas, E.A. Focal nature of neurological disorders necessitates isotype-selective histone deacetylase (HDAC) inhibitors. Mol. Neurobiol. 2009, 40, 33–45. [Google Scholar] [CrossRef]
- Kalinin, S.; Polak, P.E.; Lin, S.X.; Braun, D.; Guizzetti, M.; Zhang, X.; Rubinstein, I.; Feinstein, D.L. Dimethyl fumarate regulates histone deacetylase expression in astrocytes. J. Neuroimmunol. 2013, 263, 13–19. [Google Scholar] [CrossRef]
- Ye, F.; Chen, Y.; Hoang, T.; Montgomery, R.L.; Zhao, X.H.; Bu, H.; Hu, T.; Taketo, M.M.; van Es, J.H.; Clevers, H.; et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat. Neurosci. 2009, 12, 829–838. [Google Scholar] [CrossRef]
- Choi, E.; Han, C.; Park, I.; Lee, B.; Jin, S.; Choi, H.; Kim do, H.; Park, Z.Y.; Eddy, E.M.; Cho, C. A novel germ cell-specific protein, SHIP1, forms a complex with chromatin remodeling activity during spermatogenesis. J. Biol. Chem. 2008, 283, 35283–35294. [Google Scholar]
- Yang, W.M.; Yao, Y.L.; Sun, J.M.; Davie, J.R.; Seto, E. Isolation and characterization of cDNAs corresponding to an additional member of the human histone deacetylase gene family. J. Biol. Chem. 1997, 272, 28001–28007. [Google Scholar]
- Takami, Y.; Nakayama, T. N-terminal region, C-terminal region, nuclear export signal, and deacetylation activity of histone deacetylase-3 are essential for the viability of the DT40 chicken B cell line. J. Biol. Chem. 2000, 275, 16191–16201. [Google Scholar] [CrossRef]
- Bhaskara, S.; Chyla, B.J.; Amann, J.M.; Knutson, S.K.; Cortez, D.; Sun, Z.W.; Hiebert, S.W. Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 2008, 30, 61–72. [Google Scholar]
- Mahlknecht, U.; Hoelzer, D.; Bucala, R.; Verdin, E. Cloning and characterization of the murine histone deacetylase (HDAC3). Biochem. Biophys. Res. Commun. 1999, 263, 482–490. [Google Scholar] [CrossRef]
- Shen, S.; Li, J.; Casaccia-Bonnefil, P. Histone modifications affect timing of oligodendrocyte progenitor differentiation in the developing rat brain. J. Cell Biol. 2005, 169, 577–589. [Google Scholar] [CrossRef]
- Debacker, K.; Frizzell, A.; Gleeson, O.; Kirkham-McCarthy, L.; Mertz, T.; Lahue, R.S. Histone deacetylase complexes promote trinucleotide repeat expansions. PLoS Biol. 2012, 10, e1001257. [Google Scholar] [CrossRef]
- Wen, Y.D.; Perissi, V.; Staszewski, L.M.; Yang, W.M.; Krones, A.; Glass, C.K.; Rosenfeld, M.G.; Seto, E. The histone deacetylase-3 complex contains nuclear receptor corepressors. Proc. Natl. Acad. Sci. USA 2000, 97, 202–7207. [Google Scholar]
- Fischle, W.; Dequiedt, F.; Fillion, M.; Hendzel, M.J.; Voelter, W.; Verdin, E. Human HDAC7 histone deacetylase activity is associated with HDAC3 in vivo. J. Biol. Chem. 2001, 276, 35826–35835. [Google Scholar]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef]
- Huang, E.Y.; Zhang, J.; Miska, E.A.; Guenther, M.G.; Kouzarides, T.; Lazar, M.A. Nuclear receptor corepressors partner with class II histone deacetylases in a Sin3-independent repression pathway. Genes Dev. 2000, 14, 45–54. [Google Scholar]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2011, 6, 238–243. [Google Scholar]
- Bardai, F.H.; Price, V.; Zaayman, M.; Wang, L.; D’Mello, S.R. Histone deacetylase (HDAC1) is a molecular switch between neuronal survival and death. J. Biol. Chem. 2012, 287, 35444–35453. [Google Scholar]
