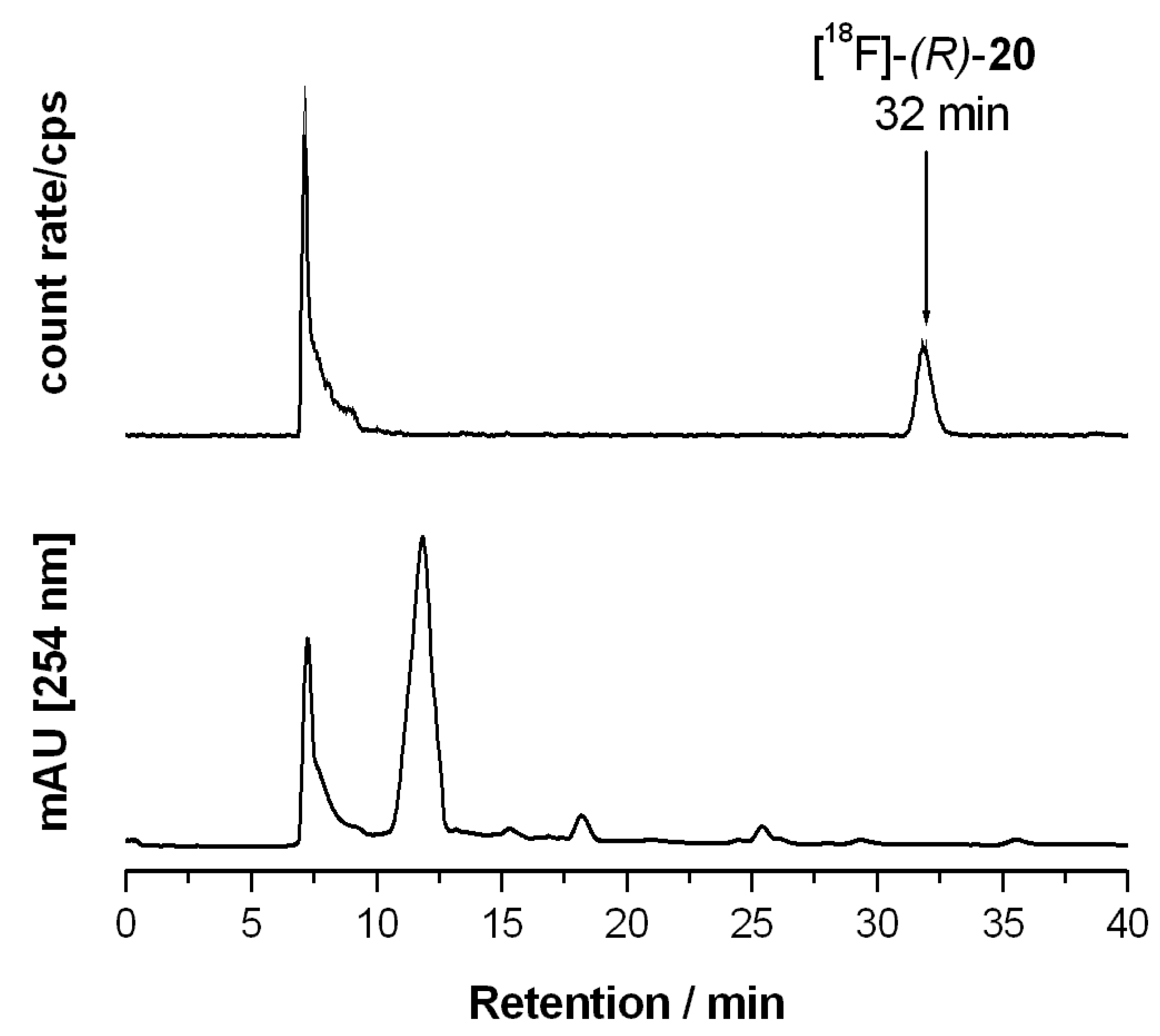
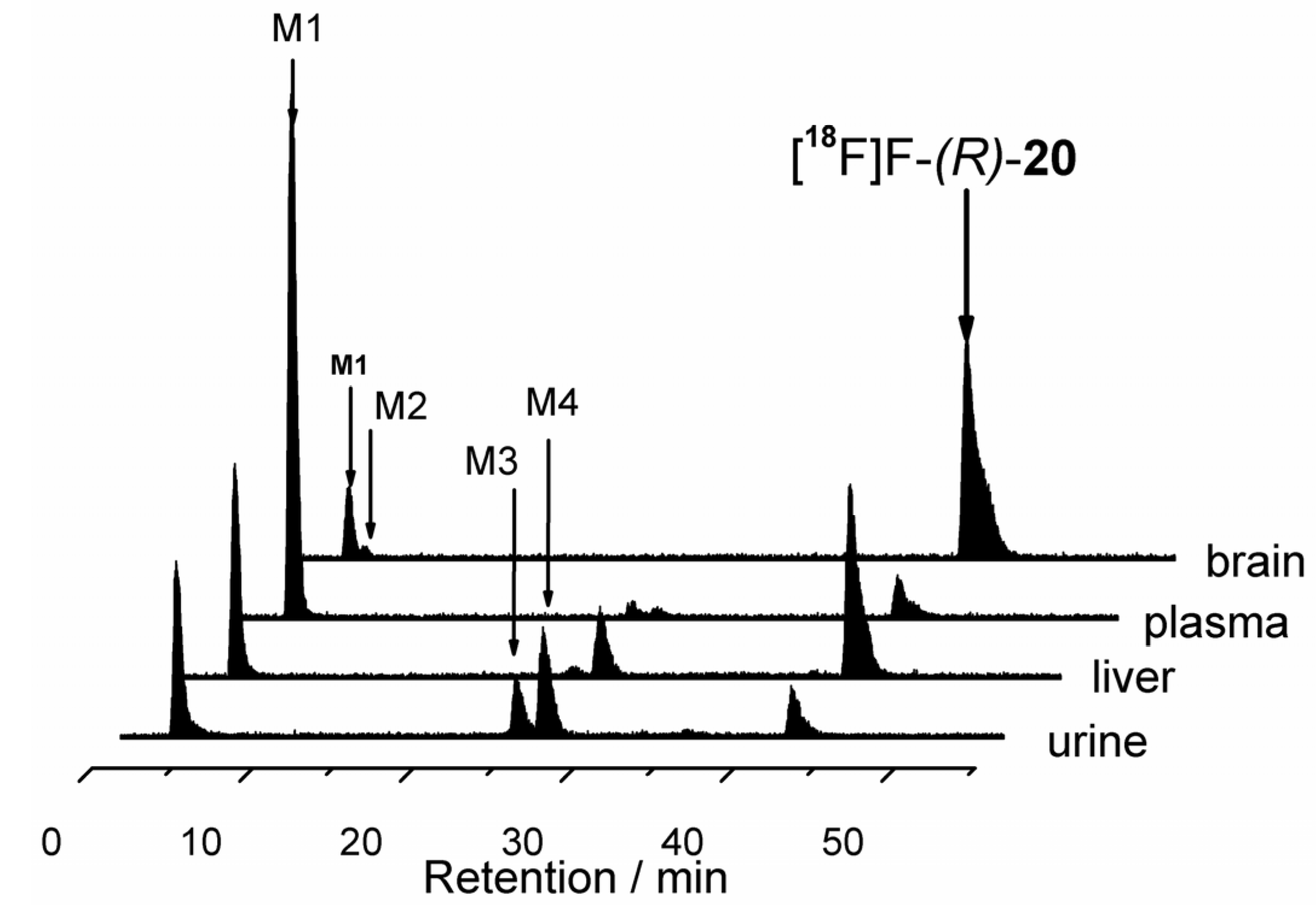
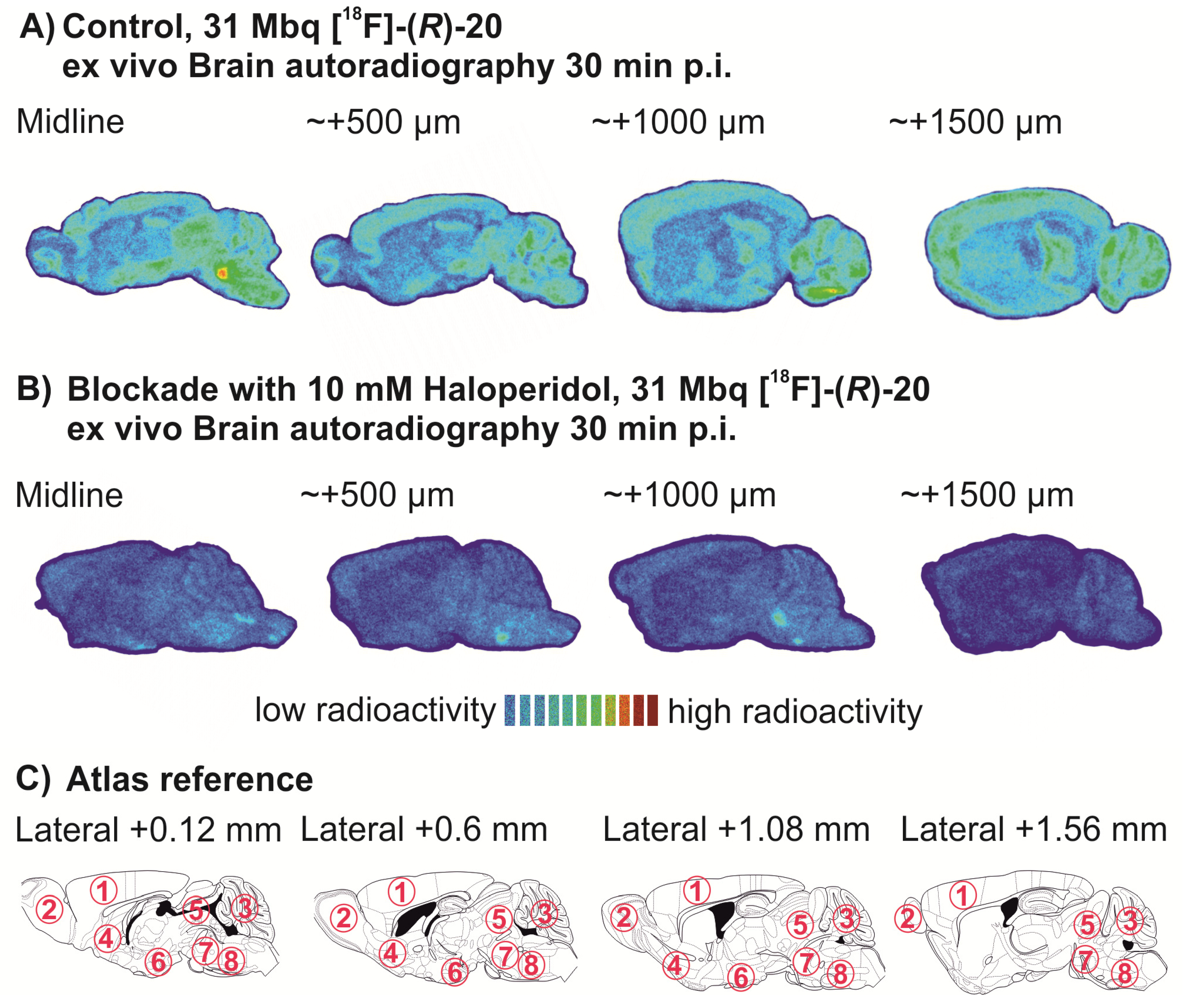
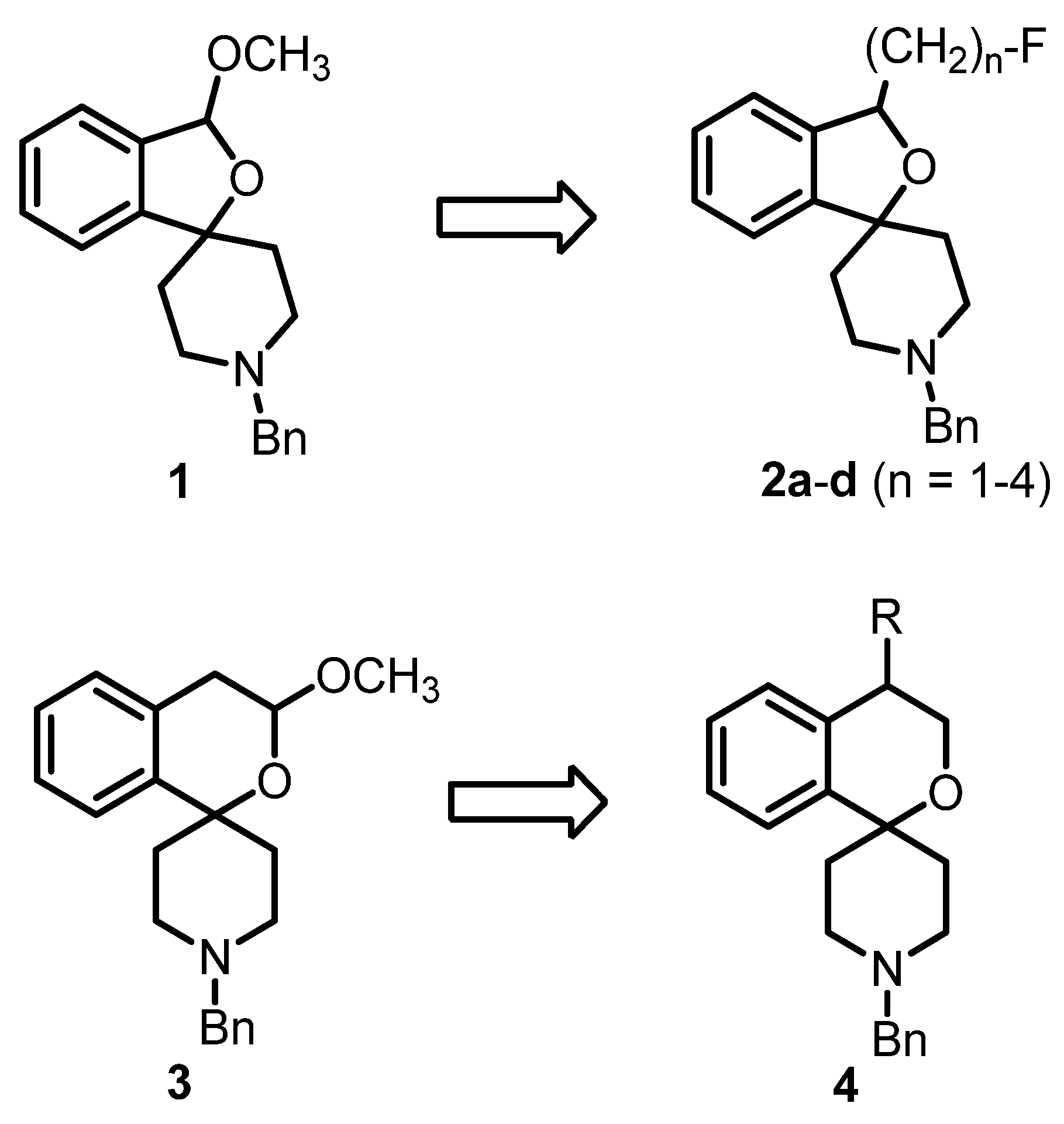
2.1.2. Synthetic Procedures
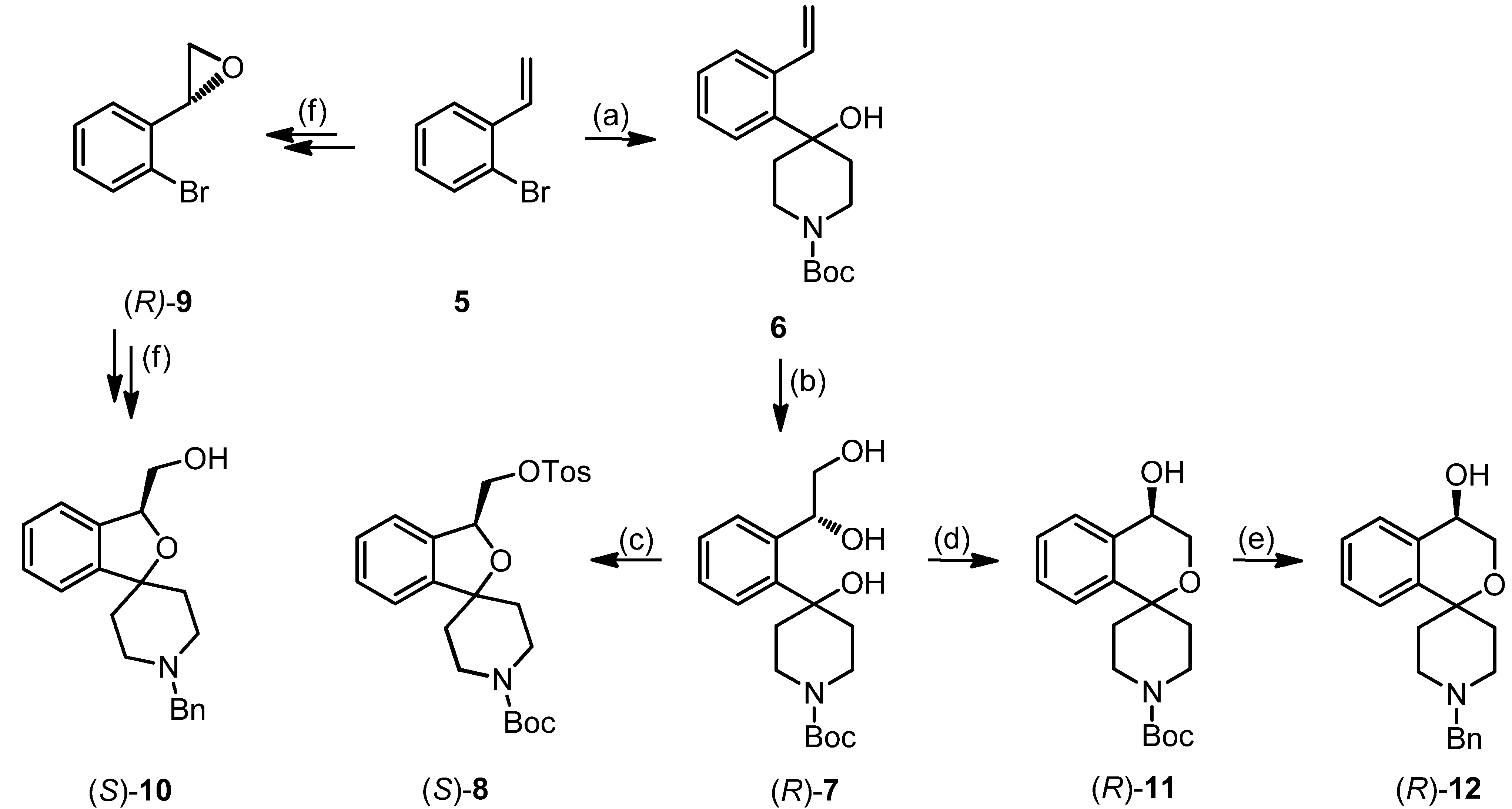
tert-Butyl-4-hydroxy-4-(2-vinylphenyl)piperidine-1-carboxylate (6)
2-Bromostyrene (
5, 3.1 g, 16.9 mmol) was dissolved in THF (125 mL). The solution was cooled to −78 °C under N
2 atmosphere. A solution of
n-butyllithium in hexanes (15 mL, 24 mmol) was added dropwise and the mixture was stirred for 15 min. Then
tert-butyl 4-oxopiperidine-1-carboxylate (4 m, 4.1 g, 20.6 mmol), dissolved in THF (50 mL), was added and the mixture was stirred at −78 °C for 2.5 h. Then the solution was warmed to ambient temperature. A solution of LiBH
4 in THF (5 mL, 20 mmol) was added dropwise and the mixture was stirred for 1 h at ambient temperature. The reaction was stopped by the addition of water and a 1 M aqueous solution of HCl. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 8 cm, h = 16 cm, cyclohexane-ethyl acetate = 9:1, V = 100 mL) to give
6 as a colorless solid (R
f = 0.34, cyclohexane-ethyl acetate = 8:2), mp 104 °C, yield 3.83 g (75%). C
18H
25NO
3 (303.4 g/mol). Purity (HPLC method 1): 99.4%, t
R = 20.4 min. Exact mass (APCI):
m/z = 304.1882 (calcd. 304.1907 for C
18H
26NO
3 [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.47 (s, 9H, CO
2C(C
H3)
3), 1.64 (s br, 1H, O
H), 1.93–2.10 (m, 4H, N(CH
2C
H2)
2), 3.32–3.36 (m, 2H, N(C
H2CH
2)
2), 3.89–4.13 (m, 2H, N(C
H2CH
2)
2), 5.28 (dd,
J = 10.9/1.8 Hz, 1H, HC=C
H2), 5.51 (dd,
J = 17.4/1.8 Hz, 1H, HC=C
H2), 7.24–7.31 (m, 2H, 3-H
arom., 4-H
arom.), 7.34–7.39 (m, 1H, 6-H
arom.)
, 7.45–7.50 (m, 1H, 5-H
arom.)
, 7.65 (dd,
J = 17.4/10.9 Hz, 1H,
HC=CH
2).
13C NMR (CDCl
3): δ (ppm) = 28.6 (3C, CO
2C(
CH
3)
3), 37.4 (br, 2C, N(CH
2CH
2)
2), 39.5 (br, 1C, N(
CH
2CH
2)
2), 40.3 (br, 1C, N(
CH
2CH
2)
2), 72.5 (1C, Ar
COH ), 79.6 (1C, CO
2C(CH
3)
3), 115.7 (1C, HC=
CH
2), 124.9 (1C, C-6
arom.), 127.7 (1C, C-4
arom.), 127.8 (1C, C-3
arom.), 129.1 (1C, C-5
arom.), 137.9 (1C, C-2
arom.), 138.0 (1C, H
C=CH
2), 143.8 (1C, C-1
arom.), 155.0 (1C,
CO
2C(CH
3)
3). FT-IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 3387 (O-H), 2967, 2932, (C-H), 1655 (C=O), 756 (1,2-disubst. arom.).
tert-Butyl (R)-4-[2-(1,2-dihydroxyethyl)phenyl]-4-hydroxypiperidine-1-carboxylate ((R)-7)
AD-mix-β (27.1 g) was added to a mixture of
tert-butyl alcohol (600 mL) and water (600 mL). The mixture was cooled to 0 °C,
6 (5.9 g, 19.5 mmol) was added and the reaction mixture was stirred at 0 °C for 3 d. Then sodium sulfite (29 g) was added and the mixture was allowed to warm to room temperature and stirred for 20 min. Ethyl acetate was added to the reaction mixture, and after separation of the layers, the aqueous layer was extracted with ethyl acetate (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 8 cm, h = 18 cm, cyclohexane-ethyl acetate = 1:2 → ethyl acetate, V = 100 mL) to give
(R)-7 as a colorless solid (R
f = 0.11, cyclohexane-ethyl acetate = 5:5), mp 93 °C, yield 5.4 g (82%). C
18H
27NO
5 (337.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = ‒24.5 (3.5; CH
2Cl
2). Purity (HPLC method 1): 99.5%, t
R = 15.3 min.
tert-Butyl (S)-4-(2-[1,2-dihydroxyethyl)phenyl]-4-hydroxypiperidine-1-carboxylate ((S)-7)
AD-mix-α (15.2 g) was added to a mixture of
tert-butyl alcohol (325 mL) and water (325 mL). The mixture was cooled to 0 °C,
