From Single Target to Multitarget/Network Therapeutics in Alzheimer’s Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
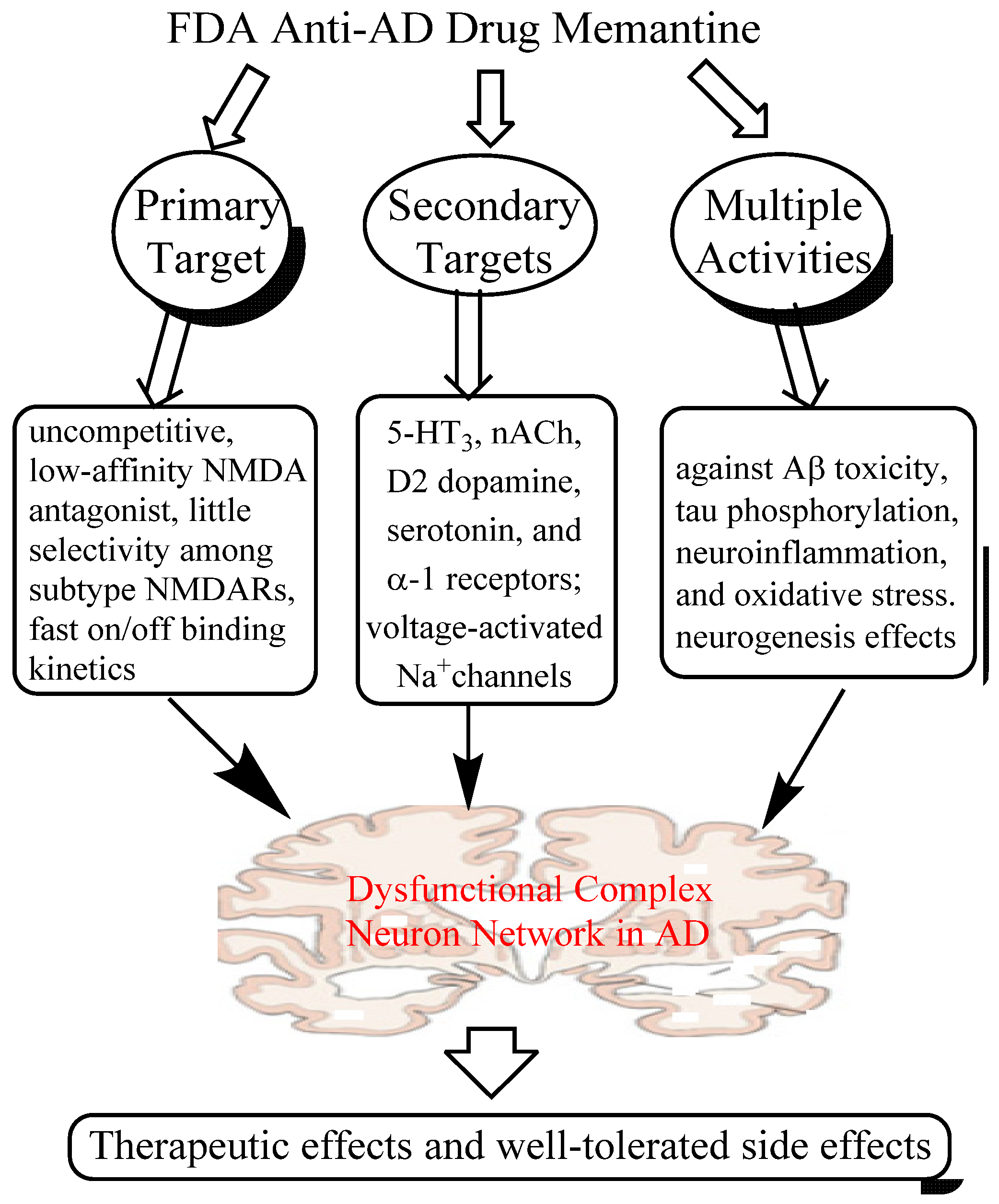
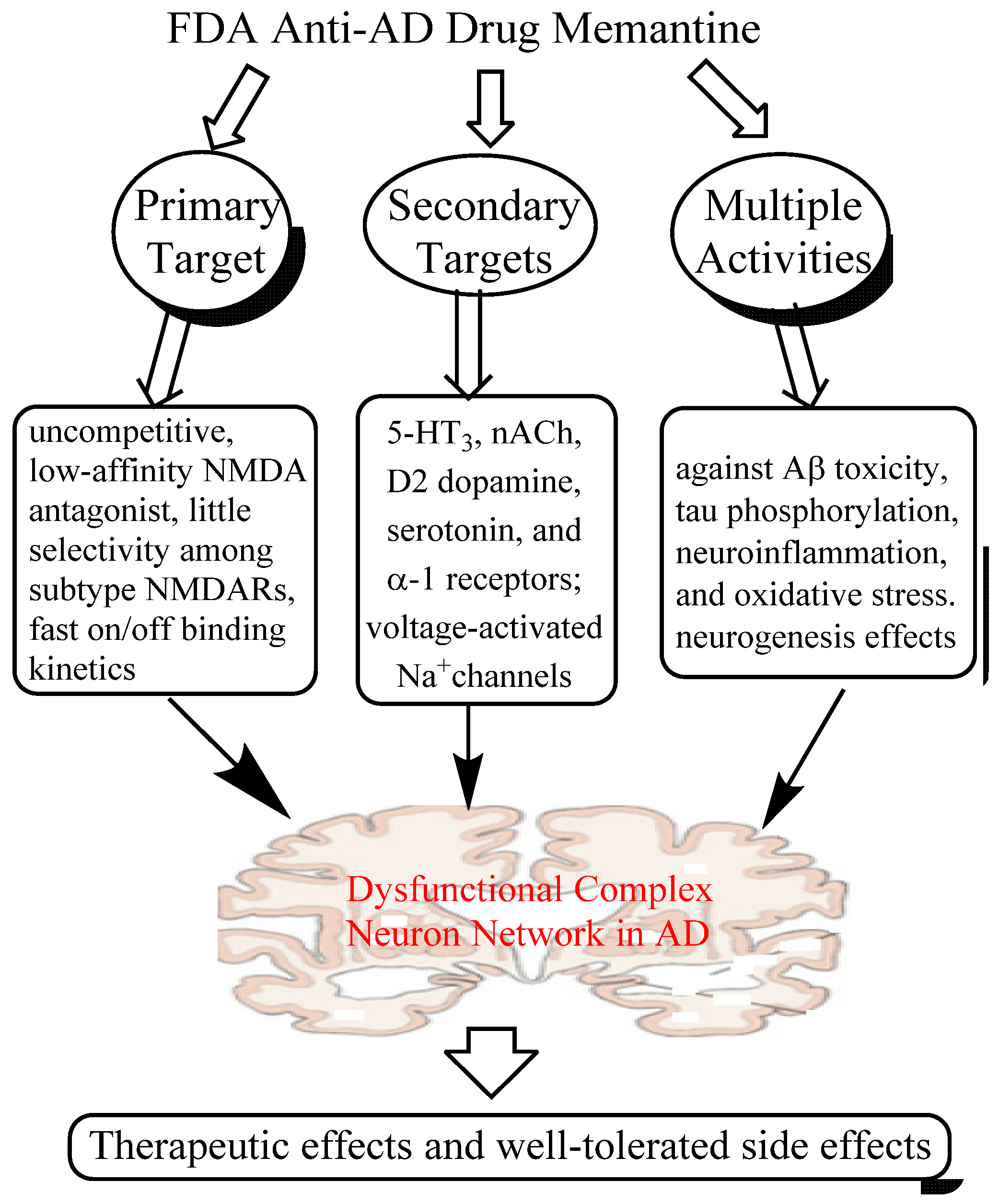
2. Memantine, An Anti-AD Drug that Breaks Conventional Rules
2.1. Mechanisms of Action of Memantine
2.1.1. Low-affinity, Dirty and Uncompetitive NMDA Antagonist
2.1.2. Effects on Other CNS Targets
2.1.3. Neuroprotective Activity in Various Culture and Animal Models
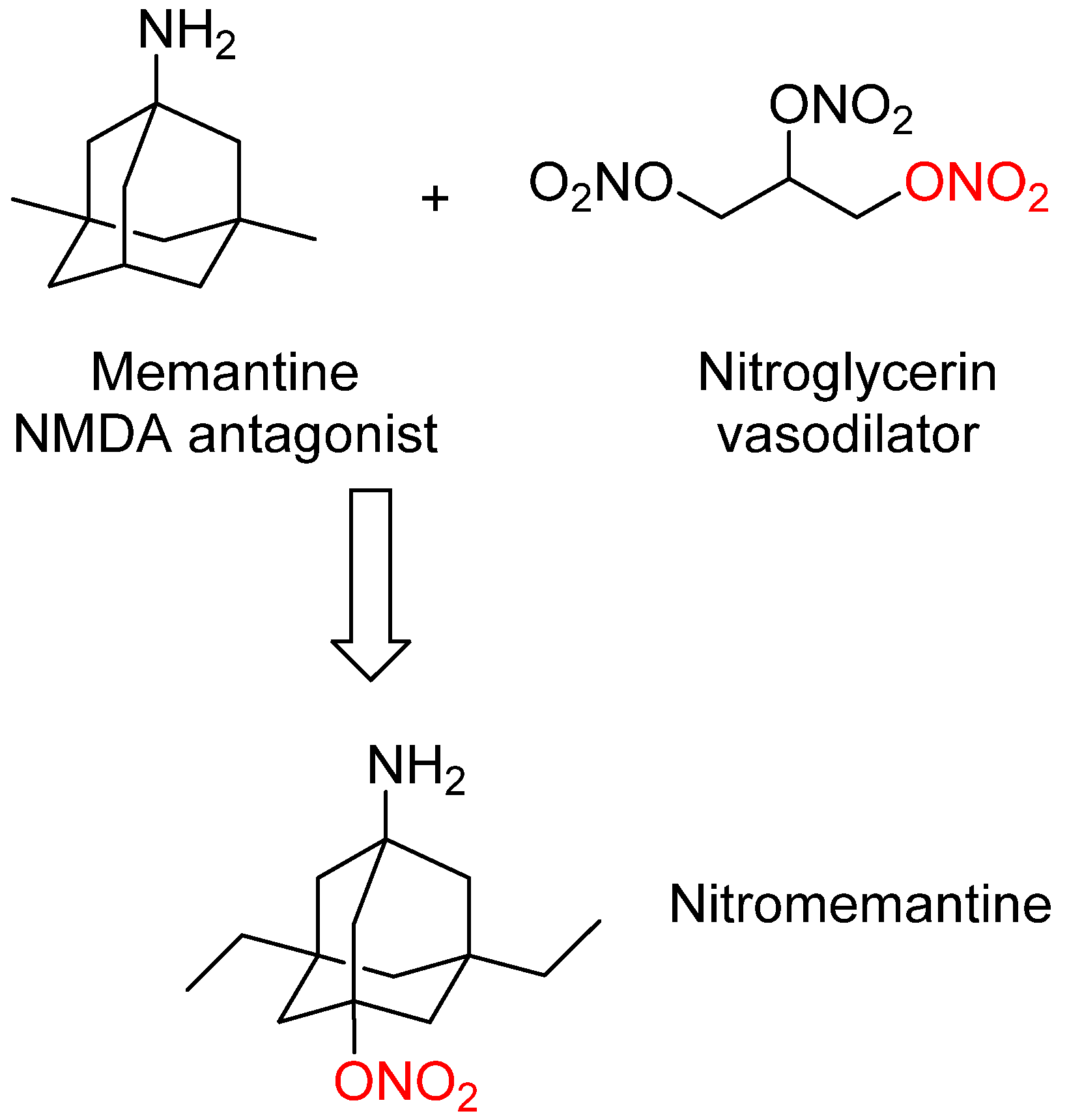
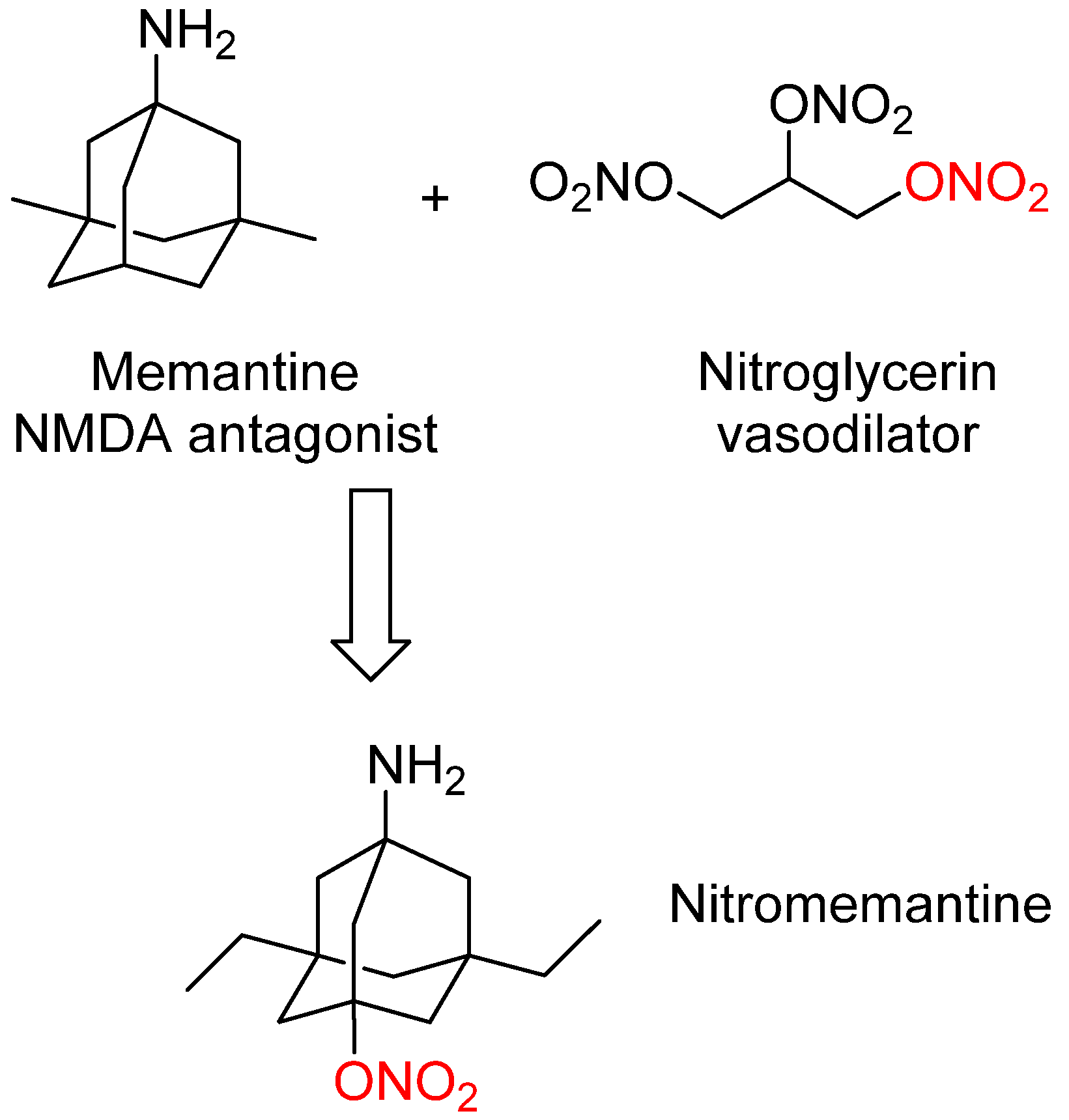
3. Nitromemantine: Second Generation Memantine Analog
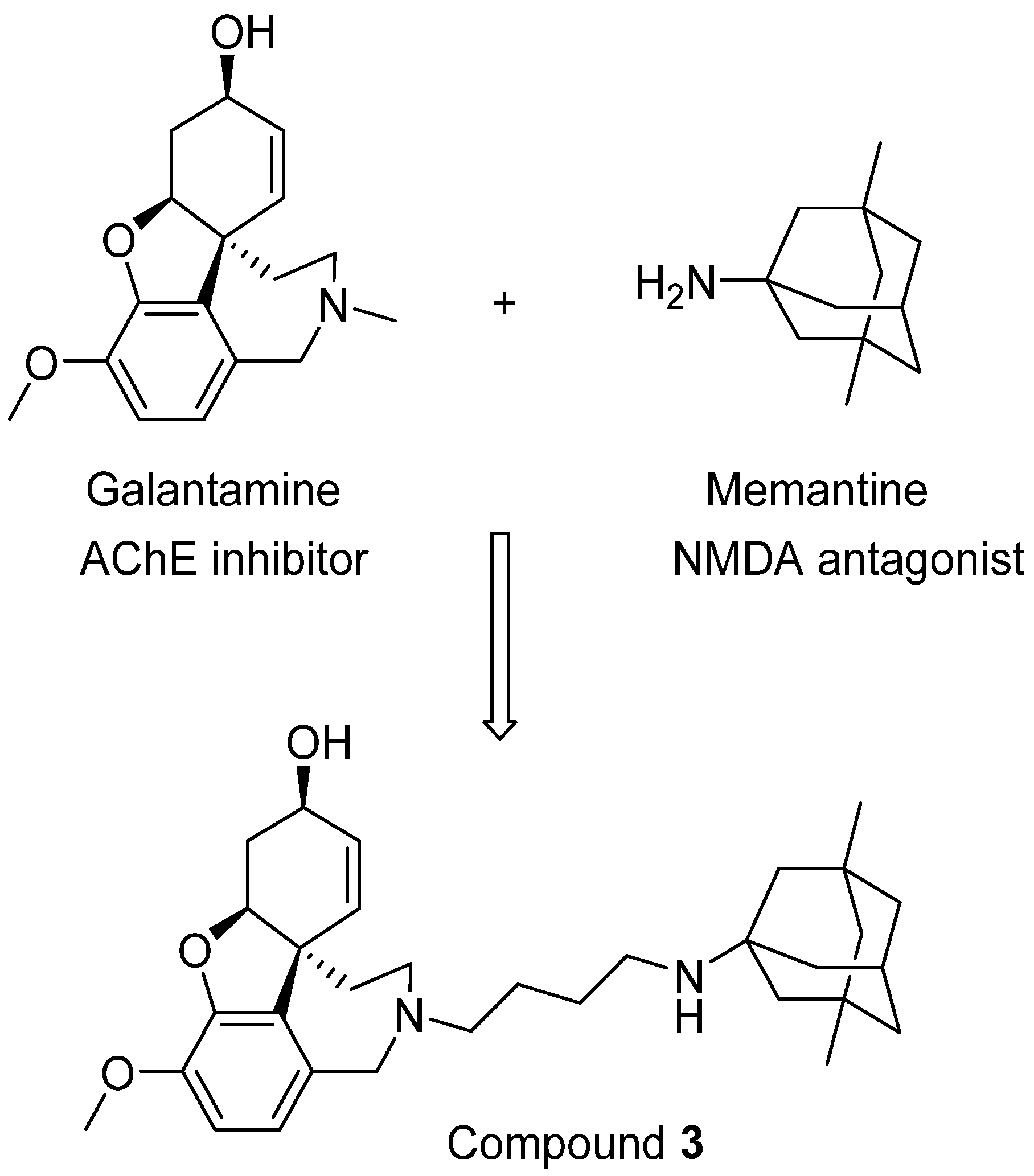
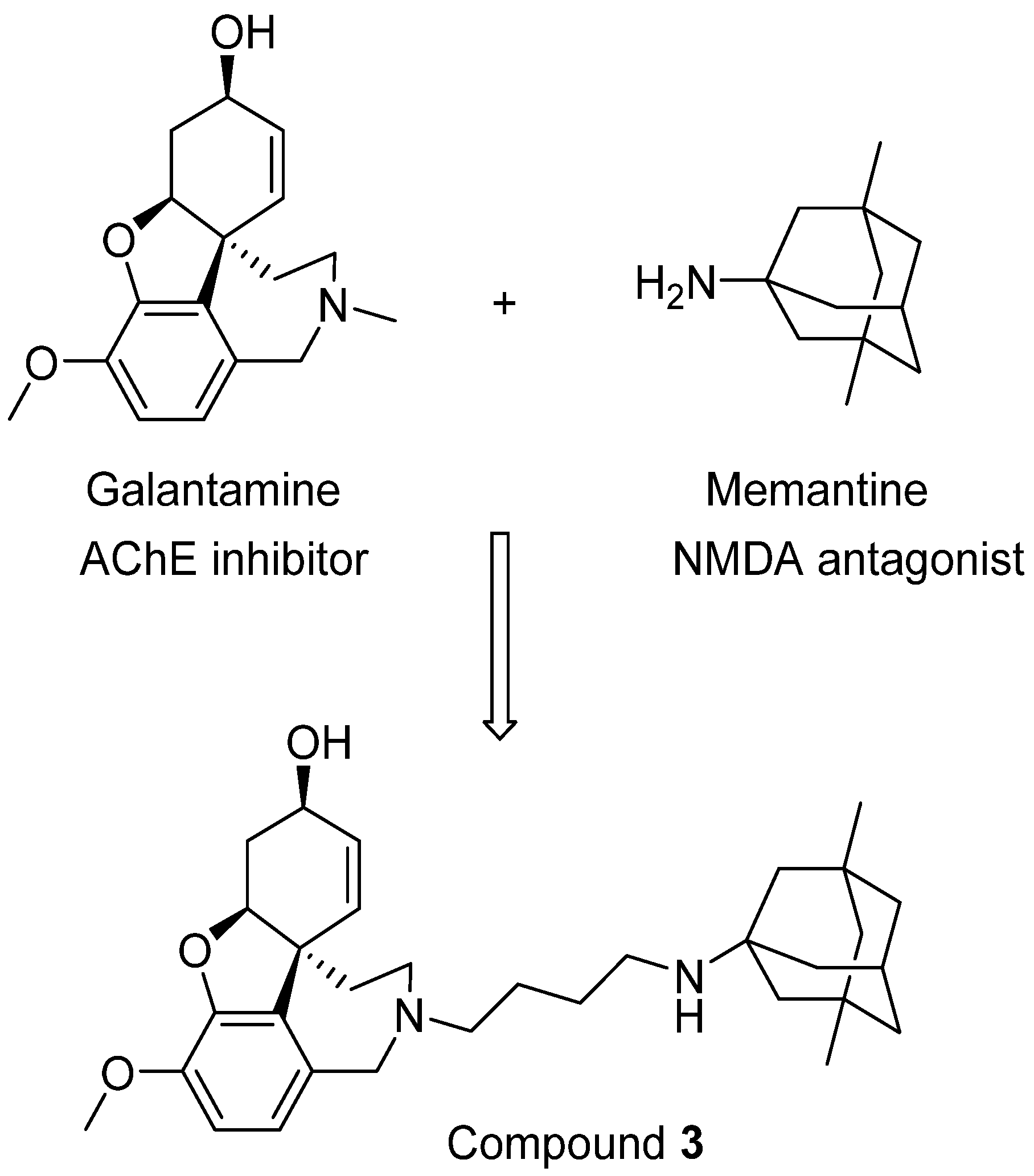
4. Drugs Targeting Both AChE and NMDA Receptors
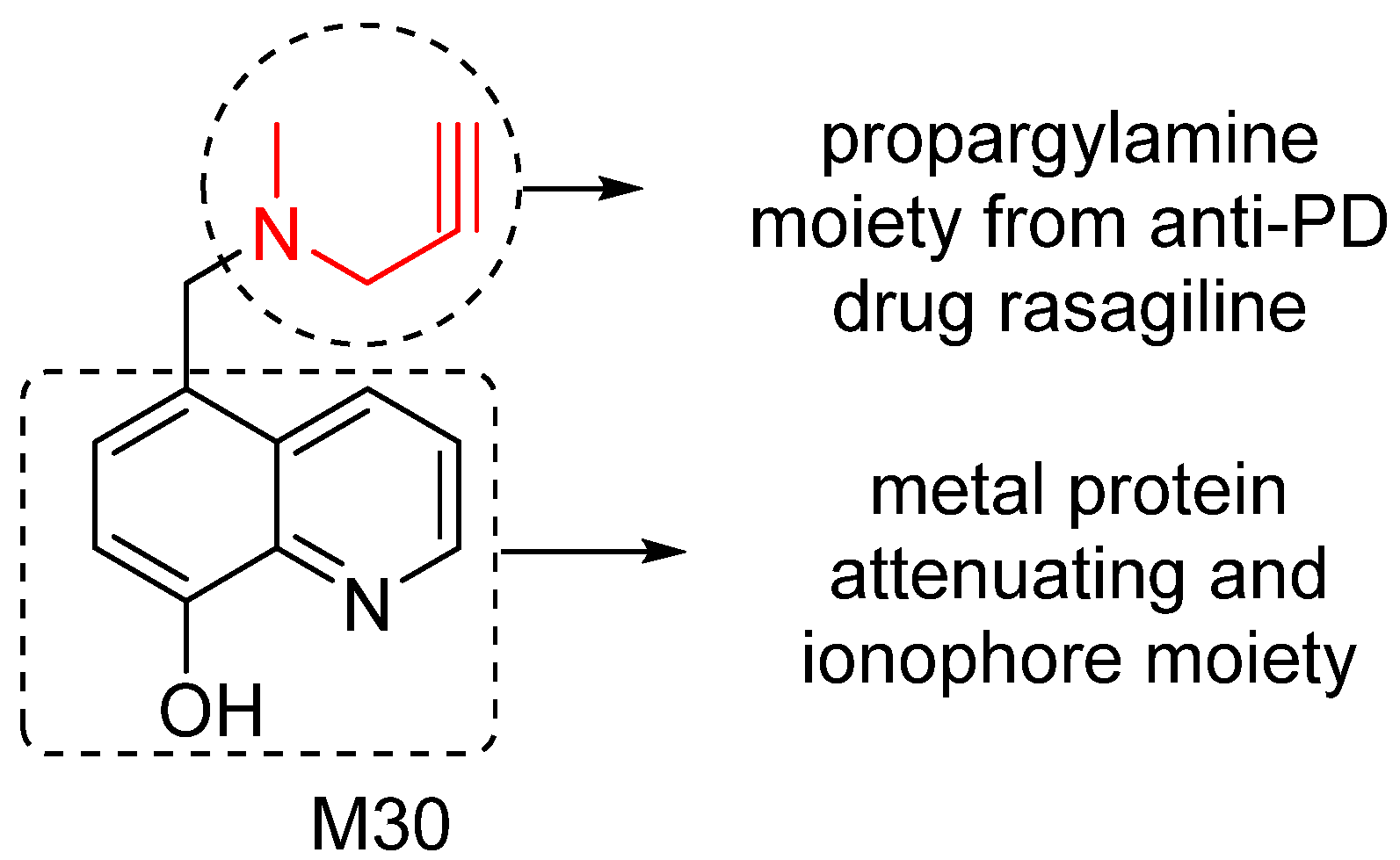
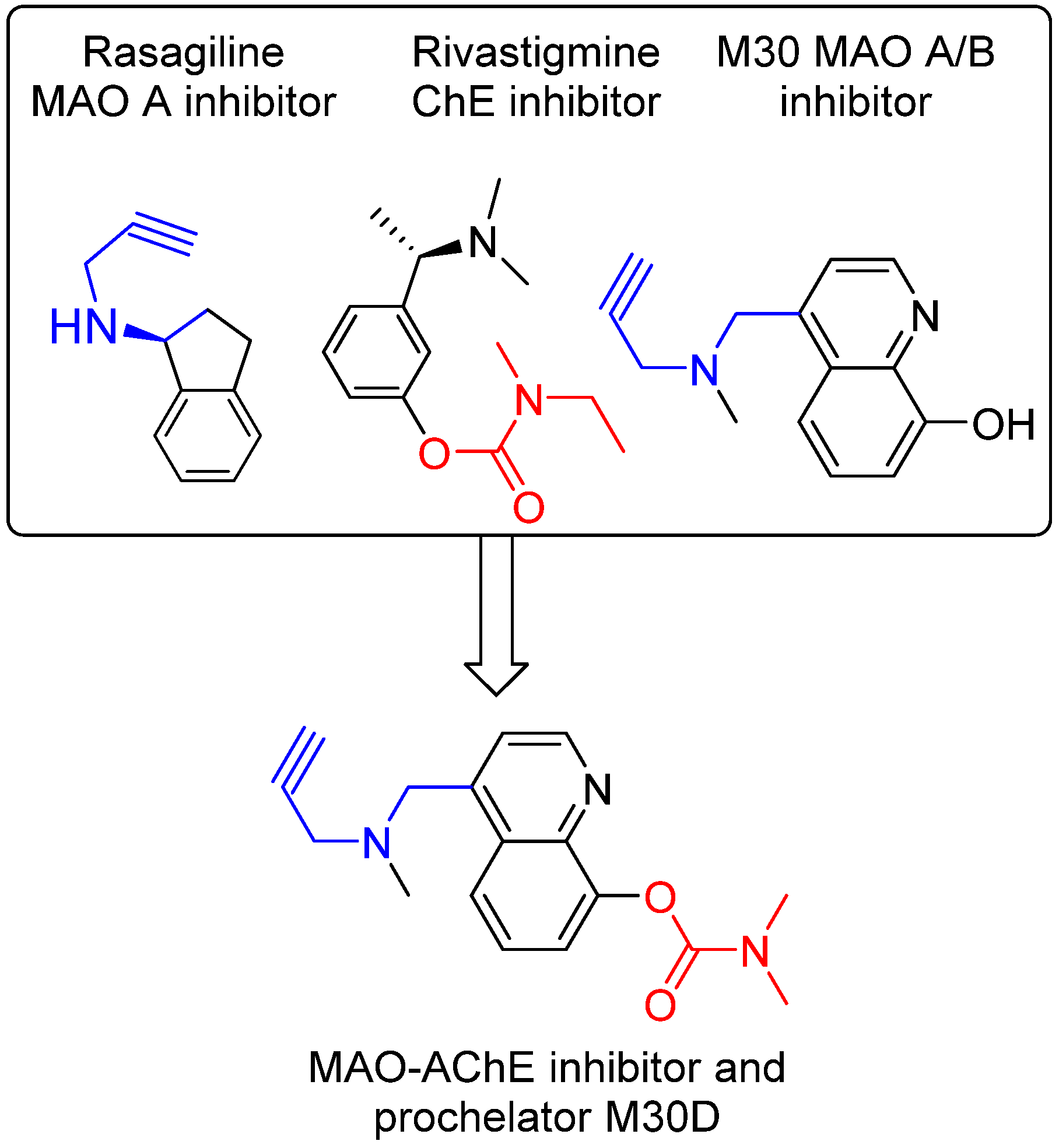
5. M30: Metal/MAO Network Dysfunction Modulator
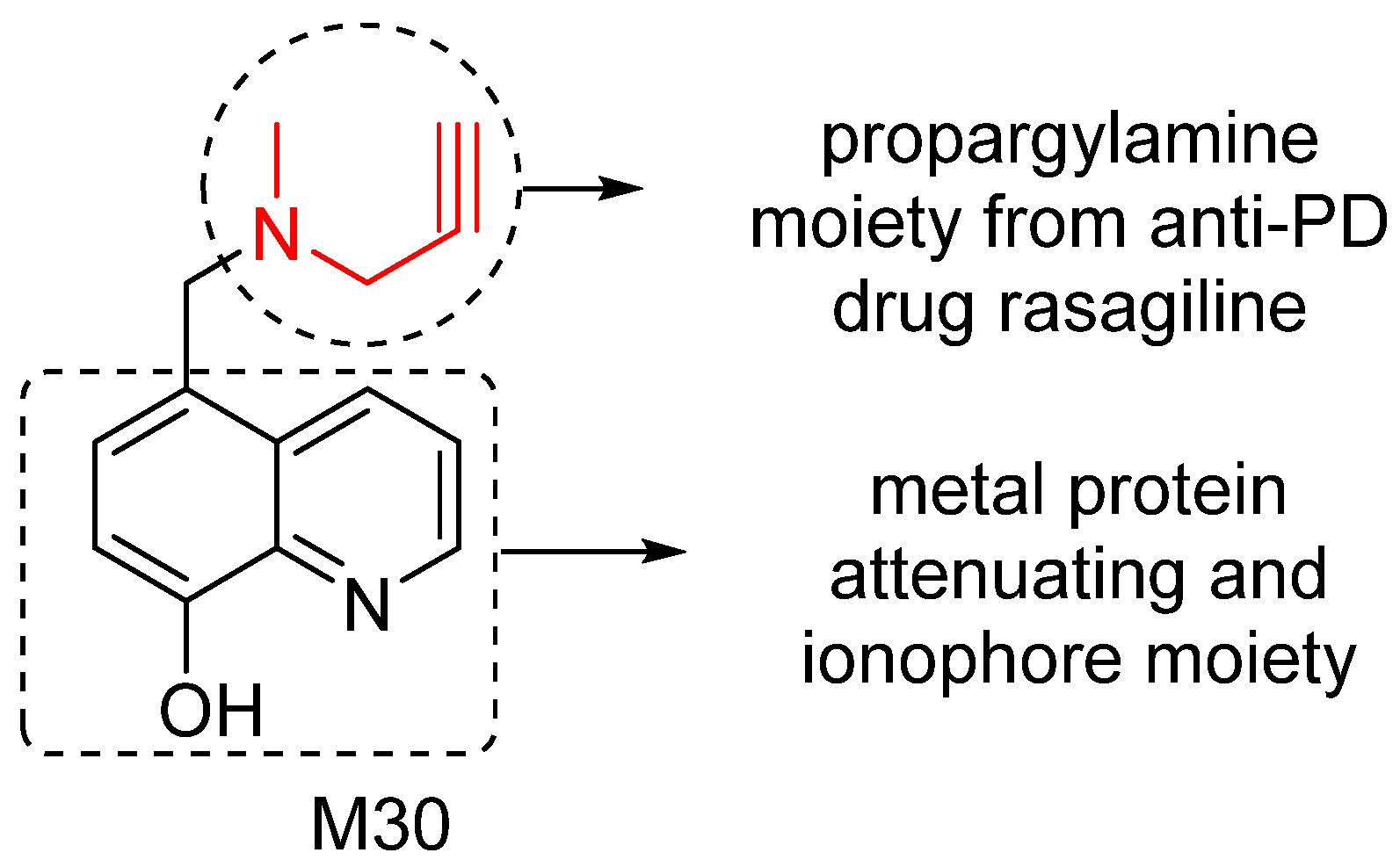
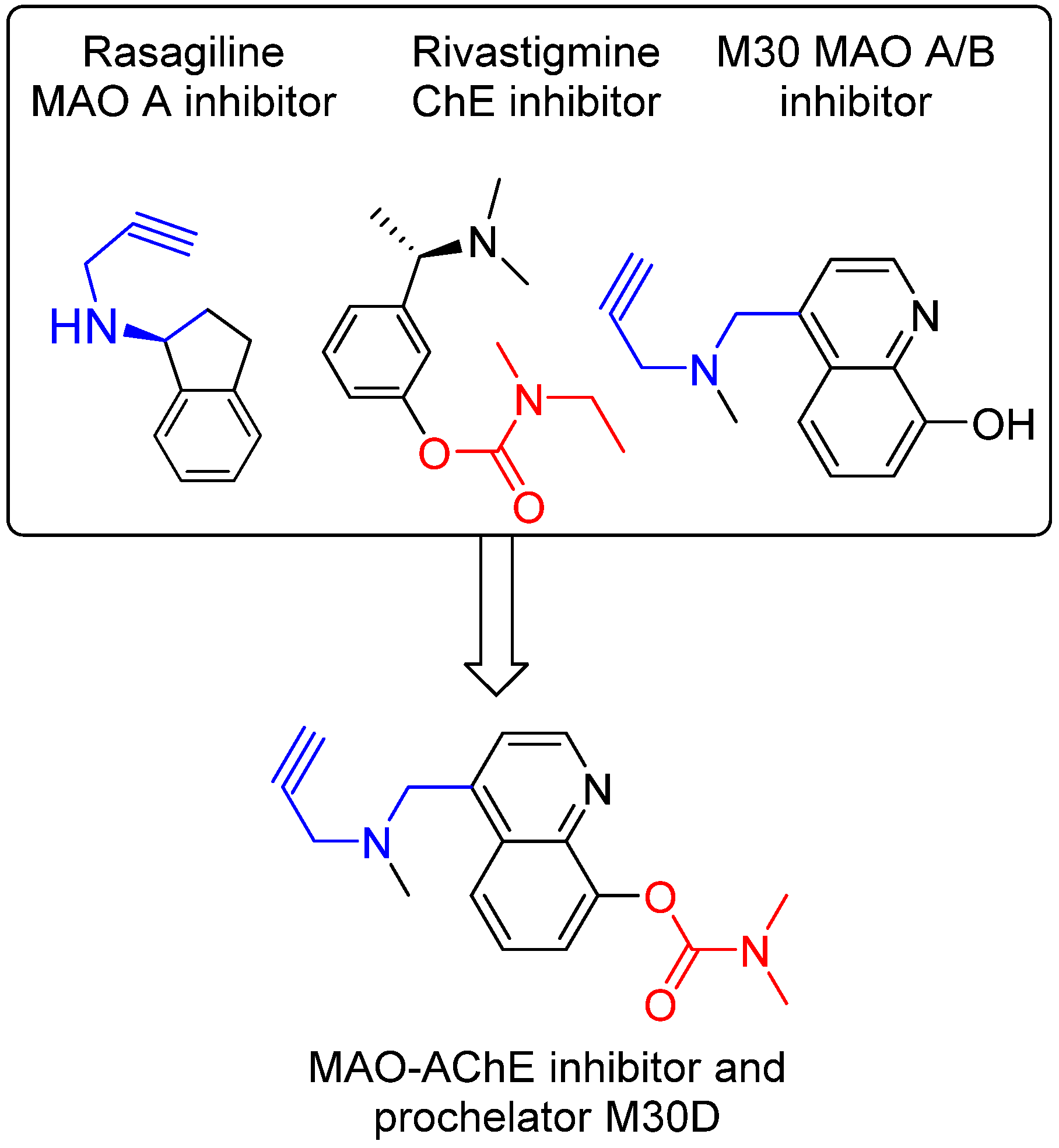
6. M30D: Second Generation M30 Analog
7. Ginkgo Biloba (GB) Extract
8. Conclusions
Author Contributions
Conflicts of Interest
References
- Medina-Franco, J.L.; Giulianotti, M.A.; Welmaker, G.S.; Houghten, R.A. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov. Today 2013, 18, 495–501. [Google Scholar] [CrossRef]
- Opar, A. Mixed results for disease-modification strategies for Alzheimer’s disease. Nat. Rev. Drug Discov. 2008, 7, 717–718. [Google Scholar] [CrossRef]
- Barabasi, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef]
- Kitano, H. A robustness-based approach to systems-oriented drug design. Nat. Rev. Drug Discov. 2007, 6, 202–210. [Google Scholar] [CrossRef]
- Darreh-Shori, T.; Soininen, H. Effects of cholinesterase inhibitors on the activities and protein levels of cholinesterases in the cerebrospinal fluid of patients with Alzheimer’s disease: A review of recent clinical studies. Curr. Alzheimer Res. 2010, 7, 67–73. [Google Scholar] [CrossRef]
- Ooi, S.L.; Pan, X.; Peyser, B.D.; Ye, P.; Meluh, P.B.; Yuan, D.S.; Irizarry, R.A.; Bader, J.S.; Spencer, F.A.; Boeke, J.D. Global synthetic-lethality analysis and yeast functional profiling. Trends Genet. 2006, 22, 56–63. [Google Scholar] [CrossRef]
- Hartman, J.L.; Garvik, B.; Hartwell, L. Principles for the buffering of genetic variation. Science 2001, 291, 1001–1004. [Google Scholar] [CrossRef]
- Britton, R.A.; Grossman, A.D. Synthetic lethal phenotypes caused by mutations affecting chromosome partitioning in Bacillus subtilis. J. Bacteriol. 1999, 181, 5860–5864. [Google Scholar]
- Li, F.; Zhao, C.; Wang, L. Molecular-targeted agents combination therapy for cancer: Developments and potentials. Int. J. Cancer 2014, 134, 1257–1269. [Google Scholar] [CrossRef]
- Chow, V.W.; Savonenko, A.V.; Melnikova, T.; Kim, H.; Price, D.L.; Li, T.; Wong, P.C. Modeling an anti-amyloid combination therapy for Alzheimer’s disease. Sci. Transl. Med. 2010, 2, 13ra1. [Google Scholar]