- Bardai, F.H.; D’Mello, S.R. Selective toxicity by HDAC3 in neurons: Regulation by Akt and GSK3beta. J. Neurosci 2011, 31, 1746–1751. [Google Scholar] [CrossRef]
- Kim, J.Y.; Shen, S.; Dietz, K.; He, Y.; Howell, O.; Reynolds, R.; Casaccia, P. HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat. Neurosci. 2010, 13, 180–189. [Google Scholar] [CrossRef]
- Morrison, B.E.; Majdzadeh, N.; Zhang, X.; Lyles, A.; Bassel-Duby, R.; Olson, E.N.; D’Mello, S.R. Neuroprotection by histone deacetylase-related protein. Mol. Cell Biol. 2006, 26, 3550–3564. [Google Scholar] [CrossRef]
- Kim, D.; Frank, C.L.; Dobbin, M.M.; Tsunemoto, R.K.; Tu, W.; Peng, P.L.; Guan, J.S.; Lee, B.H.; Moy, L.Y.; Giusti, P.; et al. Deregulation of HDAC1 by p25/Cdk5 in neurotoxicity. Neuron 2008, 60, 803–817. [Google Scholar] [CrossRef]
- Hahnen, E.; Hauke, J.; Trankle, C.; Eyupoglu, I.Y.; Wirth, B.; Blumcke, I. Histone deacetylase inhibitors: Possible implications for neurodegenerative disorders. Exp. Opin. Investig. Drugs 2008, 17, 169–184. [Google Scholar] [CrossRef]
- Sadri-Vakili, G.; Cha, J.H. Mechanisms of disease: Histone modifications in Huntington’s disease. Nat. Clin. Pract. Neurol. 2006, 2, 330–338. [Google Scholar] [CrossRef]
- Valor, L.M.; Guiretti, D.; Lopez-Atalaya, J.P.; Barco, A. Genomic landscape of transcriptional and epigenetic dysregulation in early onset polyglutamine disease. J. Neurosci. 2013, 33, 10471–10482. [Google Scholar] [CrossRef]
- Nucifora, F.C., Jr.; Sasaki, M.; Peters, M.F.; Huang, H.; Cooper, J.K.; Yamada, M.; Takahashi, H.; Tsuji, S.; Troncoso, J.; Dawson, V.L.; et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001, 291, 2423–2428. [Google Scholar] [CrossRef]
- Huntington’s Disease Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Andrew, S.E.; Goldberg, Y.P.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A.; et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403. [Google Scholar]
- Li, J.L.; Hayden, M.R.; Warby, S.C.; Durr, A.; Morrison, P.J.; Nance, M.; Ross, C.A.; Margolis, R.L.; Rosenblatt, A.; Squitieri, F.; et al. Genome-wide significance for a modifier of age at neurological onset in Huntington’s disease at 6q23–24: The HD MAPS study. BMC Med. Genet. 2006, 7, 71–79. [Google Scholar]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor, 4b, ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar]
- Kuhn, A.; Goldstein, D.R.; Hodges, A.; Strand, A.D.; Sengstag, T.; Kooperberg, C.; Becanovic, K.; Pouladi, M.A.; Sathasivam, K.; Cha, J.H.; et al. Mutant huntingtin’s effects on striatal gene expression in mice recapitulate changes observed in human Huntington’s disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 2007, 16, 1845–1861. [Google Scholar] [CrossRef]
- Hodges, A.; Strand, A.D.; Aragaki, A.K.; Kuhn, A.; Sengstag, T.; Hughes, G.; Elliston, L.A.; Hartog, C.; Goldstein, D.R.; Thu, D.; et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 2006, 15, 965–977. [Google Scholar] [CrossRef]
- Bardai, F.H.; Verma, P.; Smith, C.; Rawat, V.; Wang, L.; D'Mello, S.R. Disassociation of histone deacetylase-3 from normal huntingtin underlies mutant huntingtin neurotoxicity. J. Neurosci. 2013, 33, 11833–11838. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O'Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Yamamoto, A.; Cremona, M.L.; Rothman, J.E. Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol. 2006, 172, 719–731. [Google Scholar] [CrossRef]