6 (4.5 g, 14.9 mmol) was added and the reaction mixture was stirred overnight at 0 °C. Then methanesulfonamide (1.0 g, 10.5 mmol) was added and the mixture was stirred overnight at ambient temperature. Then sodium sulfite (16.2 g) was added and the mixture stirred for 30 min. Ethyl acetate was added to the reaction mixture, and after separation of the layers, the aqueous layer was extracted with ethyl acetate (3×). The combined organic layers were washed with a 2 M aqueous solution of NaOH, dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 8 cm, h = 15 cm, cyclohexane-ethyl acetate = 1:2 → ethyl acetate, V = 100 mL) to give
(S)-7 as a colorless solid (R
f = 0.11, cyclohexane:ethyl acetate = 5:5), mp 87 °C, yield 2.7 (74%) C
18H
27NO
5 (337.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +25.8 (3.7; CH
2Cl
2). Purity (HPLC method 1): 95.5%, t
R = 15.5 min.
Spectroscopic data for (R)-7 and (S)-7
LC-HRMS:
m/z = 360.1800 (calcd. 360.1781 for C
18H
27NNaO
5 [M+Na]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.47 (s, 9H, CO
2(C
H3)
3), 1.82–2.11 (m, 4H, N(CH
2C
H2)
2), 3.24 (t,
J = 12.6 Hz, 2H, N(C
H2CH
2)
2), 3.78 (dd,
J = 10.9/4.2 Hz, 1H, HOCHC
H2OH), 3.86 (dd,
J = 10.9/7.7 Hz, 1H, HOCHC
H2OH), 3.95–4.05 (m, 2H, N(C
H2CH
2)
2), 5.64 (dd,
J = 7.7/4.2 Hz, 1H, HOC
HCH
2OH), 7.23–7.32 (m, 3H, H
arom.), 7.48–7.52 (m, 1H, H
arom.). Signals for the OH protons are not visible in the spectrum.
13C-NMR (CDCl
3): δ (ppm) = 28.6 (3C, CO
2C(
CH
3)
3), 38.3 (br, 1C, N(CH
2CH
2)
2), 38.6 (br, 1C, N(CH
2CH
2)
2), 39.4 (br, 1C, N(
CH
2CH
2)
2), 40.0 (br, 1C, N(
CH
2CH
2)
2), 68.0 (1C, HOCH
CH
2OH), 72.1 (1C, HO
CHCH
2OH), 72.7 (1C, Ar
COH), 79.8 (1C, CO
2C(CH
3)
3), 125.6 (1C, C
arom.), 127.8 (1C, C
arom.), 127.9 (1C, C
arom.), 129.1 (1C, C
arom.), 139.5 (1C, C
arom.), 145.0 (1C, C
arom.), 155.1 (1C,
CO
2C(CH
3)
3). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 3387 (O-H), 2974, 2925, (C-H), 1663 (C=O), 1246, 1161 (C-O-C ester), 756 (1,2-disubst. arom.).
tert-Butyl (S)-3-[(tosyloxy)methyl]-3H-spiro[[2]benzofuran-1,4′-piperidine]-1′-carboxylate ((S)-8)
(R)-7 (98 mg, 0.29 mmol) was dissolved in CH
2Cl
2 (10 mL). 4-Dimethylaminopyridine (12 mg, 0.10 mmol), triethylamine (210 μL, 1.5 mmol) and 4-toulenesulfonyl chloride (110 mg, 0.58 mmol) were added and the mixture was stirred for 3 h at ambient temperature. Then water was added and after separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 2 cm, h = 16 cm, cyclohexane-ethyl acetate = 9:1, V = 10 mL) to give
(S)-8 as a colorless oil (R
f = 0.28, cyclohexane-ethyl acetate = 8:2), mp 124 °C, yield 69 mg (50%). C
25H
31NO
6S (473.6 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +19.2 (7.6; CH
2Cl
2). Purity (HPLC method 1): 98.8%, t
R = 22.8 min. Exact mass (APCI):
m/z = 474.1969 (calcd. 474.1945 for C
25H
32NO
6S [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.48 (s, 9H, CO
2C(C
H3)
3), 1.51–1.70 (m, 2H, N(CH
2C
H2)
2), 1.70 (td,
J = 13.1/4.8 Hz, 1H, N(CH
2C
H2)
2), 1.82 (td,
J = 13.1/4.8 Hz, 1H, N(CH
2C
H2)
2), 2.44 (s, 3H, C
H3), 3.02 (td,
J = 12.9/2.9 Hz, 1H, N(C
H2CH
2)
2), 3.12 (td,
J = 12.9/2.9 Hz, 1H, N(C
H2CH
2)
2), 3.95–4.07 (m, 2H, N(C
H2CH
2)
2), 4.15 (dd,
J = 10.2/5.3 Hz, 1H, C
H2OTos), 4.24 (dd,
J = 10.2/4.1 Hz, 1H, C
H2OTos), 5.37 (t,
J = 4.7 Hz 1H, ArC
HO), 7.04–7.08 (m, 1H, H
arom.), 7.12–7.17 (m, 1H, H
arom.), 7.24–7.35 (m, 4H, 3-H
tosyl, 5-H
tosyl, H
arom. (2H)), 7.71–7.75 (m, 2H, 2-H
tosyl, 6-H
tosyl).