- Lehár, J.; Krueger, A.S.; Avery, W.; Heilbut, A.M.; Johansen, L.M.; Price, E.R.; Rickles, R.J.; Short, G.F.; Staunton, J.E.; Jin, X.; et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef]
- Couratier, P.; Lesort, M.; Sindou, P.; Esclaire, F.; Yardin, C.; Hugon, J. Modifications of neuronal phosphorylated tau immunoreactivity induced by NMDA toxicity. Mol. Chem. Neuropathol. 1996, 27, 259–273. [Google Scholar] [CrossRef]
- Esclaire, F.; Lesort, M.; Blanchard, C.; Hugon, J. Glutamate toxicity enhances tau gene expression in neuronal cultures. J. Neurosci. Res. 1997, 49, 309–318. [Google Scholar] [CrossRef]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef]
- Alberdi, E.; Sanchez-Gomez, M.V.; Cavaliere, F.; Perez-Samartin, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1992, 12, 376–389. [Google Scholar]
- Kabogo, D.; Rauw, G.; Amritraj, A.; Baker, G.; Kar, S. β-amyloid-related peptides potentiate K+-evoked glutamate release from adult rat hippocampal slices. Neurobiol. Aging 2010, 31, 1164–1172. [Google Scholar] [CrossRef]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef]
- Doody, R.S.; Tariot, P.N.; Pfeiffer, E.; Olin, J.T.; Graham, S.M. Meta-analysis of six-month memantine trials in Alzheimer’s disease. Alzheimers Dement. 2007, 3, 7–17. [Google Scholar] [CrossRef]
- Wong, E.H.; Kemp, J.A.; Priestley, T.; Knight, A.R.; Woodruff, G.N.; Iversen, L.L. The anticonvulsant MK-801 is a potent N-methyl-d-aspartate antagonist. Proc. Natl. Acad. Sci. USA 1986, 83, 7104–7108. [Google Scholar] [CrossRef]
- Parsons, C.G.; Danysz, W.; Bartmann, A.; Spielmanns, P.; Frankiewicz, T.; Hesselink, M.; Eilbacher, B.; Quack, G. Amino-alkyl-cyclohexanes are novel uncompetitive NMDA receptor antagonists with strong voltage-dependency and fast blocking kinetics: In vitro and in vivo characterization. Neuropharmacology 1999, 38, 85–108. [Google Scholar] [CrossRef]
- Johnson, J.W.; Kotermanski, S.E. Mechanism of action of memantine. Curr. Opin. Pharmacol. 2006, 6, 61–67. [Google Scholar] [CrossRef]
- Maskell, P.D.; Speder, P.; Newberry, N.R.; Bermudez, I. Inhibition of human alpha 7 nicotinic acetylcholine receptors by open channel blockers of N-methyl-d-aspartate receptors. Br. J. Pharmacol. 2003, 140, 1313–1319. [Google Scholar] [CrossRef]
- Aracava, Y.; Pereira, E.F.; Maelicke, A.; Albuquerque, E.X. Memantine blocks alpha7* nicotinic acetylcholine receptors more potently than n-methyl-d-aspartate receptors in rat hippocampal neurons. J. Pharmacol. Exp. Ther. 2005, 312, 1195–1205. [Google Scholar]
- Rammes, G.; Rupprecht, R.; Ferrari, U.; Zieglgansberger, W.; Parsons, C.G. The N-methyl-d-aspartate receptor channel blockers memantine, MRZ 2/579 and other amino-alkyl-cyclohexanes antagonise 5-HT(3) receptor currents in cultured HEK-293 and N1E-115 cell systems in a non-competitive manner. Neurosci. Lett. 2001, 306, 81–84. [Google Scholar] [CrossRef]