- Jeong, H.; Then, F.; Melia, T.J., Jr.; Mazzulli, J.R.; Cui, L.; Savas, J.N.; Voisine, C.; Paganetti, P.; Tanese, N.; Hart, A.C.; et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell 2009, 137, 60–72. [Google Scholar] [CrossRef]
- Wu, S.; Zheng, S.D.; Huang, H.L.; Yan, L.C.; Yin, X.F.; Xu, H.N.; Zhang, K.J.; Gui, J.H.; Chu, L.; Liu, X.Y. Lithium down-regulates histone deacetylase 1 (HDAC1) and induces degradation of mutant huntingtin. J. Biol. Chem. 2013, 288, 35500–35510. [Google Scholar] [CrossRef]
- Bates, E.A.; Victor, M.; Jones, A.K.; Shi, Y.; Hart, A.C. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J. Neurosci. 2006, 26, 2830–2838. [Google Scholar] [CrossRef]
- Pallos, J.; Bodai, L.; Lukacsovich, T.; Purcell, J.M.; Steffan, J.S.; Thompson, L.M.; Marsh, J.L. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum. Mol. Genet. 2008, 17, 3767–3775. [Google Scholar] [CrossRef]
- Moumne, L.; Campbell, K.; Howland, D.; Ouyang, Y.; Bates, G.P. Genetic knock-down of HDAC3 does not modify disease-related phenotypes in a mouse model of Huntington’s disease. PLoS One 2012, 7, e31080. [Google Scholar]
- Underwood, B.R.; Rubinsztein, D.C. Spinocerebellar ataxias caused by polyglutamine expansions: A review of therapeutic strategies. Cerebellum 2008, 7, 215–221. [Google Scholar]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I.; et al. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef]
- Orr, H.T.; Chung, M.Y.; Banfi, S.; Kwiatkowski, T.J., Jr.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.; Zoghbi, H.Y. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef]
- Fernandez-Funez, P.; Nino-Rosales, M.L.; de Gouyon, B.; She, W.C.; Luchak, J.M.; Martinez, P.; Turiegano, E.; Benito, J.; Capovilla, M.; Skinner, P.J.; et al. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 2000, 408, 101–106. [Google Scholar] [CrossRef]
- Tsai, C.C.; Kao, H.Y.; Mitzutani, A.; Banayo, E.; Rajan, H.; McKeown, M.; Evans, R.M. Ataxin 1, a SCA1 neurodegenerative disorder protein, is functionally linked to the silencing mediator of retinoid and thyroid hormone receptors. Proc. Natl. Acad. Sci. USA 2004, 101, 4047–4052. [Google Scholar]
- Riess, O.; Rub, U.; Pastore, A.; Bauer, P.; Schols, L. SCA3: Neurological features, pathogenesis and animal models. Cerebellum 2008, 7, 125–137. [Google Scholar] [CrossRef]
- Evert, B.O.; Araujo, J.; Vieira-Saecker, A.M.; de Vos, R.A.; Harendza, S.; Klockgether, T.; Wullner, U. Ataxin-3 represses transcription via chromatin binding, interaction with histone deacetylase 3, and histone deacetylation. J. Neurosci. 2006, 26, 11474–11486. [Google Scholar] [CrossRef]
- Chou, A.H.; Chen, S.Y.; Yeh, T.H.; Weng, Y.H.; Wang, H.L. HDAC inhibitor sodium butyrate reverses transcriptional downregulation and ameliorates ataxic symptoms in a transgenic mouse model of SCA3. Neurobiol. Dis. 2011, 41, 481–488. [Google Scholar] [CrossRef]
- Duncan, C.E.; An, M.C.; Papanikolaou, T.; Rugani, C.; Vitelli, C.; Ellerby, L.M. Histone deacetylase-3 interacts with ataxin-7 and is altered in a spinocerebellar ataxia type 7 mouse model. Mol. Neurodegener 2013, 8, 42–56. [Google Scholar]