13C-NMR (CDCl
3): δ (ppm) = 21.8 (1C,
CH
3), 28.6 (3C, CO
2C(
CH
3)
3), 37.2 (1C, N(CH
2CH
2)
2), 37.9 (1C, N(CH
2CH
2)
2), 40.4 (1C, N(
CH
2CH
2)
2), 40.6 (1C, N(
CH
2CH
2)
2), 72.2 (1C,
CH
2OTos), 79.4 (1C, Ar
CHO), 79.6 (1C, CO
2C(CH
3)
3), 85.4 (1C, Ar
CO), 121.1 (1C, C
arom.), 122.0 (1C, C
arom.), 128.1 (2C, C-2
tosyl, C-6
tosyl), 128.3 (1C, C
arom.), 128.8 (1C, C
arom.), 130.0 (2C, C-3
tosyl, C-5
tosyl), 133.0 (1C, C
arom.), 136.9 (1C, C
arom.), 145.1 (1C, C
arom.), 145.8 (1C, C
arom.), 155.1 (1C,
CO
2C(CH
3)
3). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 2978, 2870 (C-H), 1686 (C=O), 1362 (O=S=O), 1234, 1173 (C-O-C, ester), 1069 (C-O-C, ether), 768 (1,2-disubst. arom.).
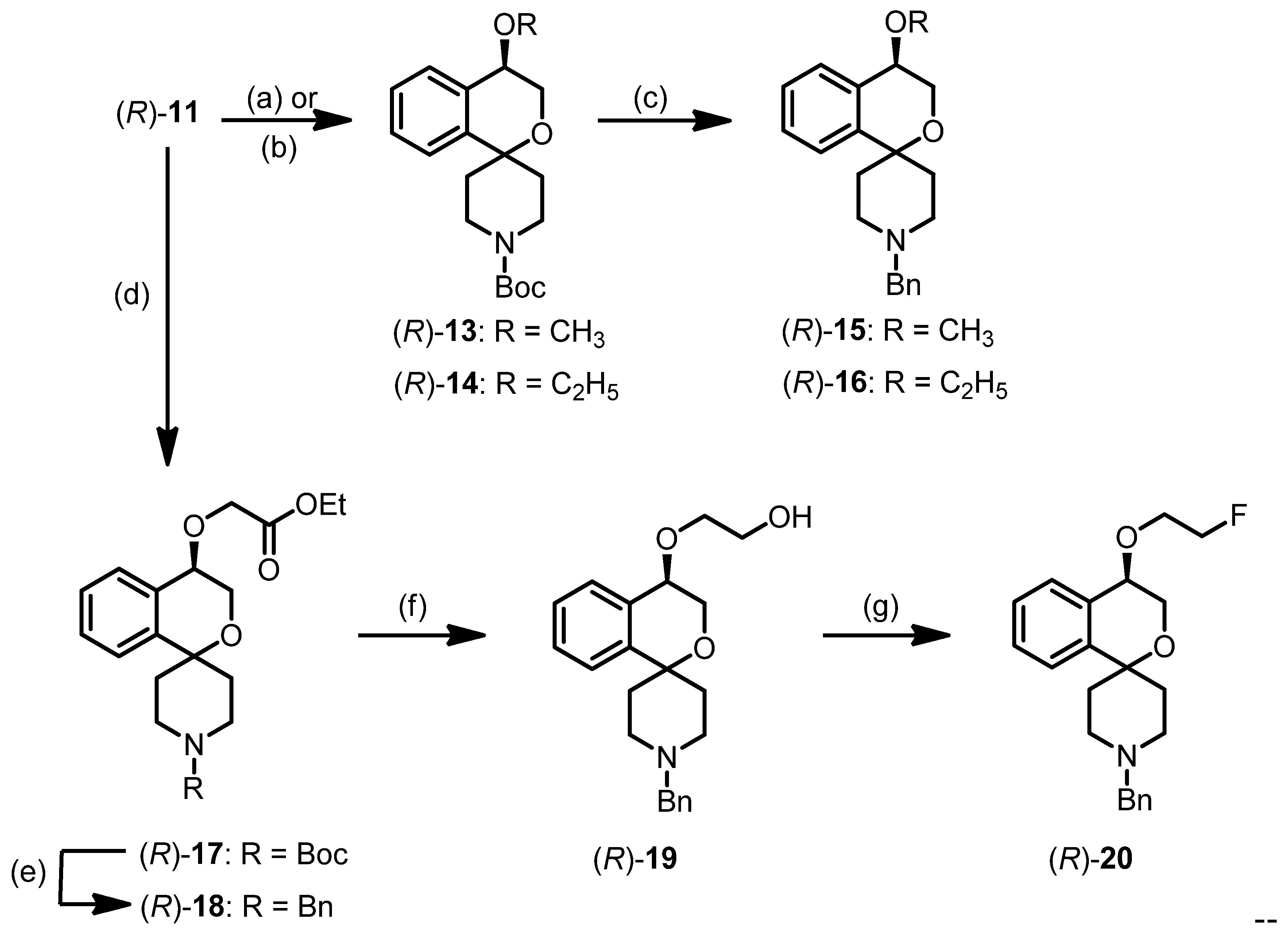
tert-Butyl (R)-4-hydroxy-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidine]-1′-carboxylate ((R)-11)
(R)-7 (908 mg, 2.7 mmol) was dissolved in THF (25 mL). Dibutyltin oxide (75 mg, 0.30 mmol), triethylamine (744 μL, 5.4 mmol) and toluene-4-sulfonyl chloride (1.0 g, 5.3 mmol) were added and the mixture was stirred for 3 h at ambient temperature. Then water and CH
2Cl
2 were added. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 5 cm, h = 15 cm, cyclohexane-ethyl acetate = 3:1, V = 30 mL) to give
(R)-11 as a colorless solid (R
f = 0.25, cyclohexane-ethyl acetate = 2:1), mp 161 °C, yield 538 mg (62%). C
18H
25NO
4 (319.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = −4.9 (11.0; CH
2Cl
2). Purity (HPLC method 1): 96.0%, t
R = 18.6 min. Enantiomeric ratio (HPLC method 2, Daicel Chiralpak AD-H, 5 μm, 250 mm/4.6 mm, isohexane:methanol = 95:5, flow rate: 1.0 mL/min, injection volume: 10 μL): (
R):(
S) = 92.5:7.5, t
R = 11.4 min.
tert-Butyl (S)-4-hydroxy-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidine]-1′-carboxylate ((S)-11)
(S)-7 (996 mg, 3.0 mmol) was dissolved in THF (15 mL). Dibutyltin oxide (86 mg, 0.35 mmol), triethylamine (2.0 mL, 14.8 mmol) and toluene-4-sulfonyl chloride (2.1 g, 6.3 mmol) were added and the mixture was stirred for 3 days at ambient temperature. Then water and CH
2Cl
2 were added. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 5 cm, h = 15 cm, cyclohexane-ethyl acetate = 3:1, V = 65 mL) to give
(S)-11 as a colorless solid (R
f = 0.25, cyclohexane-ethyl acetate = 2:1), mp 158 °C, yield 447 mg (47%). C
18H
25NO
4 (319.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +5.2 (4.4; CH
2Cl
2). Purity (HPLC method 1): 98.3%, t
R = 18.3 min. Enantiomeric ratio (HPLC method 2, Daicel Chiralpak AD-H, 5 μm, 250 mm/4.6 mm, isohexane:methanol = 95:5, flow rate: 1.0 mL/min, injection volume: 10 μL): (
R):(
S) = 11.4:88.6, t
R = 20.7 min.
Spectroscopic data for (R)-11 and (S)-11
Exact mass (APCI):
m/z = 320.1894 (calcd. 320.1856 for C
18H
26NO
4 [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.49 (s, 9H, CO
2(C
H3)
3), 1.69–1.83 (m, 2H, N(CH
2C
H2)
2), 1.90–2.05 (m, 2H, N(CH
2C
H2)
2), 3.11 (t,
J = 13.0 Hz, 1H, N(C
H2CH
2)
2), 3.22 (t,
J = 13.0 Hz, 1H, N(C
H2CH
2)
2), 3.92 (dd,
J = 12.1/3.3 Hz, 1H, HOCHC
H2O), 3.98 (dd,
J = 12.1/2.7 Hz, 1H, HOCHC
H2O), 3.99–4.07 (m, 2H, N(C
H2CH
2)
2), 4.54 (t,
J = 3.0 Hz, 1H, HOC
HCH
2O), 7.10 (dd,
J = 7.4/1.6 Hz, 1H, 8-H
arom.), 7.25–7.34 (m, 2H, 6-H
arom., 7-H
arom.), 7.42 (dd,
J = 7.2/1.8 Hz, 1H, 5-H
arom.). A signal for the OH proton is not visible in the spectrum.
13C-NMR (CDCl
3): δ (ppm) = 28.6 (3C, CO
2C(
CH
3)
3), 34.3 (br, 1C, N(CH
2CH
2)
2), 37.6 (br, 1C, N(CH
2CH
2)
2), 39.5 (br, 1C, N(
CH
2CH
2)
2), 40.1 (br, 1C, N(
CH
2CH
2)
2), 64.8 (1C, HOCH
CH
2O), 66.0 (1C, HO
CHCH
2O), 73.8 (1C, Ar
CO), 79.7 (1C, CO
2C(CH
3)
3), 125.2 (1C, C-8
arom.), 127.3 (1C, C-6
arom.), 128.6 (1C, C-7
arom.), 129.1 (1C, C-5
arom.), 135.3 (1C, C-8a
arom.), 141.2 (1C, C-4a
arom.), 155.1 (1C,
CO
2C(CH
3)
3). IR (neat):
![Pharmaceuticals 07 00078 i002]()
[cm
−1] = 3314 (O-H), 2974, 2928 (C-H), 1686 (C=O), 1169 (C-O-C ether), 768 (1,2-disubst. arom.).