- Seeman, P.; Caruso, C.; Lasaga, M. Memantine agonist action at dopamine D2High receptors. Synapse 2008, 62, 149–153. [Google Scholar] [CrossRef]
- Bickford-Wimer, P.; Kim, M.; Boyajian, C.; Cooper, D.M.; Freedman, R. Effects of pertussis toxin on caudate neuron electrophysiology: Studies with dopamine D1 and D2 agonists. Brain Res. 1990, 533, 263–267. [Google Scholar]
- George, S.R.; Watanabe, M.; Di Paolo, T.; Falardeau, P.; Labrie, F.; Seeman, P. The functional state of the dopamine receptor in the anterior pituitary is in the high affinity form. Endocrinology 1985, 117, 690–697. [Google Scholar] [CrossRef]
- Tremblay, R.; Chakravarthy, B.; Hewitt, K.; Tauskela, J.; Morley, P.; Atkinson, T.; Durkin, J.P. Transient NMDA receptor inactivation provides long-term protection to cultured cortical neurons from a variety of death signals. J. Neurosci. 2000, 20, 7183–7192. [Google Scholar]
- Hu, M.; Schurdak, M.E.; Puttfarcken, P.S.; El Kouhen, R.; Gopalakrishnan, M.; Li, J. High content screen microscopy analysis of A beta 1-42-induced neurite outgrowth reduction in rat primary cortical neurons: Neuroprotective effects of alpha 7 neuronal nicotinic acetylcholine receptor ligands. Brain Res. 2007, 1151, 227–235. [Google Scholar]
- Miguel-Hidalgo, J.J.; Alvarez, X.A.; Cacabelos, R.; Quack, G. Neuroprotection by memantine against neurodegeneration induced by beta-amyloid(1–40). Brain Res. 2002, 958, 210–221. [Google Scholar]
- Minkeviciene, R.; Rheims, S.; Dobszay, M.B.; Zilberter, M.; Hartikainen, J.; Fulop, L.; Penke, B.; Zilberter, Y.; Harkany, T.; Pitkanen, A.; et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci. 2009, 29, 3453–3462. [Google Scholar] [CrossRef]
- Dong, H.; Yuede, C.M.; Coughlan, C.; Lewis, B.; Csernansky, J.G. Effects of memantine on neuronal structure and conditioned fear in the Tg2576 mouse model of Alzheimer’s disease. Neuropsychopharmacology 2008, 33, 3226–3236. [Google Scholar] [CrossRef]
- Unger, C.; Svedberg, M.M.; Yu, W.F.; Hedberg, M.M.; Nordberg, A. Effect of subchronic treatment of memantine, galantamine, and nicotine in the brain of Tg2576 (APPswe) transgenic mice. J. Pharmacol. Exp. Ther. 2006, 317, 30–36. [Google Scholar]
- Song, M.S.; Rauw, G.; Baker, G.B.; Kar, S. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur. J. Neurosci. 2008, 28, 1989–2002. [Google Scholar] [CrossRef]
- Thornton, C.; Bright, N.J.; Sastre, M.; Muckett, P.J.; Carling, D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem. J. 2011, 434, 503–512. [Google Scholar]
- Li, L.; Sengupta, A.; Haque, N.; Grundke-Iqbal, I.; Iqbal, K. Memantine inhibits and reverses the Alzheimer type abnormal hyperphosphorylation of tau and associated neurodegeneration. FEBS Lett. 2004, 566, 261–269. [Google Scholar] [CrossRef]
- Degerman Gunnarsson, M.; Kilander, L.; Basun, H.; Lannfelt, L. Reduction of phosphorylated tau during memantine treatment of Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2007, 24, 247–252. [Google Scholar] [CrossRef]