- Harrison, I.F.; Dexter, D.T. Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson’s disease? Pharmacol. Ther. 2013, 140, 34–52. [Google Scholar] [CrossRef]
- Kalin, J.H.; Bergman, J.A. Development and therapeutic implications of selective histone deacetylase 6 inhibitors. J. Med. Chem 2013, 56, 6297–6313. [Google Scholar] [CrossRef]
- Pan, H.; Cao, J.; Xu, W. Selective histone deacetylase inhibitors. Anticancer Agents Med. Chem. 2012, 12, 247–270. [Google Scholar] [CrossRef]
- Rajak, H.; Singh, A.; Dewangan, P.K.; Patel, V.; Jain, D.K.; Tiwari, S.K.; Veerasamy, R.; Sharma, P.C. Peptide based macrocycles: selective histone deacetylase inhibitors with antiproliferative activity. Curr. Med. Chem. 2013, 20, 1887–1903. [Google Scholar] [CrossRef]
- Soragni, E.; Xu, C.; Plasterer, H.L.; Jacques, V.; Rusche, J.R.; Gottesfeld, J.M. Rationale for the development of 2-aminobenzamide histone deacetylase inhibitors as therapeutics for Friedreich ataxia. J. Child. Neurol. 2012, 27, 1164–1173. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Miller, T.; Kelly, W.K. Histone deacetylase inhibitors. Adv. Cancer Res. 2004, 91, 137–168. [Google Scholar] [CrossRef]
- Grayson, D.R.; Kundakovic, M.; Sharma, R.P. Is there a future for histone deacetylase inhibitors in the pharmacotherapy of psychiatric disorders? Mol. Pharmacol. 2010, 77, 126–135. [Google Scholar] [CrossRef]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef]
- McCampbell, A.; Taye, A.A.; Whitty, L.; Penney, E.; Steffan, J.S.; Fischbeck, K.H. Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc. Natl. Acad. Sci. USA 2001, 98, 15179–15184. [Google Scholar]
- Hockly, E.; Richon, V.M.; Woodman, B.; Smith, D.L.; Zhou, X.; Rosa, E.; Sathasivam, K.; Ghazi-Noori, S.; Mahal, A.; Lowden, P.A.; et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2041–2046. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R.; et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar]
- Gardian, G.; Browne, S.E.; Choi, D.K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J.; et al. Neuroprotective effects of phenylbutyrate in the N171–82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar]
- Steffan, J.S.; Bodai, L.; Pallos, J.; Poelman, M.; McCampbell, A.; Apostol, B.L.; Kazantsev, A.; Schmidt, E.; Zhu, Y.Z.; Greenwald, M.; et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001, 413, 739–743. [Google Scholar]
- Minamiyama, M.; Katsuno, M.; Adachi, H.; Waza, M.; Sang, C.; Kobayashi, Y.; Tanaka, F.; Doyu, M.; Inukai, A.; Sobue, G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2004, 13, 1183–1192. [Google Scholar] [CrossRef]
- Palhan, V.B.; Chen, S.; Peng, G.H.; Tjernberg, A.; Gamper, A.M.; Fan, Y.; Chait, B.T.; La Spada, A.R.; Roeder, R.G. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 8472–8477. [Google Scholar] [CrossRef]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef]
- Gurvich, N.; Tsygankova, O.M.; Meinkoth, J.L.; Klein, P.S. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer Res. 2004, 64, 1079–1086. [Google Scholar]
- Zadori, D.; Geisz, A.; Vamos, E.; Vecsei, L.; Klivenyi, P. Valproate ameliorates the survival and the motor performance in a transgenic mouse model of Huntington’s disease. Pharmacol. Biochem. Behav. 2009, 94, 148–153. [Google Scholar] [CrossRef]