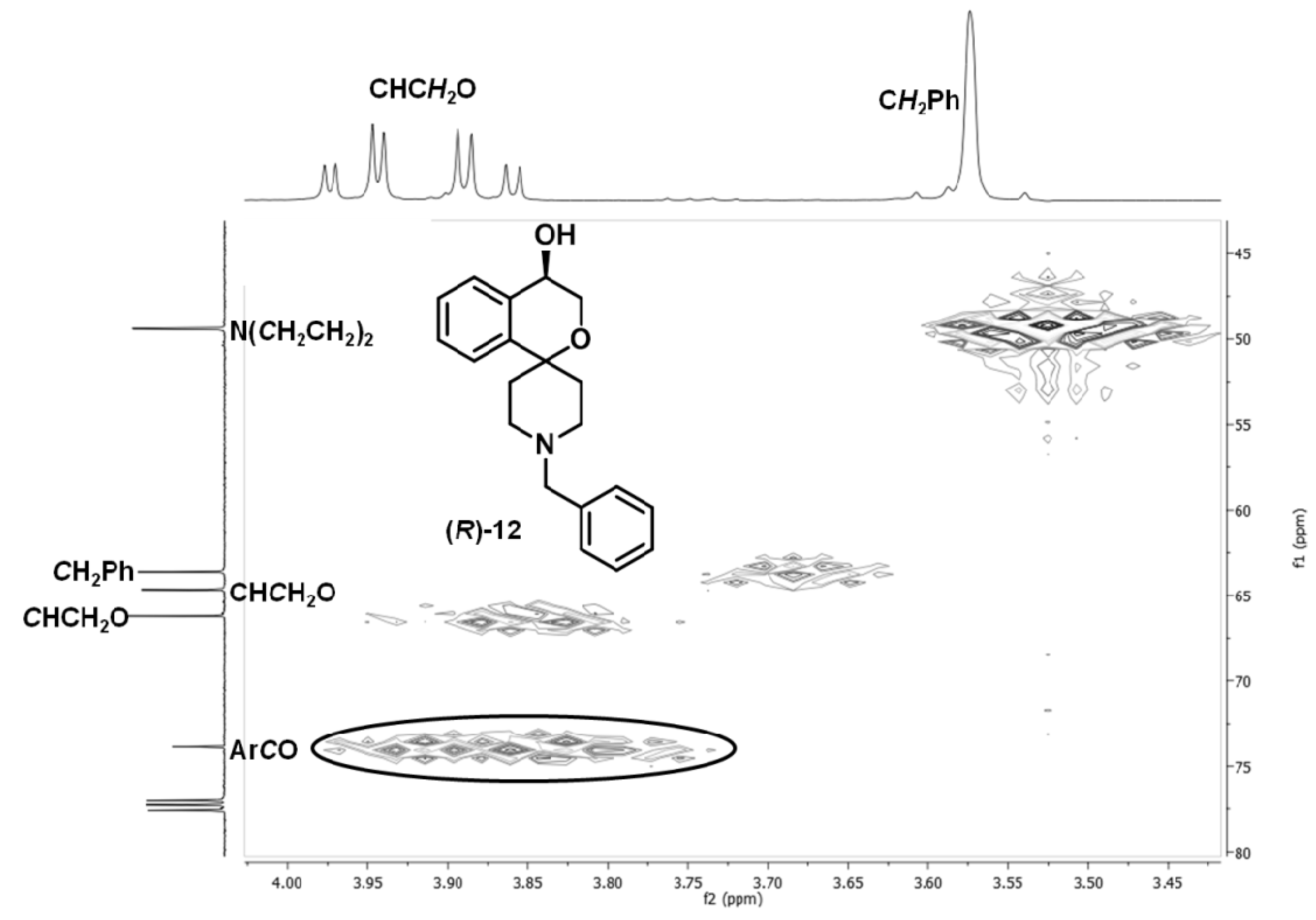
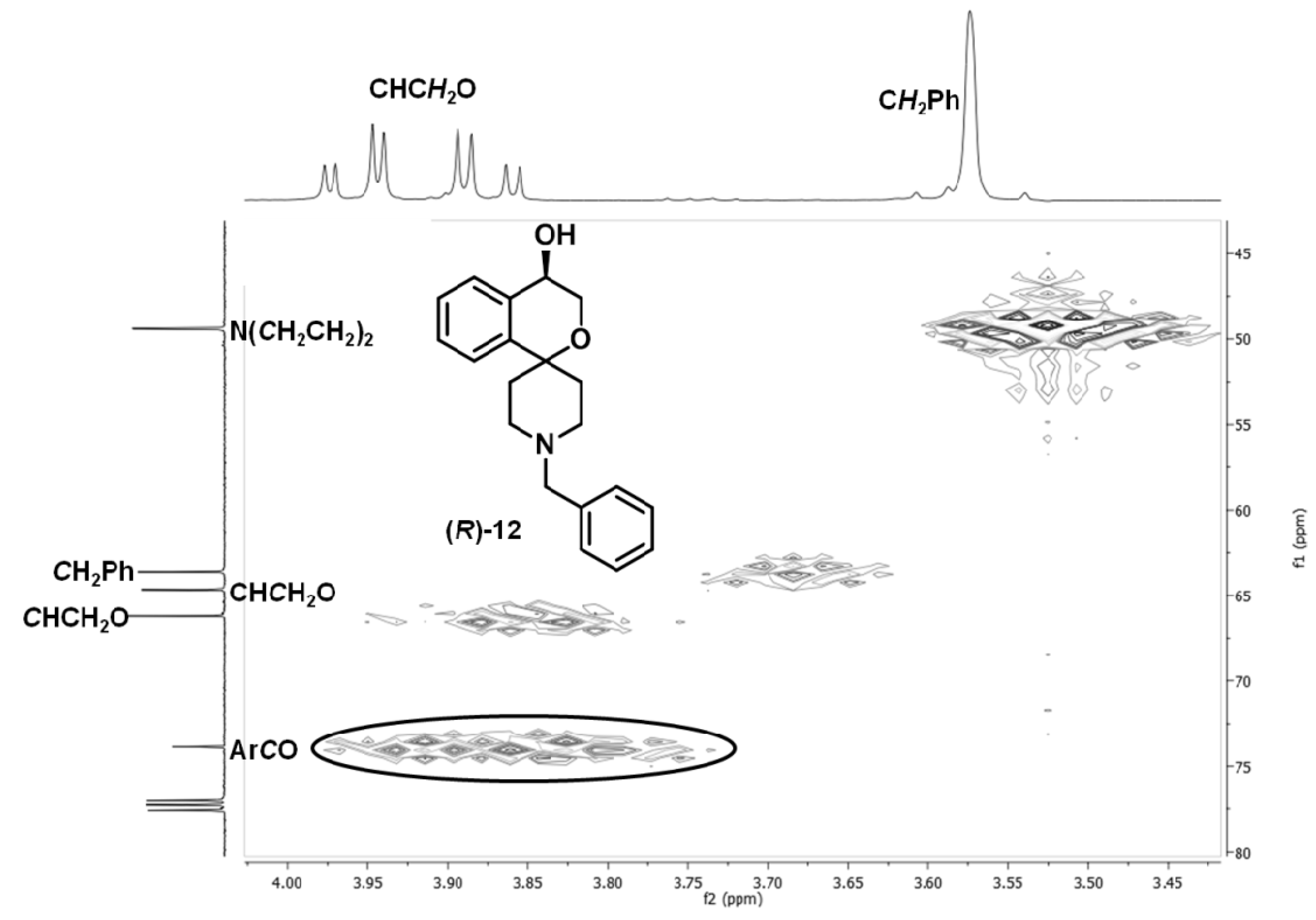
(R)-1′-Benzyl-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidin]-4-ol ((R)-12)
(R)-11 (105 mg, 0.33 mmol) was dissolved in CH
2Cl
2 (4 mL). The solution was cooled to 0 °C. Then trifluoroacetic acid (200 μL) was added and the mixture was stirred for 3.5 h at 0 °C. Then a 2
m aqueous solution of sodium hydroxide (4 mL) was added, and after separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The residue was dissolved in CH
2Cl
2 (5 mL), benzaldehyde (35 μL, 0.35 mmol) and sodium triacetoxyborohydride (85 mg, 0.40 mmol) were added and the mixture was stirred overnight at ambient temperature. The reaction was stopped by the addition of a 2 M aqueous solution of sodium hydroxide, and after separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (4×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 1.5 cm, h = 16 cm, cyclohexane-ethyl acetate = 5:1 + 1%
N,N-dimethylethylamine, V = 5 mL) to give
(R)-12 as a colorless solid (R
f = 0.14, cyclohexane-ethyl acetate = 5:5), mp 55 °C), yield 57 mg (56%). C
20H
23NO
2 (309.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = −8.4 (2.3; CH
2Cl
2). Purity (HPLC method 1): 95.3%, t
R = 13.5 min. Enantiomeric ratio (HPLC method 2, Daicel Chiralpak AD-H, 5 μm, 250 mm/4.6 mm, isohexane-isopropanol = 95:5, flow rate: 1.0 mL/min, injection volume: 10 μL): (
R):(
S) = 96.1:3.9, t
R = 9.7 min).
(S)-1′-Benzyl-3,4-dihydrospiro[2-benzopyran-1,4′-piperidin]-4-ol ((S)-12)
(R)-11 (56 mg, 0.18 mmol) was dissolved in CH
2Cl
2 (10 mL). Trifluoroacetic acid (200 μL) was added and the mixture was stirred overnight at ambient temperature. Then water was added, and after separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The residue was dissolved in CH
2Cl
2 (10 mL), benzaldehyde (50 μL, 0.45 mmol) and sodium triacetoxyborohydride (50 mg, 0.24 mmol) were added and the mixture was stirred for 5.5 h at ambient temperature. Then benzaldehyde (50 μL, 0.35 mmol) and sodium triacetoxyborohydride (60 mg, 0.28 mmol) were added and the mixture was stirred overnight at ambient temperature). The reaction was stopped by the addition of a 2 M aqueous solution of sodium hydroxide, and after separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 1 cm, h = 15 cm, cyclohexane-ethyl acetate = 5:1 + 1%
N,N-dimethylethylamine, V = 5 mL) to give
(S)-12 as a colorless solid (R
f = 0.14, cyclohexane-ethyl acetate = 5:5) mp 53 °C, yield 12 mg (22%). C
20H
23NO
2 (309.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +8.8 (2.2; CH
2Cl
2). Purity (HPLC method 1): 98.3%, t
R = 13.4 min. Enantiomeric ratio (HPLC method 2, Daicel Chiralpak AD-H, 5 μm, 250 mm/4.6 mm, isohexane:isopropanol = 95:5, flow rate: 1.0 mL/min, injection volume: 10 μL): (
R):(
S) = 11.9:88.1, t
R = 12.9 min.
Spectroscopic data for (R)-12 and (S)-12
Exact mass (APCI):
m/z = 310.1802 (calcd. 310.1802 for C
20H
24NO
2 [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.78–1.98 (m, 3H, N(CH
2C
H2)
2), 2.18 (td,
J = 13.1/4.6 Hz, 1H, N(CH
2C
H2)
2), 2.39 (dd,
J = 11.9/3.0 Hz, 1H, N(C
H2CH
2)
2), 2.46–2.54 (m, 1H, N(C
H2CH
2)
2), 2.75 (t,
J = 13.1/Hz, 2H, N(C
H2CH
2)
2), 3.58 (s, 2H, NC
H2Ph), 3.89 (dd,
J = 12.1/3.3 Hz, 1H, CHC
H2O), 3.97 (dd,
J = 12.1/2.6 Hz, 1H, CHC
H2O), 4.51 (t,
J = 2.9 Hz, 1H, C
HCH
2O), 7.20–7.41 (m, 9H, H
arom.). A signal for the OH proton is not visible in the spectrum.
13C-NMR (CDCl
3): δ (ppm) = 34.7 (1C, N(CH
2CH
2)
2), 38.0 (1C, N(CH
2CH
2)
2), 49.3 (1C, N(
CH
2CH
2)
2), 49.3 (1C, N(
CH
2CH
2)
2), 63.5 (1C, N
CH
2Ph), 64.5 (1C, CH
CH
2O ), 66.1 (1C,
CHCH
2O), 73.9 (1C, Ar
CO), 125.3 (1C, C
arom.), 127.0 (1C, C
arom.), 127.1 (1C, C
arom.), 128.3 (2C, C
arom.), 128.5 (1C, C
arom.), 128.9 (1C, C
arom.), 129.4 (2C, C
arom.), 135.5 (1C, C
arom.), 138.6 (1C, C
arom.), 141.8 (1C, C
arom.). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 3329 (O-H), 2924, 2817 (C-H), 1072 (C-O-C), 733 (1,2-disubst. arom.), 698 (monosubst. arom.).
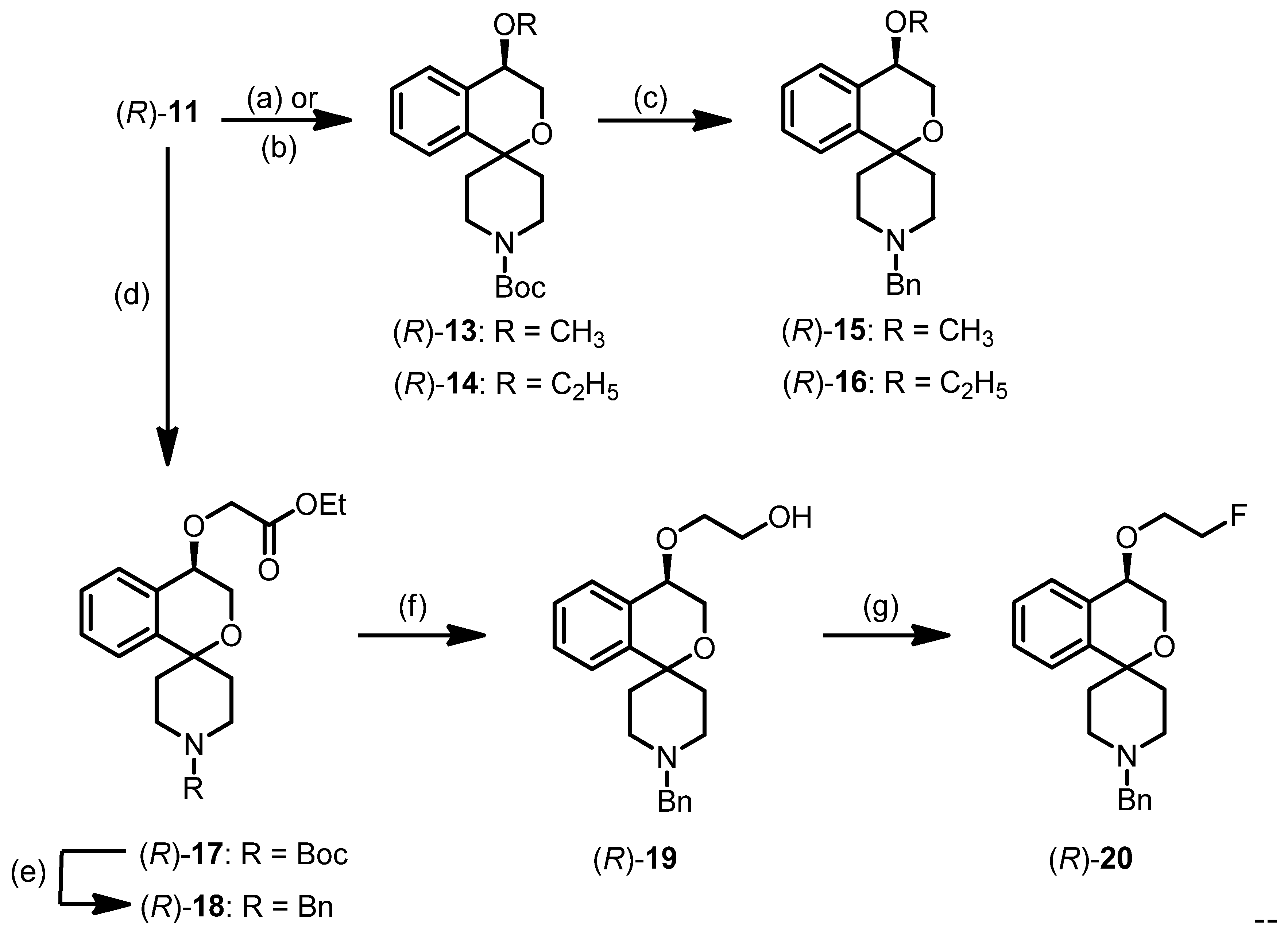
tert-Butyl (R)-4-methoxy-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidine]-1′-carboxylate ((R)-13)
(R)-10 (450 mg, 1.4 mmol) was dissolved in THF (12 mL). NaH (60% dispersion in paraffin liquid, 112 mg, 2.8 mmol) was added and the mixture was stirred for 1 h at ambient temperature. Then iodomethane (176 μL, 2.8 mmol) was added dropwise and the mixture was stirred for 1 h at ambient temperature. The solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 2.5 cm, h = 16.5 cm, cyclohexane-ethyl acetate = 9:1, V = 10 mL) to give
(R)-13 as a colorless oil (R
f = 0.28, cyclohexane-ethyl acetate = 8:2), yield 465 mg (99%). C
19H
27NO
4 (333.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
= −8.0 (4.4; CH
2Cl
2). Purity (HPLC method 1): 96.6%, t
R = 20.4 min.
tert-Butyl (S)-4-methoxy-3,4-dihydrospiro[2-benzopyran-1,4′-piperidine]-1′-carboxylate ((S)-13)
(S)-10 (180 mg, 0.56 mmol) was dissolved in THF (2.5 mL). NaH (60% dispersion in paraffin liquid, 50 mg, 1.3 mmol) was added and the mixture was stirred for 1 h at ambient temperature. Then iodomethane (77 μL, 1.2 mmol) was added dropwise and the mixture was stirred overnight at ambient temperature. The solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 2 cm, h = 15 cm, cyclohexane:ethyl acetate = 9:1, V = 10 mL) to give
(S)-13 as a pale yellow oil (R
f = 0.28, cyclohexane:ethyl acetate = 8:2), yield 128 mg (69%). C
19H
27NO
4 (333.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +7.4 (7.9; CH
2Cl
2). Purity (HPLC method 1): 97.7%, t
R = 20.4 min.