- Willard, L.B.; Hauss-Wegrzyniak, B.; Danysz, W.; Wenk, G.L. The cytotoxicity of chronic neuroinflammation upon basal forebrain cholinergic neurons of rats can be attenuated by glutamatergic antagonism or cyclooxygenase-2 inhibition. Exp. Brain Res. 2000, 134, 58–65. [Google Scholar] [CrossRef]
- Rosi, S.; Vazdarjanova, A.; Ramirez-Amaya, V.; Worley, P.F.; Barnes, C.A.; Wenk, G.L. Memantine protects against LPS-induced neuroinflammation, restores behaviorally-induced gene expression and spatial learning in the rat. Neuroscience 2006, 142, 1303–1315. [Google Scholar] [CrossRef]
- Wu, H.M.; Tzeng, N.S.; Qian, L.; Wei, S.J.; Hu, X.; Chen, S.H.; Rawls, S.M.; Flood, P.; Hong, J.S.; Lu, R.B. Novel neuroprotective mechanisms of memantine: Increase in neurotrophic factor release from astroglia and anti-inflammation by preventing microglial activation. Neuropsychopharmacology 2009, 34, 2344–2357. [Google Scholar] [CrossRef]
- Liu, W.; Xu, Z.; Deng, Y.; Xu, B.; Wei, Y.; Yang, T. Protective effects of memantine against methylmercury-induced glutamate dyshomeostasis and oxidative stress in rat cerebral cortex. Neurotox. Res. 2013, 24, 320–337. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; de Strooper, B. The toxic Abeta oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Figueiredo, C.P.; Clarke, J.R.; Ledo, J.H.; Ribeiro, F.C.; Costa, C.V.; Melo, H.M.; Mota-Sales, A.P.; Saraiva, L.M.; Klein, W.L.; Sebollela, A.; et al. Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J. Neurosci. 2013, 33, 9626–9634. [Google Scholar] [CrossRef]
- Weiner, M.W.; Sadowsky, C.; Saxton, J.; Hofbauer, R.K.; Graham, S.M.; Yu, S.Y.; Li, S.; Hsu, H.A.; Suhy, J.; Fridman, M.; et al. Magnetic resonance imaging and neuropsychological results from a trial of memantine in Alzheimer’s disease. Alzheimers Dement. 2011, 7, 425–435. [Google Scholar] [CrossRef]
- Wilkinson, D.; Andersen, H.F. Analysis of the effect of memantine in reducing the worsening of clinical symptoms in patients with moderate to severe Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2007, 24, 138–145. [Google Scholar] [CrossRef]
- Lipton, S.A.; Choi, Y.B.; Pan, Z.H.; Lei, S.Z.; Chen, H.S.; Sucher, N.J.; Loscalzo, J.; Singel, D.J.; Stamler, J.S. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 1993, 364, 626–632. [Google Scholar] [CrossRef]
- Choi, Y.B.; Tenneti, L.; Le, D.A.; Ortiz, J.; Bai, G.; Chen, H.S.; Lipton, S.A. Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation. Nat. Neurosci. 2000, 3, 15–21. [Google Scholar] [CrossRef]
- Wang, Y.; Eu, J.; Washburn, M.; Gong, T.; Chen, H.S.; James, W.L.; Lipton, S.A.; Stamler, J.S.; Went, G.T.; Porter, S. The pharmacology of aminoadamantane nitrates. Curr. Alzheimer Res. 2006, 3, 201–204. [Google Scholar] [CrossRef]
- Dawson, V.L.; Dawson, T.M.; London, E.D.; Bredt, D.S.; Snyder, S.H. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. USA 1991, 88, 6368–6371. [Google Scholar] [CrossRef]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef]