- Chiu, C.T.; Liu, G.; Leeds, P.; Chuang, D.M. Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease. Neuropsychopharmacology 2011, 36, 2406–2421. [Google Scholar] [CrossRef]
- Yi, J.; Zhang, L.; Tang, B.; Han, W.; Zhou, Y.; Chen, Z.; Jia, D.; Jiang, H. Sodium valproate alleviates neurodegeneration in SCA3/MJD via suppressing apoptosis and rescuing the hypoacetylation levels of histone H3 and H4. PLoS ONE 2013, 8, e54792. [Google Scholar]
- Pearce, I.; Heathfield, K.W.; Pearce, M.J. Valproate sodium in Huntington chorea. Arch. Neurol. 1977, 34, 308–309. [Google Scholar] [CrossRef]
- Symington, G.R.; Leonard, D.P.; Shannon, P.J.; Vajda, F.J. Sodium valproate in Huntington’s disease. Am. J. Psychiatry 1978, 135, 352–354. [Google Scholar]
- Saft, C.; Lauter, T.; Kraus, P.H.; Przuntek, H.; Andrich, J.E. Dose-dependent improvement of myoclonic hyperkinesia due to Valproic acid in eight Huntington's Disease patients: A case series. BMC Neurol. 2006, 6, 11–17. [Google Scholar] [CrossRef]
- Grove, V.E., Jr.; Quintanilla, J.; DeVaney, G.T. Improvement of Huntington’s disease with olanzapine and valproate. N. Engl. J. Med. 2000, 343, 973–974. [Google Scholar] [CrossRef]
- Lim, S.; Chesser, A.S.; Grima, J.C.; Rappold, P.M.; Blum, D.; Przedborski, S.; Tieu, K. d-beta-Hydroxybutyrate is protective in mouse models of Huntington’s disease. PLoS ONE 2011, 6, e24620. [Google Scholar]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef]
- Chou, C.J.; Herman, D.M.; Gottesfeld, J.M. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [Google Scholar] [CrossRef]
- Xu, C.; Soragni, E.; Chou, C.J.; Herman, D.; Plasterer, H.L.; Rusche, J.R.; Gottesfeld, J.M. Chemical probes identify a role for histone deacetylase 3 in Friedreich’s ataxia gene silencing. Chem. Biol. 2009, 16, 980–989. [Google Scholar] [CrossRef]
- Chen, J.Y.; Wang, E.; Galvan, L.; Huynh, M.; Joshi, P.; Cepeda, C.; Levine, M.S. Effects of the Pimelic Diphenylamide Histone Deacetylase Inhibitor HDACi 4b on the R6/2 and N171–82Q Mouse Models of Huntington’s Disease. PLoS Curr. 2013, 5–12. [Google Scholar]
- Beconi, M.; Aziz, O.; Matthews, K.; Moumne, L.; O’Connell, C.; Yates, D.; Clifton, S.; Pett, H.; Vann, J.; Crowley, L.; et al. Oral administration of the pimelic diphenylamide HDAC inhibitor HDACi 4b is unsuitable for chronic inhibition of HDAC activity in the CNS in vivo. PLoS ONE 2012, 7, e44498. [Google Scholar] [CrossRef]
- Jia, H.; Kast, R.J.; Steffan, J.S.; Thomas, E.A. Selective histone deacetylase (HDAC) inhibition imparts beneficial effects in Huntington’s disease mice: Implications for the ubiquitin-proteasomal and autophagy systems. Hum. Mol. Genet. 2012, 21, 5280–5293. [Google Scholar] [CrossRef]
- McQuown, S.C.; Barrett, R.M.; Matheos, D.P.; Post, R.J.; Rogge, G.A.; Alenghat, T.; Mullican, S.E.; Jones, S.; Rusche, J.R.; Lazar, M.A.; et al. HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci. 2011, 31, 764–774. [Google Scholar] [CrossRef]
- Jia, H.; Pallos, J.; Jacques, V.; Lau, A.; Tang, B.; Cooper, A.; Syed, A.; Purcell, J.; Chen, Y.; Sharma, S.; et al. Histone deacetylase inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in Huntington’s disease model systems. In Proceedings of the CHDI 7th Annual Huntington’s Disease Therapeutics Conference: A Forum for Drug Discovery & Development, Palm Springs, CA, USA, February 27–March 01 2012.