Spectroscopic data for (R)-13 and (S)-13
Exact mass (APCI):
m/z = 334.2009 (calcd. 334.2013 for C
19H
28NO
4 [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.49 (s, 9H, CO
2(C
H3)
3), 1.75 (td,
J = 13.2/4.9 Hz, 1H, N(CH
2C
H2)
2), 1.84–1.99 (m, 3H, N(CH
2C
H2)
2), 3.03–3.28 (m, 2H, N(C
H2CH
2)
2), 3.50 (s, 3H, OC
H3), 3.93–4.06 (m, 4H, N(C
H2CH
2)
2 (2), CHC
H2O (2)), 4.19 (t,
J = 3.4 Hz, 1H, C
HCH
2O), 7.11 (dd,
J = 7.7/1.4 Hz, 1H, H
arom.), 7.25 (td,
J = 7.4/1.4 Hz, 1H, H
arom.), 7.30 (td,
J = 7.4/1.6 Hz, 1H, H
arom.), 7.37 (dd,
J = 7.4/1.6 Hz, 1H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 28.6 (3C, CO
2C(
CH
3)
3), 34.9 (br, 1C, N(CH
2CH
2)
2), 37.0 (br, 1C, N(CH
2CH
2)
2), 39.4 (br, 1C, N(
CH
2CH
2)
2), 40.2 (br, 1C, N(
CH
2CH
2)
2), 56.9 (1C, O
CH
3), 61.6 (1C, CH
CH
2O), 73.5 (1C, Ar
CO), 74.0 (1C,
CHCH
2O), 79.5 (1C, CO
2C(CH
3)
3), 125.1 (1C, C
arom.), 126.7 (1C, C
arom.), 128.4 (1C, C
arom.), 129.1 (1C, C
arom.), 132.8 (1C, C
arom.), 141.8 (1C, C
arom.), 155.0 (1C,
CO
2C(CH
3)
3). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 2970, 2928 (
C-H), 1686 (
C=O), 1084 (
C-O-C ether), 756 (1,2-disubst. arom.).
tert-Butyl (R)-4-ethoxy-3,4-dihyrospiro[[2]benzopyran-1,4′-piperidine]-1′-carboxylate ((R)-14)
(R)-10 (210 mg, 0.66 mmol) was dissolved in THF (15 mL). NaH (60% dispersion in paraffin liquid, 80 mg, 2.0 mmol) was added and the mixture was stirred for 1 h at ambient temperature. Then iodoethane (0.53 mL, 6.6 mmol) was added dropwise and the mixture was stirred for 2.5 h at ambient temperature. A 1 m solution of lithium bis(trimethylsilyl)amide (4.5 mL) was added and the mixture was heated to reflux overnight. The mixture was allowed to cool to ambient temperature and stirred overnight. Then water and CH
2Cl
2 were added. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 2 cm, h = 15 cm, cyclohexane-ethyl acetate = 9:1, V = 10 mL) to give
(R)-14 as a pale yellow oil (R
f = 0.15, cyclo-hexane-ethyl acetate = 9:1), yield 66 mg (29%). C
20H
29NO
4 (347.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = −2.2 (3.2; CH
2Cl
2). Purity (HPLC method 1): 96.3%, t
R = 21.3 min.
tert-Butyl (S)-4-ethoxy-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidine]-1′-carboxylate ((S)-14)
(S)-10 (200 mg, 0.63 mmol) was dissolved in THF (5 mL). A 1 m solution of lithium bis(trimethylsilyl)amide (6.3 mL) was added and the mixture was stirred for 1 h at ambient temperature. Then iodoethane (500 μL, 6.3 mmol) was added dropwise and the mixture was stirred for 16 h at ambient temperature. NaH (60% dispersion in paraffin liquid, 250 mg, 6.3 mmol) and iodoethane (500 μL, 6.3 mmol) were added and the mixture was heated to reflux overnight. The mixture was allowed to cool to ambient temperature and stirred for 3 days. Then water was added. After separation of the layers, the aqueous layer was extracted with ethyl acetate (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography three times (1. Ø = 2 cm, h = 15 cm, cyclohexane-ethyl acetate = 9:1, V = 10 mL; 2. Ø = 1.5 cm, h = 15 cm, cyclohexane-ethyl acetate = 9:1, V = 5 mL; 3. Ø = 1.5 cm, h = 15 cm, cyclohexane-ethyl acetate = 95:5, V = 5 mL) to give
(S)-14 as a pale yellow oil (R
f = 0.15, cyclohexane-ethyl acetate = 9:1), yield 110 mg (50%). C
20H
29NO
4 (347.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +2.4 (3.9; CH
2Cl
2). Purity (HPLC method 1): 98.4%, t
R = 21.5 min.
Spectroscopic data for (R)-14 and (S)-14
Exact mass (APCI):
m/z = 348.2199 (calcd. 348.2169 for C
20H
30NO
4 [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.28 (t,
J = 7.0 Hz, 3H, OCH
2C
H3), 1.49 (s, 9H, CO
2(C
H3)
3), 1.76–1.86 (m, 2H, N(CH
2C
H2)
2), 1.86–1.96 (m, 2H, N(CH
2C
H2)
2), 3.04–3.26 (m, 2H, N(C
H2CH
2)
2), 3.64–3.78 (m, 2H, OC
H2CH
3), 3.86–4.08 (m, 2H, N(C
H2CH
2)
2), 3.89 (dd,
J = 12.0/5.4 Hz, 1H, CHC
H2O), 4.00 (dd,
J = 12.0/3.7 Hz, 1H, CHC
H2O), 4.35 (t,
J = 4.5 Hz, 1H, C
HCH
2O), 7.08 (dd,
J = 7.2/2.0 Hz, 1H, H
arom.), 7.20–7.32 (m, 2H, H
arom.), 7.42 (dd,
J = 6.9/2.3 Hz, 1H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 15.9 (1C, OCH
2CH
3), 28.7 (3C, CO
2C(
CH
3)
3), 35.8 (br, 1C, N(CH
2CH
2)
2), 36.4 (br, 1C, N(CH
2CH
2)
2), 39.5 (br, 1C, N(
CH
2CH
2)
2), 40.3 (br, 1C, N(
CH
2CH
2)
2), 62.1 (1C, CH
CH
2O), 64.9 (1C, O
CH
2CH
3), 72.4 (1C,
CHCH
2O), 73.7 (1C, CO
2C(CH
3)
3), 79.6 (1C, Ar
CO), 125.1 (1C, C
arom.), 126.8 (1C, C
arom.), 128.1 (1C, C
arom.), 128.5 (1C, C
arom.), 134.0 (1C, C
arom.), 141.8 (1C, C
arom.), 155.1 (1C,
CO
2C(CH
3)
3). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 2970, 2928 (C-H), 1690 (C=O), 1092 (C-O-C ether), 756 (1,2-disubst. arom.).
(R)-1′-Benzyl-4-methoxy-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidine] ((R)-15)
(
R)-
13 (360 mg, 1.1 mmol) was dissolved in CH
2Cl
2 (5 mL). The solution was cooled to 0 °C. Trifluoroacetic acid (0.7 mL) was added and the mixture was stirred for 2 h at 0 °C. Then a 2 M aqueous solution of NaOH was added. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The residue was dissolved in CH
2Cl
2 (5 mL). Benzaldehyde (30 μL, 0.30 mmol) and sodium triacetoxyborohydride (76 mg, 0.36 mmol) were added and the mixture was stirred for 26 h at ambient temperature. Then a 2 M aqueous solution of NaOH (3 mL) and water (3 mL) were added. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 0.75 cm, h = 15 cm, cyclohexane-ethyl acetate = 4:1, V = 5 mL) to give
(R)-15 as a colorless oil (R
f = 0.27, cyclohexane-ethyl acetate = 5:5), yield 31 mg (9%). C
21H
25NO
2 (323.4 G/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = −8.6 (2.8; CH
2Cl
2). Purity (HPLC method 1): 98.2%, t
R = 15.8 min. Enantiomeric ratio (HPLC method 2, Daicel Chiralpak IB, 5 μm, 250 mm/4.6 mm, isohexane-methanol = 97:3, flow rate: 1.0 mL/min, injection volume: 5 μL): (
R):(
S) = 94.8:5.2, t
R = 7.2 min.
(S)-1′-Benzyl-4-methoxy-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidine] ((S)-15)
(S)-13 (50 mg, 0.15 mmol) was dissolved in CH
2Cl
2 (5 mL). Trifluoroacetic acid (200 μL) was added and the mixture was stirred for 4.5 h at ambient temperature. Then a 2 M aqueous solution of NaOH was added. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The residue was dissolved in CH
2Cl
2 (10 mL). Benzaldehyde (70 μL, 0.69 mmol) and sodium triacetoxyborohydride (96 mg, 0.45 mmol) were added and the mixture was stirred overnight at ambient temperature. The reaction was stopped by the addition of a 2 M aqueous solution of NaOH. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography twice (1. Ø = 1.5 cm, h = 16 cm, cyclohexane-ethyl acetate = 4:1, V = 5 mL; 2. Ø = 1.5 cm, h = 15 cm, cyclohexane-ethyl acetate = 6:1, V = 5 mL) to give
(S)-15 as a yellowish oil (R
f = 0.27, cyclohexane-ethyl acetate = 5:5), yield 39 mg (80%). C
21H
25NO
2 (323.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +7.7 (8.3; CH
2Cl
2). Purity (HPLC method 1): 97.3%, t
R = 15.6 min. Enantiomeric ratio (HPLC method 2, Daicel Chiralpak IB, 5 μm, 250 mm/4.6 mm, isohexane:methanol = 97:3, flow rate: 1.0 mL/min, injection volume: 5 μL): (
R):(
S) = 9.0:91.0, t
R = 8.6 min.
Spectroscopic data for (R)-15 and (S)-15
Exact mass (APCI):
m/z = 324.1950 (calcd. 324.1958 for C
21H
26NO
2 [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.87–1.95 (m, 3H, N(CH
2C
H2)
2), 2.13 (td,
J = 13.0/4.6 Hz, 1H, N(CH
2C
H2)
2), 2.36–2.44 (m, 1H, N(C
H2CH
2)
2), 2.51 (td,
J = 13.0/2.5 Hz, 1H, N(C
H2CH
2)
2), 2.70–2.79 (m, 2H, N(C
H2CH
2)
2), 3.50 (s, 3H, OC
H3), 3.56 (d,
J = 13.0 Hz, 1H, NC
H2Ph), 3.60 (d,
J = 13.0 Hz, 1H, NC
H2Ph), 3.93–4.06 (d,
J = 3.6 Hz, 2H, CHC
H2O), 4.19 (t,
J = 3.6 Hz, 1H, C
HCH
2O), 7.21–7.39 (m, 9H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 35.5 (1C, N(CH
2CH
2)
2), 37.3 (1C, N(CH
2CH
2)
2), 49.4 (1C, N(
CH
2CH
2)
2), 49.4 (1C, N(
CH
2CH
2)
2), 56.9 (1C, O
CH
3), 61.3 (1C, CH
CH
2O), 63.5 (1C, N
CH
2Ph), 73.5 (1C, Ar
CO), 74.1 (1C,
CHCH
2O), 125.3 (1C, C
arom.), 126.5 (1C, C
arom.), 127.1 (1C, C
arom.), 128.3 (1C, C
arom.), 128.3 (2C, 3-C
benzyl, 5-C
benzyl), 129.0 (1C, 6-C
arom.), 129.4 (2C, 2-C
benzyl, 6-C
benzyl), 133.1 (1C, 2-C
arom.), 138.7 (1C, 1-Cbenzyl), 142.5 (1C, 1-Carom.). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 2924, 2816 (C-H), 1088 (C-O-C), 737 (1,2-disubst. arom.), 737 (monosubst. arom.).
(R)-1′-Benzyl-4-ethoxy-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidine] ((R)-16)
(R)-14 (49 mg, 0.14 mmol) was dissolved in CH
2Cl
2 (10 mL). Trifluoroacetic acid (200 μL) was added and the mixture was stirred for 3 h at ambient temperature. Then a 2 M aqueous solution of NaOH (10 mL) was added. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The residue was dissolved in CH
2Cl
2 (10 mL). Benzaldehyde (60 μL, 0.59 mmol) and (after 45 min) sodium triacetoxyborohydride (181 mg, 0.85 mmol) were added and the mixture was stirred overnight at ambient temperature. The reaction was stopped by the addition of a 2 M aqueous solution of NaOH. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (2×) and ethyl acetate (1×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography twice (1. Ø = 1.5 cm, h = 17 cm, cyclohexane-ethyl acetate = 4:1, V = 5 mL; 2. Ø = 1.5 cm, h = 17 cm, cyclohexane-ethyl acetate = 6:1, V = 5 mL) to give
(R)-16 as a pale yellow oil (R
f = 0.31, cyclohexane-ethyl acetate = 5:5), yield 33 mg (70%). C
22H
27NO
2 (337.5 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = −3.3 (7.7; CH
2Cl
2). Purity (HPLC method 1): 99.1%, t
R = 17.0 min.
(S)-1′-Benzyl-4-ethoxy-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidine] ((S)-16)
(S)-14 (63 mg, 0.18 mmol) was dissolved in CH
2Cl
2 (5 mL). Trifluoroacetic acid (300 μL) was added and the mixture was stirred for 2 h at ambient temperature. Then a 2 M aqueous solution of NaOH was added. After separation of the layers, the aqueous layer was extracted with ethyl acetate (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The residue was dissolved in CH
2Cl
2 (5 mL). Benzaldehyde (40 μL, 0.39 mmol) and after 15 min, sodium triacetoxyborohydride (120 mg, 0.57 mmol) were added and the mixture was stirred at ambient temperature for 8 h. The reaction was stopped by the addition of a 2 M aqueous solution of NaOH. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 1.25 cm, h = 15 cm, cyclohexane-ethyl acetate = 6:1, V = 5 mL) to give (
S)-16 as a pale yellow oil (R
f = 0.31, cyclohexane-ethyl acetate = 5:5), yield 28 mg (46%). C
22H
27NO
2 (337.5 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +2.6 (11.7; CH
2Cl
2). Purity (HPLC method 1): 96.6%, t
R = 17.0 min.