- Zheng, H.; Fridkin, M.; Youdim, M.B. Novel chelators targeting cell cycle arrest, acetylcholinesterase, and monoamine oxidase for Alzheimer’s therapy. Curr. Drug Targets 2012, 13, 1089–1106. [Google Scholar] [CrossRef]
- Atri, A.; Shaughnessy, L.W.; Locascio, J.J.; Growdon, J.H. Long-term course and effectiveness of combination therapy in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2008, 22, 209–221. [Google Scholar] [CrossRef]
- Atri, A.; Molinuevo, J.L.; Lemming, O.; Wirth, Y.; Pulte, I.; Wilkinson, D. Memantine in patients with Alzheimer’s disease receiving donepezil: New analyses of efficacy and safety for combination therapy. Alzheimers Res. Ther. 2013, 5, 6. [Google Scholar] [CrossRef]
- Simoni, E.; Daniele, S.; Bottegoni, G.; Pizzirani, D.; Trincavelli, M.L.; Goldoni, L.; Tarozzo, G.; Reggiani, A.; Martini, C.; Piomelli, D.; et al. Combining galantamine and memantine in multitargeted, new chemical entities potentially useful in Alzheimer’s disease. J. Med. Chem. 2012, 55, 9708–9721. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.X.; Tammer, A.; et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef]
- Wikipedia, Selegiline. Available online: http://en.wikipedia.org/wiki/Selegiline (accessed on 15 September 2013).
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s disease cooperative study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef]
- Birks, J.; Flicker, L. Selegiline for Alzheimer’s disease. Cochrane Database Syst. Rev. 2003, 1, CD000442. [Google Scholar]
- Sung, S.; Yao, Y.; Uryu, K.; Yang, H.; Lee, V.M.; Trojanowski, J.Q.; Pratico, D. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer’s disease. FASEB J. 2004, 18, 323–325. [Google Scholar]
- Petersen, R.C.; Bennett, D. Mild cognitive impairment: Is it Alzheimer’s disease or not? J. Alzheimers Dis. 2005, 7, 241–245. [Google Scholar]
- Lee, H.P.; Zhu, X.; Casadesus, G.; Castellani, R.J.; Nunomura, A.; Smith, M.A.; Lee, H.G.; Perry, G. Antioxidant approaches for the treatment of Alzheimer’s disease. Expert Rev. Neurother. 2010, 10, 1201–1208. [Google Scholar] [CrossRef]
- Olanow, C.W.; Rascol, O.; Hauser, R.; Feigin, P.D.; Jankovic, J.; Lang, A.; Langston, W.; Melamed, E.; Poewe, W.; Stocchi, F.; et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1268–1278. [Google Scholar] [CrossRef]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B. Rasagiline: A novel anti-Parkinsonian monoamine oxidase-B inhibitor with neuroprotective activity. Prog. Neurobiol. 2010, 92, 330–344. [Google Scholar] [CrossRef]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar]
- Ritchie, C.W.; Bush, A.I.; Masters, C.L. Metal-protein attenuating compounds and Alzheimer’s disease. Expert Opin. Investig. Drugs 2004, 13, 1585–1592. [Google Scholar] [CrossRef]