- Peart, M.J.; Smyth, G.K.; van Laar, R.K.; Bowtell, D.D.; Richon, V.M.; Marks, P.A.; Holloway, A.J.; Johnstone, R.W. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 3697–3702. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef]
- Finkbeiner, S.; Mitra, S. The ubiquitin-proteasome pathway in Huntington’s disease. Sci. World J. 2008, 8, 421–433. [Google Scholar] [CrossRef]
- De Pril, R.; Fischer, D.F.; Maat-Schieman, M.L.; Hobo, B.; de Vos, R.A.; Brunt, E.R.; Hol, E.M.; Roos, R.A.; van Leeuwen, F.W. Accumulation of aberrant ubiquitin induces aggregate formation and cell death in polyglutamine diseases. Hum. Mol. Genet. 2004, 13, 1803–1813. [Google Scholar] [CrossRef]
- Bennett, E.J.; Shaler, T.A.; Woodman, B.; Ryu, K.Y.; Zaitseva, T.S.; Becker, C.H.; Bates, G.P.; Schulman, H.; Kopito, R.R. Global changes to the ubiquitin system in Huntington’s disease. Nature 2007, 448, 704–708. [Google Scholar] [CrossRef]
- Diaz-Hernandez, M.; Hernandez, F.; Martin-Aparicio, E.; Gomez-Ramos, P.; Moran, M.A.; Castano, J.G.; Ferrer, I.; Avila, J.; Lucas, J.J. Neuronal induction of the immunoproteasome in Huntington’s disease. J. Neurosci. 2003, 23, 11653–11661. [Google Scholar]
- Wang, J.; Wang, C.E.; Orr, A.; Tydlacka, S.; Li, S.H.; Li, X.J. Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J. Cell. Biol. 2008, 180, 1177–1189. [Google Scholar] [CrossRef]
- Jana, N.R.; Dikshit, P.; Goswami, A.; Kotliarova, S.; Murata, S.; Tanaka, K.; Nukina, N. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 2005, 280, 11635–11640. [Google Scholar]
- Kalchman, M.A.; Graham, R.K.; Xia, G.; Koide, H.B.; Hodgson, J.G.; Graham, K.C.; Goldberg, Y.P.; Gietz, R.D.; Pickart, C.M.; Hayden, M.R. Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme. J. Biol. Chem. 1996, 271, 19385–19394. [Google Scholar] [CrossRef]
- De Pril, R.; Fischer, D.F.; Roos, R.A.; van Leeuwen, F.W. Ubiquitin-conjugating enzyme E2–25K increases aggregate formation and cell death in polyglutamine diseases. Mol. Cell. Neurosci. 2007, 34, 10–19. [Google Scholar] [CrossRef]
- Langley, B.; Gensert, J.M.; Beal, M.F.; Ratan, R.R. Remodeling chromatin and stress resistance in the central nervous system: histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord 2005, 4, 41–50. [Google Scholar] [CrossRef]
- Wu, X.; Chen, P.S.; Dallas, S.; Wilson, B.; Block, M.L.; Wang, C.C.; Kinyamu, H.; Lu, N.; Gao, X.; Leng, Y.; et al. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int. J. Neuropsychopharmacol. 2008, 11, 1123–1134. [Google Scholar] [CrossRef]
- Hasan, M.R.; Kim, J.H.; Kim, Y.J.; Kwon, K.J.; Shin, C.Y.; Kim, H.Y.; Han, S.H.; Choi, D.H.; Lee, J. Effect of HDAC inhibitors on neuroprotection and neurite outgrowth in primary rat cortical neurons following ischemic insult. Neurochem. Res. 2013, 38, 1921–1934. [Google Scholar] [CrossRef]
- Chen, P.S.; Peng, G.S.; Li, G.; Yang, S.; Wu, X.; Wang, C.C.; Wilson, B.; Lu, R.B.; Gean, P.W.; Chuang, D.M.; et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol. Psychiatry 2006, 11, 1116–1125. [Google Scholar] [CrossRef]
- Zhang, Z.Z.; Gong, Y.Y.; Shi, Y.H.; Zhang, W.; Qin, X.H.; Wu, X.W. Valproate promotes survival of retinal ganglion cells in a rat model of optic nerve crush. Neuroscience 2012, 224, 282–293. [Google Scholar] [CrossRef]
- Fukuchi, M.; Nii, T.; Ishimaru, N.; Minamino, A.; Hara, D.; Takasaki, I.; Tabuchi, A.; Tsuda, M. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci. Res. 2009, 65, 35–43. [Google Scholar] [CrossRef]
- Yasuda, S.; Liang, M.H.; Marinova, Z.; Yahyavi, A.; Chuang, D.M. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry 2009, 14, 51–59. [Google Scholar] [CrossRef]
- Timmusk, T.; Palm, K.; Lendahl, U.; Metsis, M. Brain-derived neurotrophic factor expression in vivo is under the control of neuron-restrictive silencer element. J. Biol. Chem. 1999, 274, 1078–1084. [Google Scholar]
