Rationale and Means to Target Pro-Inflammatory Interleukin-8 (CXCL8) Signaling in Cancer

Abstract
:1. Introduction
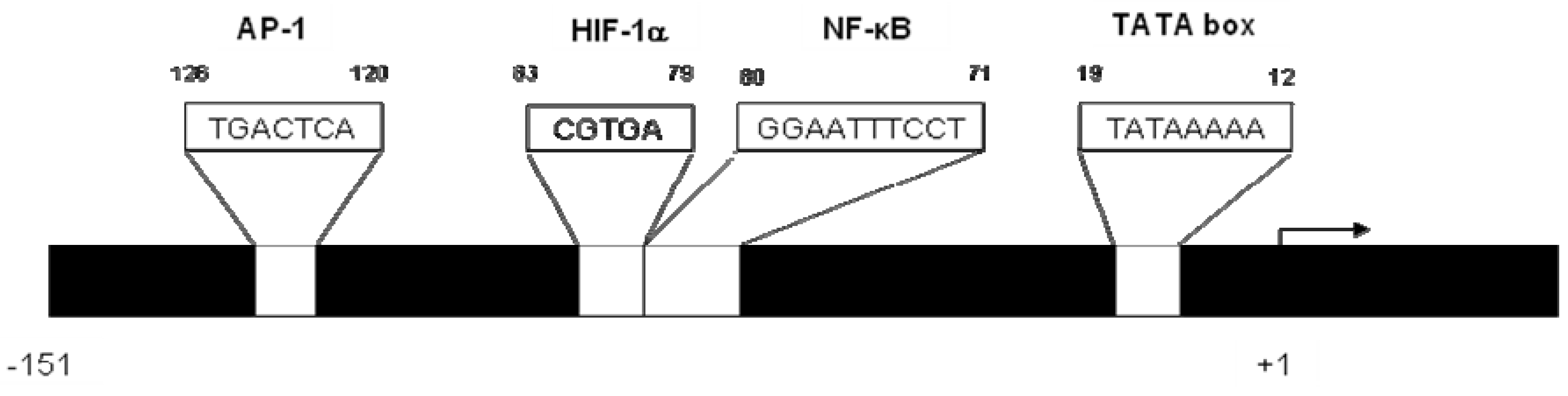
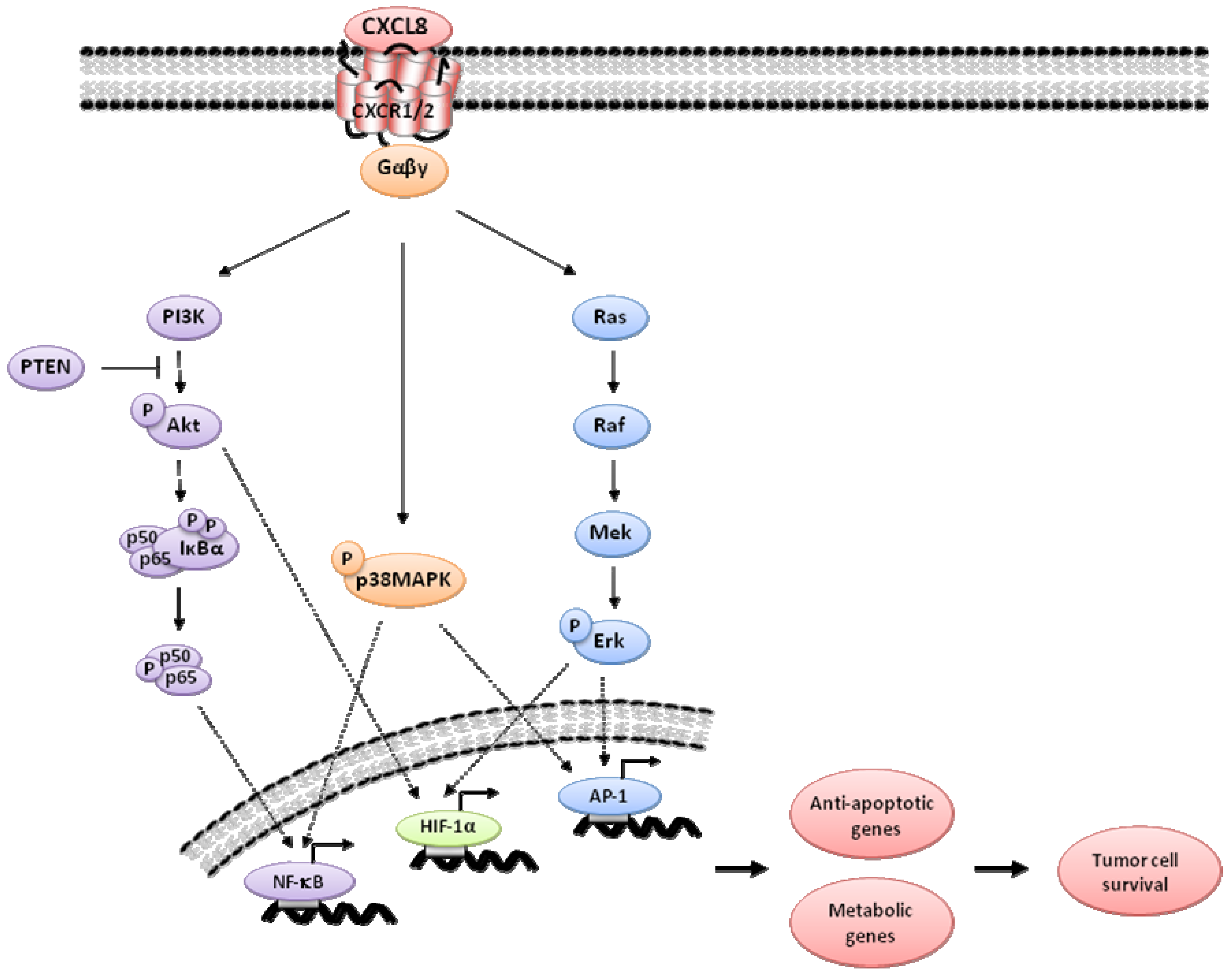
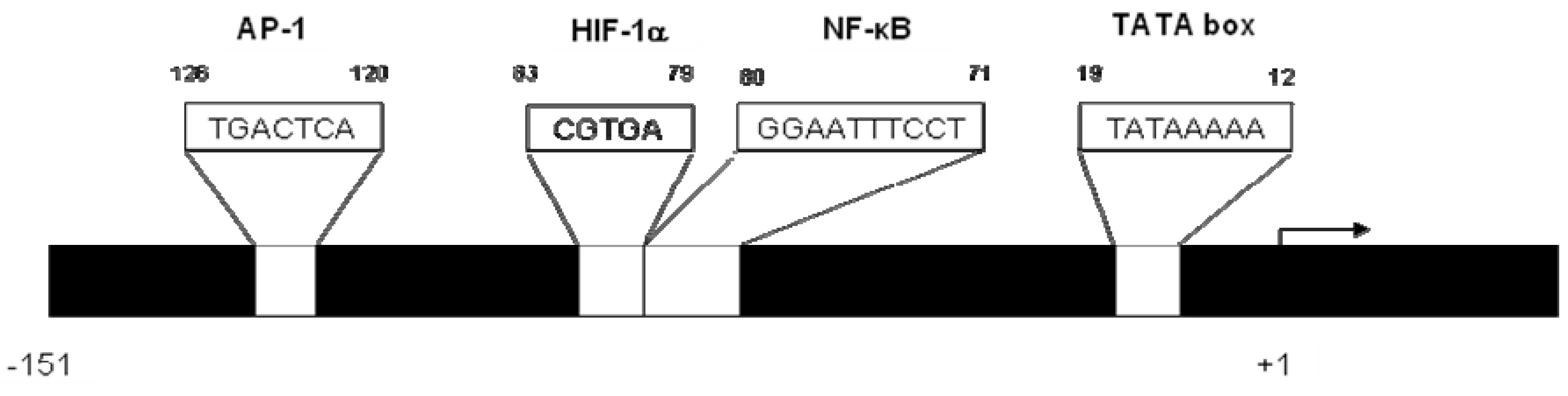
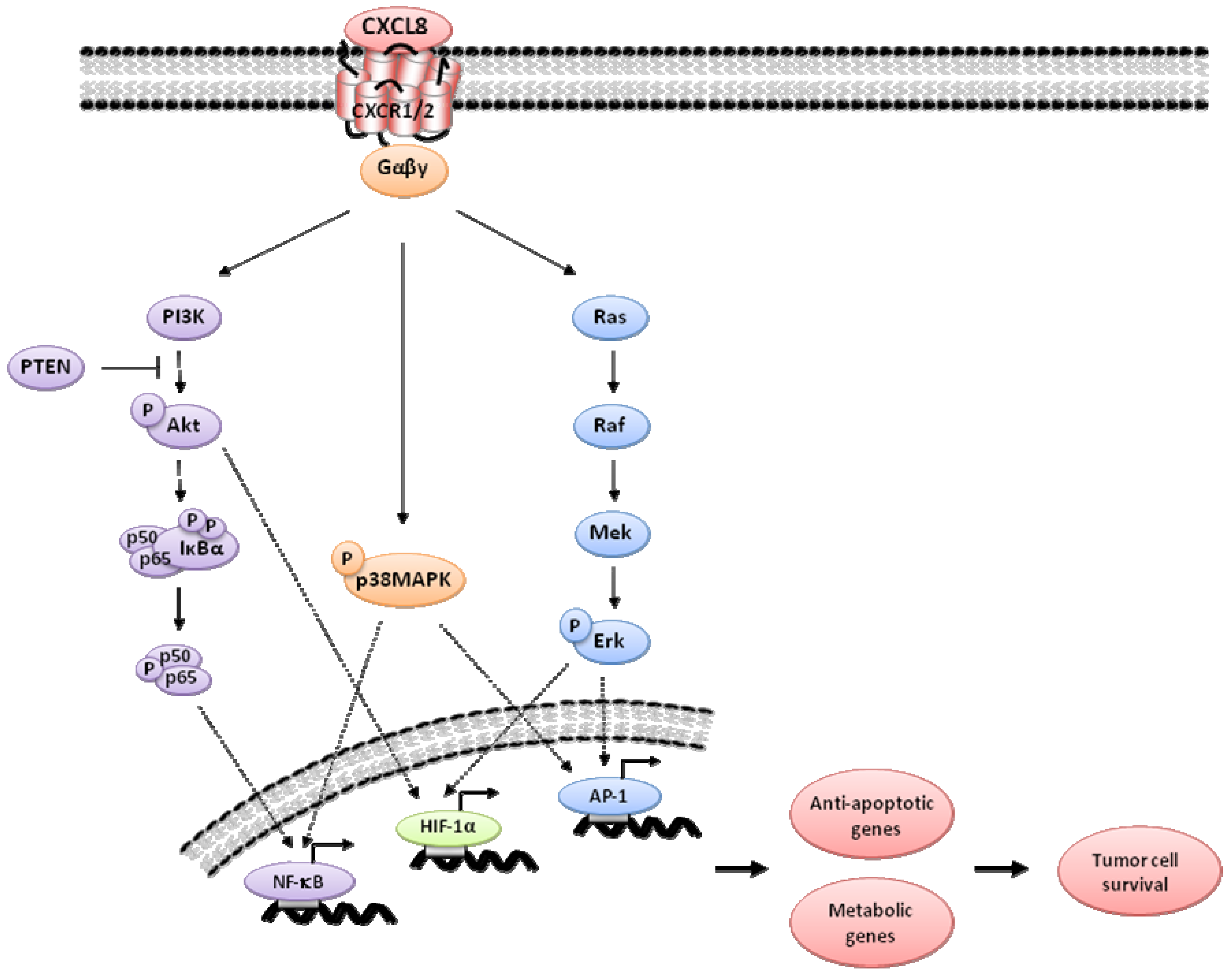
2. CXCL8 Signaling in Cancer
3. Indirect Targeting
3.1. Signal Transduction Pathway Inhibitors
3.1.1. MAPK Inhibitors
3.1.2. Phosphatidylinositol-3-Kinase (PI3K)/Akt Inhibitors
3.2. NFκB Inhibitors
3.3. NSAIDS
4. Direct Targeting
4.1. CXCL8 Neutralizing Antibodies
4.2. CXCR1/2 Neutralizing Antibodies
4.3. Small Molecule CXCR1/2 Antagonists
4.3.1. Dompé
4.3.2. Schering-Plough
4.3.3. GlaxoSmith Kline (GSK)
4.3.4. AstraZeneca
4.4. Pepducin CXCR1/2 Inhibitors

{kind=link}
{kind=link}
| Company | Dompé | Schering-Plough | GlaxoSmith Kline | AstraZeneca | ||||