Spectroscopic data for (R)-16 and (S)-16
Exact mass (APCI):
m/z = 338.2130 (calcd. 338.2115 for C
22H
28NO
2 [M+H]
+)
1H-NMR (CDCl
3): δ (ppm) = 1.20 (t,
J = 6.9 Hz, 3H, OCH
2C
H3), 1.74–2.04 (m, 4H, N(CH
2C
H2)
2), 2.28–2.24 (m, 2H, N(C
H2CH
2)
2), 2.62–2.70 (m, 2H, N(C
H2CH
2)
2), 3.50 (s, 2H, NC
H2Ph), 3.58–3.68 (m, 2H, OC
H2CH
3), 3.79 (dd,
J = 11.9/5.4 Hz, 1H, CHC
H2O), 3.93 (dd,
J = 11.9/3.9 Hz, 1H, CHC
H2O), 4.28 (t,
J = 4.6 Hz, 1H, C
HCH
2O), 7.11–7.35 (m, 9H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 15.9 (1C, OCH
2CH
3), 36.3 (1C, N(CH
2CH
2)
2), 36.7 (1C, N(CH
2CH
2)
2), 49.3 (1C, N(
CH
2CH
2)
2), 49.5 (1C, N(
CH
2CH
2)
2), 61.8 (1C, CH
CH
2O), 63.6 (1C, N
CH
2Ph), 64.8 (1C, O
CH
2CH
3), 72.5 (1C,
CHCH
2O), 73.7 (1C, Ar
CO), 125.2 (1C, C
arom.), 126.6 (1C, C
arom.), 127.1 (1C, C
arom.), 128.0 (1C, C
arom.), 128.2 (1C, C
arom.), 128.3 (2C, C
arom.), 129.4 (2C, C
arom.), 134.2 (1C, C
arom.), 138.7 (1C, C
arom.), 142.4 (1C, C
arom.). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 2928, 2812 (C-H), 1092 (C-O-C ether), 737 (1,2-disubst. arom.), 698 (monosubst. arom
.).
tert-Butyl (R)-4-(2-ethoxy-2-oxoethoxy)-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidine]-1′-carboxylate ((R)-17)
(R)-11 (1.6 g, 5.0 mmol) was dissolved in THF (60 mL). A 1 M solution of lithium bis(trimethyl-silyl)amide (41 mL, 41 mmol) was added and the mixture was stirred for 1 h at ambient temperature. Then ethyl 2-bromoacetate (4.6 mL, 41.5 mmol) and tetrabutylammonium iodide (191 mg, 0.52 mmol) were added and the mixture was heated to reflux overnight. The solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 5.5 cm, h = 15 cm, cyclohexane-ethyl acetate = 9:1, V = 65 mL) to give
(R)-17 as a pale yellow oil (R
f = 0.17, cyclohexane-ethyl acetate = 5:1), yield 1.2 g (59%). C
22H
31NO
6 (405.5 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = −14.7 (5.5; CH
2Cl
2). Purity (HPLC method 1): 97.3%, t
R = 21.3 min.
tert-Butyl (S)-4-(2-ethoxy-2-oxoethoxy)-3,4-dihydrospiro[2-benzopyran-1,4′-piperidine]-1′-carboxylate ((S)-17)
(S)-11 (2.0 g, 6.3 mmol) was dissolved in THF (50 mL). A 1 M solution of lithium bis(trimethyl-silyl)amide (50 mL, 50 mmol) was added and the mixture was stirred for 45 min at ambient temperature. Then ethyl 2-bromoacetate (50 mL, 50.5 mmol) and tetrabutylammonium iodide (247 mg, 0.67 mmol) were added and the mixture was heated to reflux overnight. The solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 5 cm, h = 17 cm, cyclohexane-ethyl acetate = 9:1, V = 30 mL) to give
(S)-17 as a pale yellow oil (R
f = 0.17, cyclo-hexane-ethyl acetate = 5:1), yield 1.4 g (55%). C
22H
31NO
6 (405.5 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +14.3 (3.6; CH
2Cl
2). Purity (HPLC method 1): 96.0%, t
R = 20.9 min.
Spectroscopic data for (R)-17 and (S)-17
Exact mass (APCI):
m/z = 406.2224 (calcd. 406.2224 for C
22H
32NO
6 [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.29 (t,
J = 7.1 Hz, 3H, CH
2C
H3), 1.49 (s, 9H, CO
2C(C
H3)
3), 1.68–1.79 (m, 1H, N(CH
2C
H2)
2), 1.83–2.02 (m, 3H, N(CH
2C
H2)
2), 3.01–3.29 (m, 2H, N(C
H2CH
2)
2), 3.93–4.11 (m, 4H, N(C
H2CH
2)
2 (2H), CHC
H2O (2H)), 4.17-4.28 (m, 4H, C
H2CH
3 (2), OC
H2CO
2 (2)), 4.52 (t,
J = 3.4 Hz, 1H, C
HCH
2O), 7.11 (dd,
J = 7.6/1.5 Hz, 1H, H
arom.), 7.25–7.35 (m, 2H, H
arom.), 7.55 (dd,
J = 7.5/1.7 Hz, 1H, H
arom).
13C-NMR (CDCl
3): δ (ppm) = 14.4 (1C, CH
2CH
3), 28.7 (3C, CO
2C(
CH
3)
3), 34.7 (br, 1C, N(CH
2CH
2)
2), 37.1 (br, 1C, N(CH
2CH
2)
2), 39.5 (br, 1C, N(
CH
2CH
2)
2), 40.1 (br, 1C, N(
CH
2CH
2)
2), 61.0 (1C,
CH
2CH
3), 62.0 (1C, CH
CH
2O), 65.7 (1C, O
CH
2CO
2), 72.6 (1C,
CHCH
2O), 73.6 (1C, CO
2C(CH
3)
3), 79.6 (1C, Ar
CO), 125.0 (1C, C
arom.), 127.0 (1C, C
arom.), 128.8 (1C, C
arom.), 129.5 (1C, C
arom.), 131.8 (1C, C
arom.), 142.1 (1C, C
arom.), 155.0 (1C,
CO
2C(CH
3)
3), 170.9 (1C, OCH2
CO2). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 2974, 2928 (C-H), 1751, 1690 (C=O), 1165, 1099 (C-O-C ether), 759 (1,2-disubst. arom.).
Ethyl (R)-2-[(1′-benzyl-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidin]-4-yl)oxy]acetate ((R)-18)
(R)-17 (86 mg, 0.21 mmol) was dissolved in CH
2Cl
2 (4 mL). Trifluoroacetic acid (200 μL) was added and the mixture was stirred overnight at ambient temperature. Then water was added. After separation of the layers, the aqueous layer was extracted with ethyl acetate (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The residue was dissolved in CH
2Cl
2 (2 mL). Benzaldehyde (103 μL, 1.0 mmol) and sodium triacetoxyborohydride (161 mg, 0.76 mmol) were added and the mixture was stirred at ambient temperature for 4 days. The reaction was stopped by the addition of a 2 M aqueous solution of NaOH. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 1.5 cm, h = 14 cm, cyclohexane-ethyl acetate = 3:1, V = 5 mL) to give
(R)-18 as a yellowish oil (R
f = 0.16, cyclohexane-ethyl acetate = 5:5), yield 36 mg (43%). C
24H
29NO
4 (395.5 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = −16.0 (3.3; CH
2Cl
2). Purity (HPLC method 1): 95.4%, t
R = 17.7 min.
Ethyl (S)-2-[(1′-benzyl-3,4-dihydro-3,4-dihydrospiro[2-benzopyran-1,4′-piperidin]-4-yl)oxy]acetate ((S)-18)
(S)-17 (1.3 mg, 3.2 mmol) was dissolved in CH
2Cl
2 (60 mL). Trifluoroacetic acid (3.5 mL) was added and the mixture was stirred for 7 h at ambient temperature. Then a 2 M aqueous solution of NaOH was added. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The residue was dissolved in CH
2Cl
2 (50 mL). Benzaldehyde (1.0 mL, 9.9 mmol) and, after 15 min, sodium triacetoxyborohydride (2.0 g, 9.4 mmol) were added and the mixture was stirred overnight at ambient temperature. The reaction was stopped by the addition of a 2 M aqueous solution of NaOH and worked up as described for
(R)-18. The crude product was purified by flash column chromatography (Ø = 5 cm, h = 15 cm, cyclohexane-ethyl acetate = 3:1, V = 30 mL) to give
(S)-18 as a yellowish oil (R
f = 0.16, cyclohexane-ethyl acetate = 5:5), yield 36 mg (43%). C
24H
29NO
4 (395.5 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +15.3 (3.4; CH
2Cl
2). Purity (HPLC method 1): 93.1%, t
R = 17.3 min.
Spectroscopic data for (R)-18 and (S)-18
Exact mass (APCI):
m/z = 396.2177 (calcd. 396.2169 for C
24H
30NO
4 [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.30 (t,
J = 7.1 Hz, 3H, CH
2C
H3), 1.87–1.95 (m, 3H, N(CH
2C
H2)
2), 2.16 (td,
J = 13.1/4.6 Hz, 1H, N(CH
2C
H2)
2), 2.34–2.45 (m, 1H, N(C
H2CH
2)
2), 2.47–2.55 (m, 1H, N(C
H2CH
2)
2), 2.70–2.81 (m, 2H, N(C
H2CH
2)
2), 3.57 (d,
J = 13.1 Hz, 1H, NC
H2Ph), 3.61 (d,
J = 13.1 Hz, 1H, NC
H2Ph), 3.98 (dd,
J = 12.4/3.3 Hz, 1H, CHC
H2O), 4.05 (dd,
J = 12.4/3.8 Hz, 1H, CHC
H2O), 4.16–4.29 (m, 4H, C
H2CH
3 (2H), OC
H2CO
2 (2H)), 4.53 (t,
J = 3.5 Hz, C
HCH
2O), 7.22–7.41 (m, 8H, H
arom.), 7.53–7.57 (m, 1H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 14.4 (1C, CH
2CH
3), 35.1 (1C, N(CH
2CH
2)
2), 37.4 (1C, N(CH
2CH
2)
2), 49.3 (1C, N(
CH
2CH
2)
2), 49.4 (1C, N(
CH
2CH
2)
2), 61.0 (1C,
CH
2CH
3), 61.7 (1C, CH
CH
2O), 63.5 (1C, N
CH
2Ph), 65.6 (1C, O
CH
2CO
2), 72.7 (1C,
CHCH
2O), 73.6 (1C, Ar
CO), 125.2 (1C, C
arom.), 126.8 (1C, C
arom.), 127.1 (1C, C
arom.), 128.3 (2C, C
arom.), 128.6 (1C, C
arom.), 129.4 (1C, C
arom.), 129.4 (2C, C
arom.), 132.0 (1C, 4a-C
arom.), 138.6 (1C, 1-C
benzyl), 142.8 (1C, 8a-C
arom.), 170.9 (1C, OCH2
CO2). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 2920, 2866 (C-H), 1748 (C=O), 1099, 1053 (C-O-C ether), 737 (1,2-disubst. arom.), 698 (monosubst. arom.).