- Zheng, H.; Gal, S.; Weiner, L.M.; Bar-Am, O.; Warshawsky, A.; Fridkin, M.; Youdim, M.B. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases: In vitro studies on antioxidant activity, prevention of lipid peroxide formation and monoamine oxidase inhibition. J. Neurochem. 2005, 95, 68–78. [Google Scholar] [CrossRef]
- Gal, S.; Zheng, H.; Fridkin, M.; Youdim, M.B. Novel multifunctional neuroprotective iron chelator-monoamine oxidase inhibitor drugs for neurodegenerative diseases. In vivo selective brain monoamine oxidase inhibition and prevention of MPTP-induced striatal dopamine depletion. J. Neurochem. 2005, 95, 79–88. [Google Scholar] [CrossRef]
- Gal, S.; Abassi, Z.A.; Youdim, M.B. Limited potentiation of blood pressure in response to oral tyramine by the anti-Parkinson brain selective multifunctional monoamine oxidase-AB inhibitor, M30. Neurotox. Res. 2010, 18, 143–150. [Google Scholar] [CrossRef]
- Zheng, H.; Weiner, L.M.; Bar-Am, O.; Epsztejn, S.; Cabantchik, Z.I.; Warshawsky, A.; Youdim, M.B.; Fridkin, M. Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. Bioorg. Med. Chem. 2005, 13, 773–783. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Amit, T.; Bar-Am, O.; Youdim, M.B.; Weinreb, O. Neuroprotection by the multitarget iron chelator M30 on age-related alterations in mice. Mech. Ageing Dev. 2012, 133, 267–274. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Weinreb, O.; Amit, T.; Mandel, S.; Carri, M.T.; Youdim, M.B. Neuroprotective and neuritogenic activities of novel multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 2009, 23, 3766–3779. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Amit, T.; Bar-Am, O.; Weinreb, O.; Youdim, M.B. Multi-target, neuroprotective and neurorestorative M30 improves cognitive impairment and reduces Alzheimer’s-like neuropathology and age-related alterations in mice. Mol. Neurobiol. 2012, 46, 217–220. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Amit, T.; Bar-Am, O.; Youdim, M.B.; Weinreb, O. The novel multi-target iron chelating-radical scavenging compound M30 possesses beneficial effects on major hallmarks of Alzheimer’s disease. Antioxid. Redox Signal. 2012, 17, 860–877. [Google Scholar] [CrossRef]
- Avramovich-Tirosh, Y.; Bar-Am, O.; Amit, T.; Youdim, M.B.; Weinreb, O. Up-regulation of hypoxia-inducible factor (HIF) 1alpha and HIF-target genes in cortical neurons by the novel multifunctional iron chelator anti-Alzheimer drug, M30. Curr. Alzheimer Res. 2010, 7, 300–306. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Weinreb, O.; Amit, T.; Mandel, S.; Bar-Am, O.; Youdim, M.B. Novel molecular targets of the neuroprotective/neurorescue multimodal iron chelating drug M30 in the mouse brain. Neuroscience 2011, 189, 345–358. [Google Scholar] [CrossRef]
- Bartolini, M.; Bertucci, C.; Cavrini, V.; Andrisano, V. beta-Amyloid aggregation induced by human acetylcholinesterase: Inhibition studies. Biochem. Pharmacol. 2003, 65, 407–416. [Google Scholar]