- Kim, S.J.; Lee, B.H.; Lee, Y.S.; Kang, K.S. Defective cholesterol traffic and neuronal differentiation in neural stem cells of Niemann-Pick type C disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 2007, 360, 593–599. [Google Scholar] [CrossRef]
- Checler, F.; da Costa, C.A. p53 in neurodegenerative diseases and brain cancers. Pharmacol. Ther. 2013, 142, 99–113. [Google Scholar] [CrossRef]
- Murphy, M.; Ahn, J.; Walker, K.K.; Hoffman, W.H.; Evans, R.M.; Levine, A.J.; George, D.L. Transcriptional repression by wild-type p53 utilizes histone deacetylases, mediated by interaction with mSin3a. Genes Dev. 1999, 13, 2490–2501. [Google Scholar] [CrossRef]
- Juan, L.J.; Shia, W.J.; Chen, M.H.; Yang, W.M.; Seto, E.; Lin, Y.S.; Wu, C.W. Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 2000, 275, 20436–20443. [Google Scholar]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vincente, M.; Arrasate, M.; O’Rourke, J.G.; Khashwji, H.; et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell. Biol. 2009, 187, 1083–1099. [Google Scholar] [CrossRef]
- Chou, D.H.; Holson, E.B.; Wagner, F.F.; Tang, A.J.; Maglathlin, R.L.; Lewis, T.A.; Schreiber, S.L.; Wagner, B.K. Inhibition of histone deacetylase 3 protects beta cells from cytokine-induced apoptosis. Chem. Biol. 2012, 19, 669–673. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Zhang, Z.; Schluesener, H.J. MS-275, an histone deacetylase inhibitor, reduces the inflammatory reaction in rat experimental autoimmune neuritis. Neuroscience 2010, 169, 370–377. [Google Scholar] [CrossRef]
- Gillespie, J.; Savic, S.; Wong, C.; Hempshall, A.; Inman, M.; Emery, P.; Grigg, R.; McDermott, M.F. Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthr. Rheum 2012, 64, 418–422. [Google Scholar] [CrossRef]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–E2874. [Google Scholar] [CrossRef]
- Gregoire, S.; Xiao, L.; Nie, J.; Zhang, X.; Xu, M.; Li, J.; Wong, J.; Seto, E.; Yang, X.J. Histone deacetylase 3 interacts with and deacetylates myocyte enhancer factor 2. Mol. Cell. Biol. 2007, 27, 1280–1295. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef]
- Jeong, Y.; Du, R.; Zhu, X.; Yin, S.; Wang, J.; Cui, H.; Cao, W.; Lowenstein, C.J. Histone deacetylase isoforms regulate innate immune responses by deacetylating mitogen-activated protein kinase phosphatase-1. J. Leukoc Biol. 2013, 95, 651–659. [Google Scholar]
- Memet, S. NF-kappaB functions in the nervous system: from development to disease. Biochem. Pharmacol. 2006, 72, 1180–1195. [Google Scholar] [CrossRef]
- Ziesche, E.; Kettner-Buhrow, D.; Weber, A.; Wittwer, T.; Jurida, L.; Soelch, J.; Muller, H.; Newel, D.; Kronich, P.; Schneider, H.; et al. The coactivator role of histone deacetylase 3 in IL-1-signaling involves deacetylation of p65 NF-kappaB. Nucleic Acids Res. 2013, 41, 90–109. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Thomas, E.A. Involvement of HDAC1 and HDAC3 in the Pathology of Polyglutamine Disorders: Therapeutic Implications for Selective HDAC1/HDAC3 Inhibitors. Pharmaceuticals 2014, 7, 634-661. https://doi.org/10.3390/ph7060634
Thomas EA. Involvement of HDAC1 and HDAC3 in the Pathology of Polyglutamine Disorders: Therapeutic Implications for Selective HDAC1/HDAC3 Inhibitors. Pharmaceuticals. 2014; 7(6):634-661. https://doi.org/10.3390/ph7060634
Chicago/Turabian StyleThomas, Elizabeth A. 2014. "Involvement of HDAC1 and HDAC3 in the Pathology of Polyglutamine Disorders: Therapeutic Implications for Selective HDAC1/HDAC3 Inhibitors" Pharmaceuticals 7, no. 6: 634-661. https://doi.org/10.3390/ph7060634
APA StyleThomas, E. A. (2014). Involvement of HDAC1 and HDAC3 in the Pathology of Polyglutamine Disorders: Therapeutic Implications for Selective HDAC1/HDAC3 Inhibitors. Pharmaceuticals, 7(6), 634-661. https://doi.org/10.3390/ph7060634