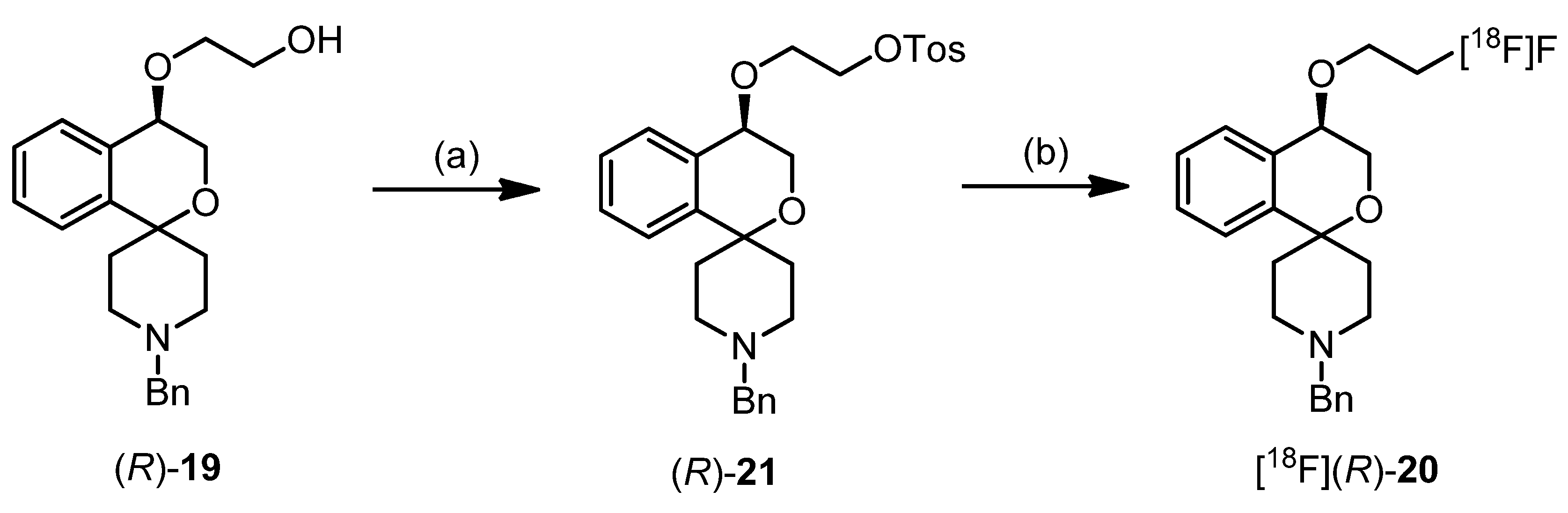
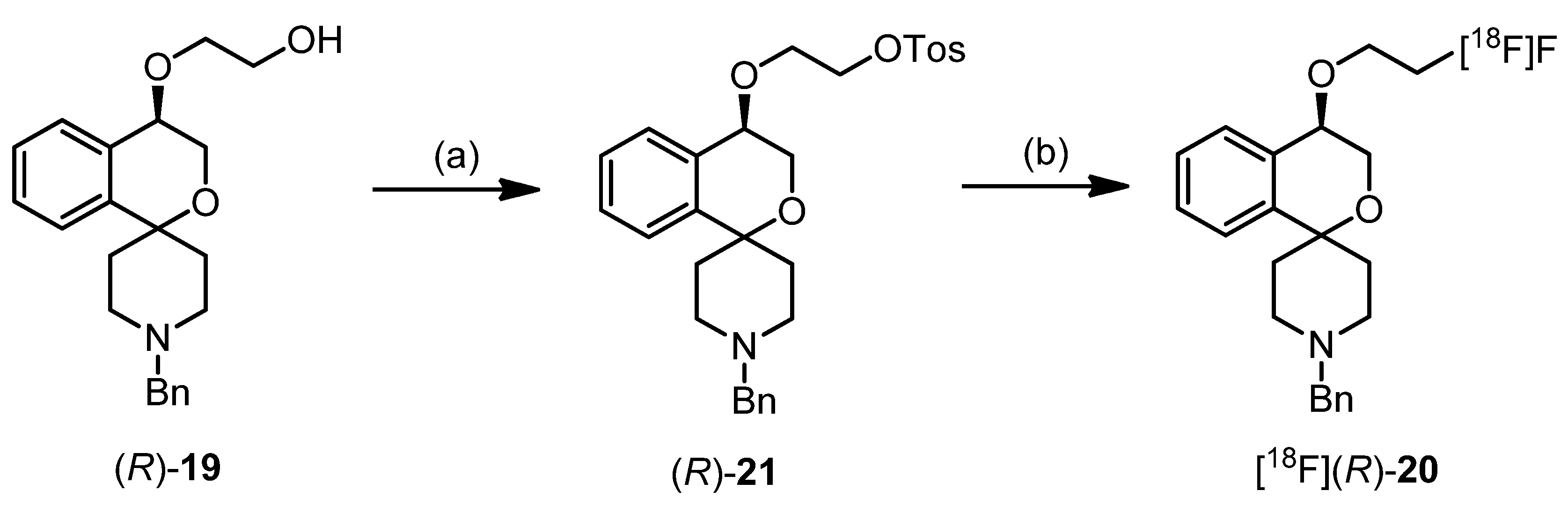
(R)-2-[(1′-Benzyl-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidin]-4-yl)oxy]ethanol ((R)-19)
(R)-18 (480 mg, 1.21 mmol) was dissolved in THF (5 mL). A 1 m solution of LiAlH
4 in THF (6 mL, 6 mmol) was added and the mixture was stirred overnight at ambient temperature. Then water was added. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were washed with water (2×) and brine (1×), dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 3 cm, h = 15 cm, cyclohexane-ethyl acetate = 5:5, V = 20 mL) to give
(R)-19 as a yellowish oil (R
f = 0.06, ethyl acetate), yield 254 mg (59%). C
22H
27NO
3 (353.5 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = −2.2 (1.7; CH
2Cl
2). Purity (HPLC method 1): 95.1%, t
R = 13.8 min.
(S)-2-[(1′-Benzyl-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidin]-4-yl)oxy]ethanol ((S)-19)
(S)-18 (451 mg, 1.14 mmol) was dissolved in THF (10 mL). A 1 M solution of LiAlH
4 in THF (2.5 mL, 2.5 mmol) was added and the mixture was stirred overnight at ambient temperature. Then water and CH
2Cl
2 were added. The reaction was worked up as described for
(R)-19. The crude product was purified by flash column chromatography (Ø = 2 cm, h = 15 cm, cyclohexane-ethyl acetate = 5:5, V = 10 mL) to give
(S)-19 as a pale yellow oil (R
f = 0.06, ethyl acetate), yield 302 mg (75%). C
22H
27NO
3 (353.5 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +2.1 (1.8; CH
2Cl
2). Purity (HPLC method 1): 94.3%, t
R = 13.9 min.
Spectroscopic data for (R)-19 and (S)-19
Exact mass (APCI):
m/z = 354.2039 (calcd. 354.2064 for C
22H
28NO
3 [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.85–1.99 (m, 3H, N(CH
2C
H2)
2), 2.13 (td,
J = 13.3/4.6 Hz, 1H, N(CH
2C
H2)
2), 2.30–2.44 (s br, 1H, O
H), 2.40 (td,
J = 10.9/4.6 Hz, 1H, N(C
H2CH
2)
2), 2.45–2.53 (m, 1H, N(C
H2CH
2)
2), 2.70–2.79 (m, 2H, N(C
H2CH
2)
2), 3.58 (s, 2H, NC
H2Ph), 3.72–3.81 (m, 4H, OC
H2C
H2OH), 3.94 (dd,
J = 12.3/3.3 Hz, 1H, CHC
H2O), 4.01 (dd,
J = 12.3/3.9 Hz, 1H, CHC
H2O), 4.36 (t,
J = 3.6 Hz, 1H, C
HCH
2O), 7.22–7.39 (m, 9H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 35.4 (1C, N(CH
2CH
2)
2), 37.4 (1C, N(CH
2CH
2)
2), 49.3 (1C, N(
CH
2CH
2)
2), 49.4 (1C, N(
CH
2CH
2)
2), 61.6 (1C, CH
CH
2O), 62.2 (1C, OCH
2CH
2OH), 63.5 (1C, N
CH
2Ph), 70.1 (1C, O
CH
2CH
2OH), 73.1 (1C,
CHCH
2O), 73.7 (1C, Ar
CO),125.4 (1C, C
arom.), 126.7 (1C, C
arom.), 127.1 (1C, C
arom.), 128.3 (2C, C
arom.), 128.5 (1C, C
arom.), 128.9 (1C, C
arom.), 129.4 (2C, C
arom.), 133.1 (1C, C
arom.), 138.7 (1C, C
arom.), 142.5 (1C, C
arom.). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 3399 (O-H), 2924, 2816 (C-H), 1092, 1076 (C-O-C, ether), 741 (1,2-disubst. arom.), 698 (monosubst. arom.).
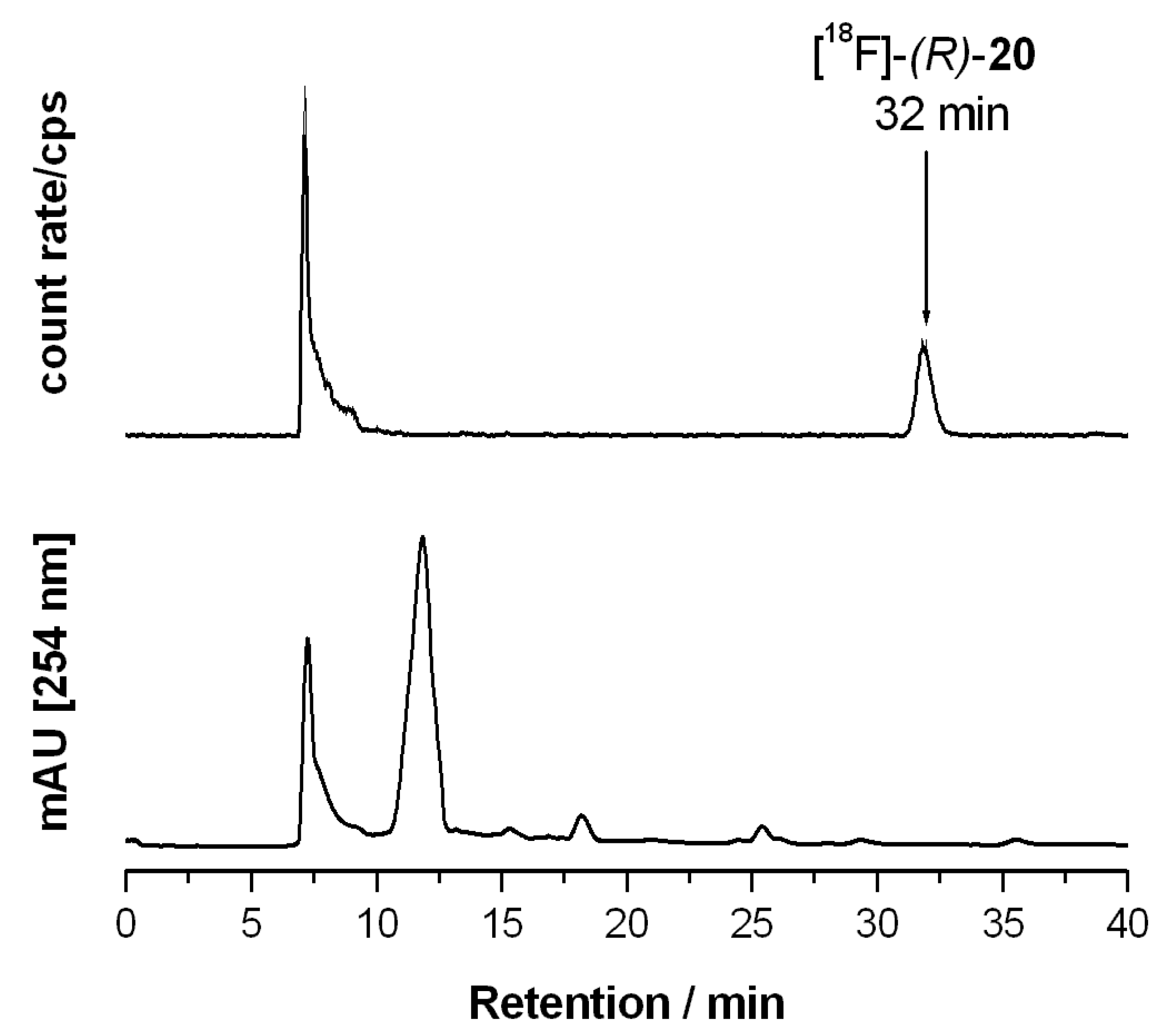
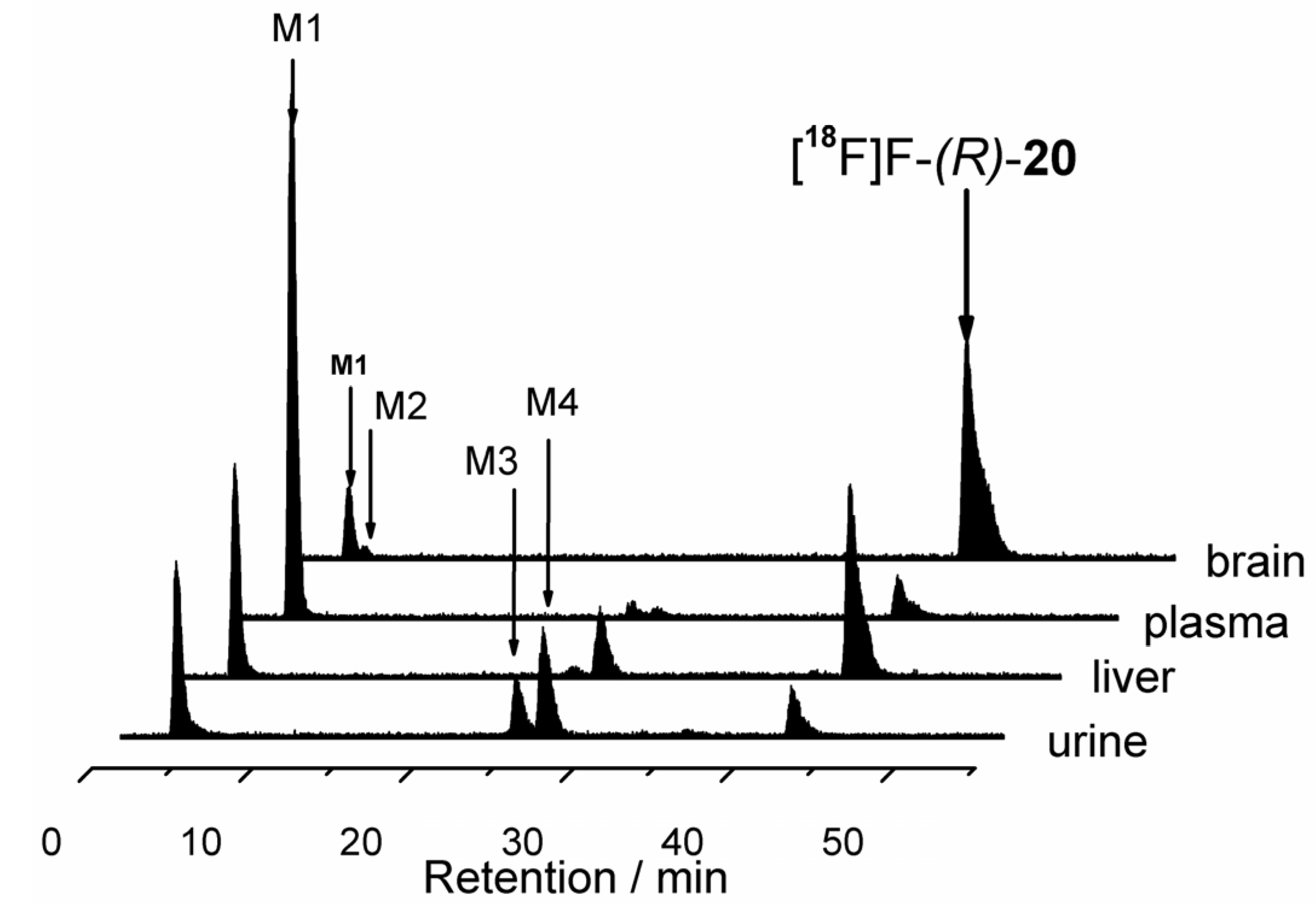
(R)-1′-Benzyl-4-(2-fluoroethoxy)-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidine] ((R)-20)
(Diethylamino)difluorosulfonium tetrafluoroborate (Xtal-Fluor E
®, 46 mg, 0.20 mmol) and triethylamine trihydrofluoride (45 μL, 0.28 mmol) were dissolved in CH
2Cl
2 (1 mL). The solution was cooled to −78 °C.