- Mancini, F.; Naldi, M.; Cavrini, V.; Andrisano, V. Multiwell fluorometric and colorimetric microassays for the evaluation of beta-secretase (BACE-1) inhibitors. Anal. Bioanal. Chem. 2007, 388, 1175–1183. [Google Scholar] [CrossRef]
- Meunier, J.; Ieni, J.; Maurice, T. Antiamnesic and neuroprotective effects of donepezil against learning impairments induced in mice by exposure to carbon monoxide gas. J. Pharmacol. Exp. Ther. 2006, 317, 1307–1319. [Google Scholar] [CrossRef]
- Kimura, M.; Akasofu, S.; Ogura, H.; Sawada, K. Protective effect of donepezil against Abeta(1–40) neurotoxicity in rat septal neurons. Brain Res. 2005, 1047, 72–84. [Google Scholar]
- Kimura, M.; Komatsu, H.; Ogura, H.; Sawada, K. Comparison of donepezil and memantine for protective effect against amyloid-beta(1–42) toxicity in rat septal neurons. Neurosci. Lett. 2005, 391, 17–21. [Google Scholar] [CrossRef]
- Meunier, J.; Ieni, J.; Maurice, T. The anti-amnesic and neuroprotective effects of donepezil against amyloid β25–35 peptide-induced toxicity in mice involve an interaction with the sigma1 receptor. Br. J. Pharmacol. 2006, 149, 998–1012. [Google Scholar] [CrossRef]
- Pepeu, G.; Giovannini, M.G. Cholinesterase inhibitors and beyond. Curr. Alzheimer Res. 2009, 6, 86–96. [Google Scholar] [CrossRef]
- Pohanka, M. Acetylcholinesterase inhibitors: A patent review (2008–present). Expert Opin. Ther. Pat. 2012, 22, 871–886. [Google Scholar] [CrossRef]
- Zheng, H.; Youdim, M.B.; Fridkin, M. Site-activated chelators targeting acetylcholinesterase and monoamine oxidase for Alzheimer’s therapy. ACS Chem. Biol. 2010, 5, 603–610. [Google Scholar] [CrossRef]
- Mahadevan, S.; Park, Y. Multifaceted therapeutic benefits of Ginkgo biloba L.: Chemistry, efficacy, safety, and uses. J. Food Sci. 2008, 73, R14–R19. [Google Scholar] [CrossRef]
- Fehske, C.J.; Leuner, K.; Muller, W.E. Ginkgo biloba extract (EGb761) influences monoaminergic neurotransmission via inhibition of NE uptake, but not MAO activity after chronic treatment. Pharmacol. Res. 2009, 60, 68–73. [Google Scholar] [CrossRef]
- Koch, E. Inhibition of platelet activating factor (PAF)-induced aggregation of human thrombocytes by ginkgolides: Considerations on possible bleeding complications after oral intake of Ginkgo biloba extracts. Phytomedicine 2005, 12, 10–16. [Google Scholar] [CrossRef]
- Koltermann, A.; Hartkorn, A.; Koch, E.; Furst, R.; Vollmar, A.M.; Zahler, S. Ginkgo biloba extract EGb 761 increases endothelial nitric oxide production in vitro and in vivo. Cell. Mol. Life Sci. 2007, 64, 1715–1722. [Google Scholar] [CrossRef]
- Winter, J.C.; Timineri, D. The discriminative stimulus properties of EGb 761, an extract of Ginkgo biloba. Pharmacol. Biochem. Behav. 1999, 62, 543–547. [Google Scholar]
- Wu, Y.; Wu, Z.; Butko, P.; Christen, Y.; Lambert, M.P.; Klein, W.L.; Link, C.D.; Luo, Y. Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J. Neurosci. 2006, 26, 13102–13113. [Google Scholar] [CrossRef]
- Bastianetto, S.; Ramassamy, C.; Dore, S.; Christen, Y.; Poirier, J.; Quirion, R. The Ginkgo biloba extract (EGb 761) protects hippocampal neurons against cell death induced by beta-amyloid. Eur. J. Neurosci. 2000, 12, 1882–1890. [Google Scholar] [CrossRef]
- Kampkotter, A.; Pielarski, T.; Rohrig, R.; Timpel, C.; Chovolou, Y.; Watjen, W.; Kahl, R. The Ginkgo biloba extract EGb761 reduces stress sensitivity, ROS accumulation and expression of catalase and glutathione S-transferase 4 in Caenorhabditis elegans. Pharmacol. Res. 2007, 55, 139–147. [Google Scholar] [CrossRef]
- Mazza, M.; Capuano, A.; Bria, P.; Mazza, S. Ginkgo biloba and donepezil: A comparison in the treatment of Alzheimer’s dementia in a randomized placebo-controlled double-blind study. Eur. J. Neurol. 2006, 13, 981–985. [Google Scholar] [CrossRef]
- McCarney, R.; Fisher, P.; Iliffe, S.; van Haselen, R.; Griffin, M.; van der Meulen, J.; Warner, J. Ginkgo biloba for mild to moderate dementia in a community setting: A pragmatic, randomised, parallel-group, double-blind, placebo-controlled trial. Int. J. Geriatr. Psychiatry 2008, 23, 1222–1230. [Google Scholar] [CrossRef]
- Snitz, B.E.; O’Meara, E.S.; Carlson, M.C.; Arnold, A.M.; Ives, D.G.; Rapp, S.R.; Saxton, J.; Lopez, O.L.; Dunn, L.O.; Sink, K.M.; et al. Ginkgo biloba for preventing cognitive decline in older adults: A randomized trial. JAMA 2009, 302, 2663–2670. [Google Scholar]
- Ihl, R.; Bachinskaya, N.; Korczyn, A.D.; Vakhapova, V.; Tribanek, M.; Hoerr, R.; Napryeyenko, O. Efficacy and safety of a once-daily formulation of Ginkgo biloba extract EGb 761 in dementia with neuropsychiatric features: A randomized controlled trial. Int. J. Geriatr. Psychiatry 2011, 26, 1186–1194. [Google Scholar]
- Brondino, N.; de Silvestri, A.; Re, S.; Lanati, N.; Thiemann, P.; Verna, A.; Emanuele, E.; Politi, P. A systematic review and meta-analysis of ginkgo biloba in neuropsychiatric disorders: From ancient tradition to modern-day medicine. Evid. Based Complement. Alternat. Med. 2013, 2013, 915691. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zheng, H.; Fridkin, M.; Youdim, M. From Single Target to Multitarget/Network Therapeutics in Alzheimer’s Therapy. Pharmaceuticals 2014, 7, 113-135. https://doi.org/10.3390/ph7020113
Zheng H, Fridkin M, Youdim M. From Single Target to Multitarget/Network Therapeutics in Alzheimer’s Therapy. Pharmaceuticals. 2014; 7(2):113-135. https://doi.org/10.3390/ph7020113
Chicago/Turabian StyleZheng, Hailin, Mati Fridkin, and Moussa Youdim. 2014. "From Single Target to Multitarget/Network Therapeutics in Alzheimer’s Therapy" Pharmaceuticals 7, no. 2: 113-135. https://doi.org/10.3390/ph7020113
APA StyleZheng, H., Fridkin, M., & Youdim, M. (2014). From Single Target to Multitarget/Network Therapeutics in Alzheimer’s Therapy. Pharmaceuticals, 7(2), 113-135. https://doi.org/10.3390/ph7020113