(R)-19 (48 mg, 0.14 mmol) was added and the mixture was stirred at ‒78 °C for 1 h, then at 0 °C for 1 h and at ambient temperature for 1 h. A 5% aqueous solution of NaHCO
3 (3 mL) was added and the mixture was stirred for 15 min. The aqueous layer was extracted with CH
2Cl
2 (2×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 0.75 cm, h = 16 cm, cyclohexane-ethyl acetate = 2:1, V = 5 mL) to give
(R)-20 as a pale yellow oil (R
f = 0.34, cyclohexane-ethyl acetate = 5:5), yield 27 mg (54%). C
22H
26FNO
2 (355.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = −6.4 (5.8; CH
2Cl
2). Purity (HPLC method 1): 97.0%, t
R = 16.4 min.
(S)-1′-Benzyl-4-(2-fluoroethoxy)-3,4-dohydrospiro[[2]benzopyran-1,4′-piperidine] ((S)-20)
(Diethylamino)difluorosulfonium tetrafluoroborate (Xtal-Fluor E
®, 120 mg, 0.52 mmol) and triethylamine trihydrofluoride (305 μL, 1.87 mmol) were dissolved in CH
2Cl
2 (2 mL). The solution was cooled to −78 °C.
(S)-19 (120 mg, 0.34 mmol) dissolved in CH
2Cl
2 (2 mL), was added and the mixture was stirred at -78 °C for 1 h, then at 0 °C for 1 h and at ambient temperature overnight. A 20% aqueous solution of NaHCO
3 (4 mL) was added and the mixture was stirred for 15 min. The aqueous layer was extracted with CH
2Cl
2 (2×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 1.5 cm, h = 18 cm, cyclohexane-ethyl acetate = 3:1, V = 5 mL) to give
(S)-20 as a pale yellow oil (R
f = 0.34, cyclohexane-ethyl acetate = 5:5), yield 86 mg (71%). C
22H
26FNO
2 (355.4 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +5.9 (4.4; CH
2Cl
2). Purity (HPLC method 1): 95.1%, t
R = 16.1 min.
Spectroscopic data for (R)-20 and (S)-20
Exact mass (APCI):
m/z = 356.2032 (calcd. 356.2020 for C
22H
27FNO
2 [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.85–2.00 (m, 3H, N(CH
2C
H2)
2), 2.10 (td,
J = 13.3/4.6 Hz, 1H, N(CH
2C
H2)
2), 2.39 (td,
J = 11.6/3.1 Hz, 1H, N(C
H2CH
2)
2), 2.48 (td,
J = 11.6/2.5 Hz, 1H, N(C
H2CH
2)
2), 2.69–2.78 (m, 2H, N(C
H2CH
2)
2), 3.57 (s, 2H, NC
H2Ph), 3.78–3.87 (m, 1H, OC
H2CH
2F), 3.87–3.94 (m, 1H, OC
H2CH
2F), 3.94 (dd,
J = 12.1/4.7 Hz, 1H, CHC
H2O), 4.01 (dd,
J = 12.1/3.7 Hz, 1H, CHC
H2O), 4.45 (t,
J = 4.2 Hz, 1H, C
HCH
2O), 4.59 (dt,
J = 47.7/4.2 Hz, 2H, OCH
2C
H2F), 7.21–7.38 (m, 8H, H
arom.), 7.41–7.44 (m, 1H, H
arom.).
13C-NMR (CDCl
3): δ (ppm) = 35.7 (1C, N(CH
2CH
2)
2), 37.0 (1C, N(CH
2CH
2)
2), 49.3 (1C, N(
CH
2CH
2)
2), 49.4 (1C, N(
CH
2CH
2)
2), 61.8 (1C, CH
CH
2O), 63.5 (1C, N
CH
2Ph), 68.0 (d,
J = 20.3 Hz, 1C, O
CH
2CH
2F), 73.1 (1C,
CHCH
2O), 73.6 (1C, Ar
CO), 83.5 (d,
J = 179.1 Hz, 1C, OCH
2CH
2F), 125.3 (1C, C
arom.), 126.7 (1C, C
arom.), 127.2 (1C, C
arom.), 128.4 (2C, C
arom.), 128.7 (1C, C
arom.), 129.5 (2C, C
arom.), 129.6 (1C, C
arom.), 133.0 (1C, C
arom.), 138.4 (1C, C
arom.), 142.5 (1C, Carom.). IR (neat):
![Pharmaceuticals 07 00078 i002]()
(cm
−1) = 2934, 2812 (C-H), 1096 (C-O-C, ether), 737 (1,2-disubst. arom.), 698 (monosubst. arom.).
{(R)-2-[(1′-Benzyl-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidin]-4-yl)oxy]ethyl} 4-methylbenzene-sulfonate ((R)-21)
(R)-19 (90 mg, 0.25 mmol) was dissolved in CH
2Cl
2 (13 mL). 4-Dimethylaminopyridine (8 mg, 0.07 mmol), triethylamine (176 μL, 1.3 mmol) and 4-toulenesulfonyl chloride (108 mg, 0.57 mmol) were added and the mixture was stirred overnight at ambient temperature. Then a 2 M aqueous solution of NaOH was added. After separation of the layers, the aqueous layer was extracted with CH
2Cl
2 (3×). The combined organic layers were dried (Na
2SO
4), filtered and the solvent was removed
in vacuo. The crude product was purified by flash column chromatography (Ø = 1.5 cm, h = 16 cm, cyclohexane-ethyl acetate = 7:3, V = 5 mL) to give
(R)-21 as a colorless oil (R
f = 0.13, cyclohexane-ethyl acetate = 5:5), yield 59 mg (46%). C
29H
33NO
5S (507.6 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = −5.9 (21.8; CH
2Cl
2). Purity (HPLC method 1): 94.5%, t
R = 20.3 min.
{(S)-2-[(1′-Benzyl-3,4-dihydrospiro[[2]benzopyran-1,4′-piperidin]-4-yl)oxy]ethyl} 4-methylbenzene-sulfonate ((S)-21)
(S)-19 (140 mg, 0.40 mmol) was dissolved in CH
2Cl
2 (20 mL). Triethylamine (274 μL, 2.0 mmol), 4-dimethylaminopyridine (16 mg, 0.13 mmol) and 4-toulenesulfonyl chloride (152 mg, 0.80 mmol) were added and the mixture was stirred overnight at ambient temperature. Then reaction was worked up as described for
(R)-21. The crude product was purified by flash column chromatography (Ø = 2 cm, h = 15 cm, cyclohexane-ethyl acetate = 7:3, V = 10 mL) to give
(S)-21 as a yellow oil (R
f = 0.13, cyclohexane-ethyl acetate = 5:5), yield 66 mg (32%). C
29H
33NO
5S (507.6 g/mol). Specific rotation:
![Pharmaceuticals 07 00078 i003]()
: = +7.5 (23.8; CH
2Cl
2). Purity (HPLC method 1): 95.1%, t
R = 19.6 min.
Spectroscopic data for (R)-21 and (S)-21
Exact mass (APCI):
m/z = 508.2151 (calcd. 508.2152 for C
29H
35NO
5S [M+H]
+).
1H-NMR (CDCl
3): δ (ppm) = 1.78–1.90 (m, 2H, N(CH
2C
H2)
2), 1.91–2.03 (m, 1H, N(CH
2C
H2)
2), 2.14 (br t,
J = 12.9 Hz, 1H, N(CH
2C
H2)
2), 2.41 (s, 3H, C
H3), 2.45–2.62 (m, 2H, N(C
H2CH
2)
2), 2.77–2.82 (m, 2H, N(C
H2CH
2)
2), 3.62 (s, 2H, NC
H2Ph), 3.70–3.87 (m, 3H, OC
H2CH
2OTos (2H), CHC
H2O (1H)), 3.91 (dd,
J = 12.2/3.4 Hz, 1H, CHC
H2O), 4.09–4.25 (m, 2H, OCH
2C
H2OTos), 4.33 (t,
J = 3.8 Hz, C
HCH
2O), 7.08–7.57 (m, 11H, H
arom.), 7.71–7.83 (m, 2H, H-2
tosyl, H-6
tosyl).
13C-NMR (CDCl
3): δ (ppm) = 21.8 (1C,
CH
3), 35.2 (1C, N(CH
2CH
2)
2), 36.8 (1C, N(CH
2CH
2)
2), 49.2 (1C, N(
CH
2CH
2)
2), 49.3 (1C, N(
CH
2CH
2)
2), 61.7 (1C, CH
CH
2O), 63.3 (1C, N
CH
2Ph), 66.1 (1C, O
CH
2CH
2OTos), 69.7 (1C, OCH
2CH
2OTos), 73.0 (1C,
CHCH
2O), 73.4 (1C, Ar
CO), 125.2 (1C, C
arom.), 126.1 (1C, C
arom.), 126.7 (1C, C
arom.), 128.1 (2C, C
arom.), 128.4 (2C, C
arom.), 128.7 (1C, C
arom.), 128.8 (2C, C
arom.), 129.6 (1C, C
arom.), 129,9 (2C, C
arom.), 132.2 (1C, C
arom.), 133.0 (1C, C
arom.), 133.9 (1C, C
arom.), 144.9 (1C, C
arom.), 146.0 (1C, Carom.). IR (neat):
![Pharmaceuticals 07 00078 i002]()
[cm
−1] = 2924, 2812 (C-H), 1358(m), 1177 (O=S=O), 1096 (C-O-C), 741 (1,2-disubst. arom.), 698 (monosubst. arom.).

 ,
, 
 (cm−1) = 3387 (O-H), 2967, 2932, (C-H), 1655 (C=O), 756 (1,2-disubst. arom.).
(cm−1) = 3387 (O-H), 2967, 2932, (C-H), 1655 (C=O), 756 (1,2-disubst. arom.). : = ‒24.5 (3.5; CH2Cl2). Purity (HPLC method 1): 99.5%, tR = 15.3 min.
: = ‒24.5 (3.5; CH2Cl2). Purity (HPLC method 1): 99.5%, tR = 15.3 min.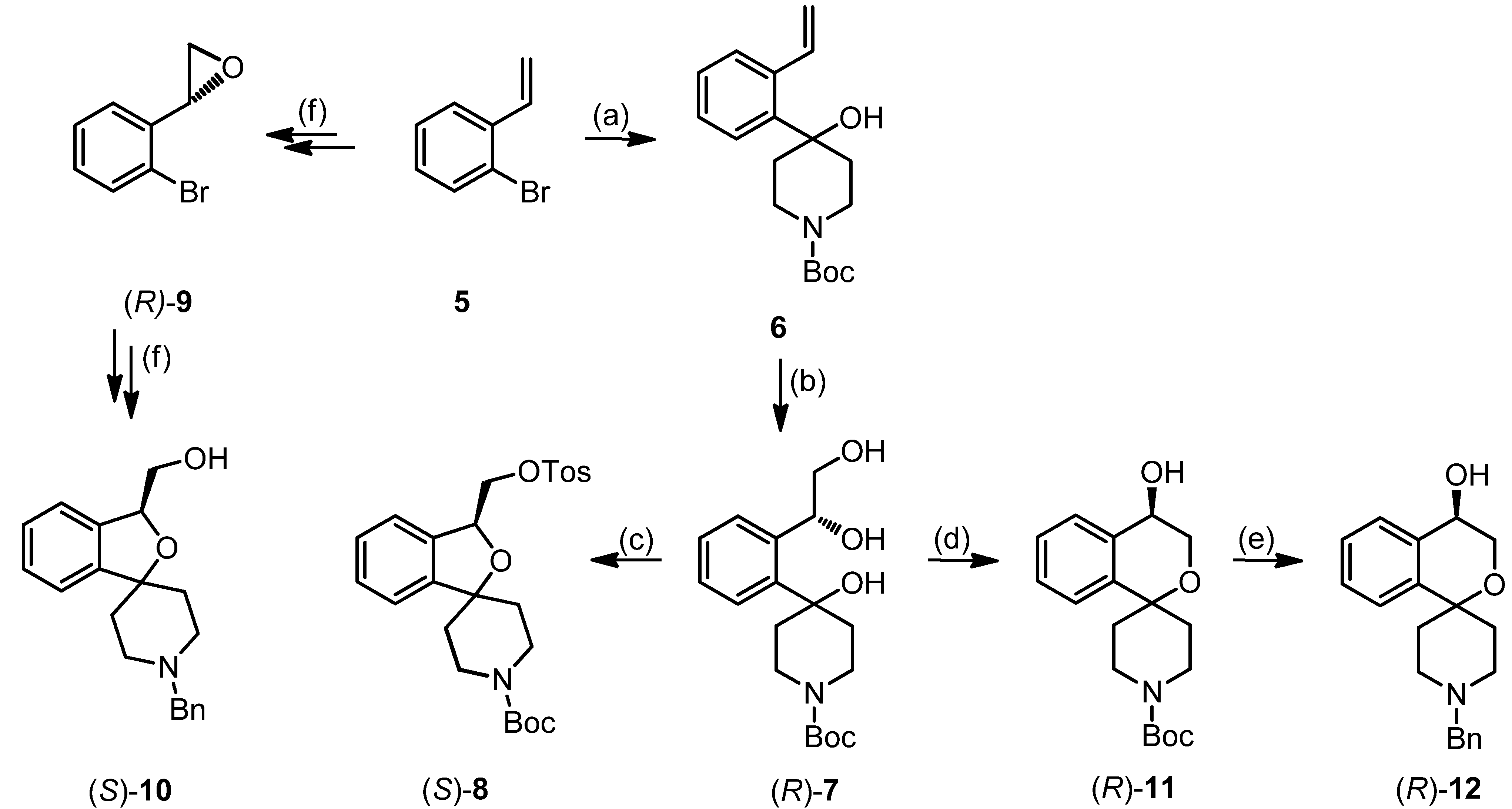


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}