|---|---|---|---|---|---|---|---|---|
| Antagonist | CXCR1 | CXCR2 | CXCR1 | CXCR2 | CXCR1 | CXCR2 | CXCR1 | CXCR2 |
| Reparixin | SCH527123 | SB225002 SB656933 * | AZD8309 AZD5069 * | |||||
| DF2162 * | ||||||||
| Cancer pre-clinical studies | Breast cancer xenografts | Colorectal cancer xenografts | Colitis | |||||
| Clinical trials | Diabetes (islet cell transplantation) Breast cancer | Ozone-induced neutrophilia COPD Asthma | Ozone-induced airway inflammation * Cystic fibrosis * COPD * Ulcerative colitis * | COPD Rheumatoid arthritis COPD * Bronchiectasis * Asthma * | ||||
5. Translational Issues
6. Conclusions
Conflict of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- De Marzo, A.M.; DeWeese, T.L.; Platz, E.A.; Meeker, A.K.; Nakayama, M.; Epstein, J.I.; Isaacs, W.B.; Nelson, W.G. Pathological and molecular mechanisms of prostate carcinogenesis: Implications for diagnosis, detection, prevention, and treatment. J. Cell. Biochem. 2004, 91, 459–477. [Google Scholar] [CrossRef]
- Gupta, R.B.; Harpaz, N.; Itzkowitz, S.; Hossain, S.; Matula, S.; Kornbluth, A.; Bodian, C.; Ullman, T. Histologic inflammation is a risk factor for progression to colorectal neoplasia in ulcerative colitis: A cohort study. Gastroenterology 2007, 133, 1099–1105. [Google Scholar] [CrossRef]
- Triantafillidis, J.K.; Nasioulas, G.; Kosmidis, P.A. Colorectal cancer and inflammatory bowel disease: Epidemiology, risk factors, mechanisms of carcinogenesis and prevention strategies. Anticancer Res. 2009, 29, 2727–2737. [Google Scholar]
- Nelson, W.G.; de Marzo, A.M.; DeWeese, T.L.; Isaacs, W.B. The role of inflammation in the pathogenesis of prostate cancer. J. Urol. 2004, 172, S6–S11. [Google Scholar] [CrossRef]
- Cohen, R.J.; Shannon, B.A.; McNeal, J.E.; Shannon, T.; Garrett, K.L. Propionibacterium acnes associated with inflammation in radical prostatectomy specimens: A possible link to cancer evolution? J. Urol. 2005, 173, 1969–1974. [Google Scholar] [CrossRef]
- Harris, R.E.; Kasbari, S.; Farrar, W.B. Prospective study of nonsteroidal anti-inflammatory drugs and breast cancer. Oncol. Rep. 1999, 6, 71–73. [Google Scholar]
- Flossmann, E.; Rothwell, P.M. Effect of aspirin on long-term risk of colorectal cancer: Consistent evidence from randomised and observational studies. Lancet 2007, 369, 1603–1613. [Google Scholar] [CrossRef]
- Maeda, H.; Akaike, T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry Mosc. 1998, 63, 854–865. [Google Scholar]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Sparmann, A.; Bar-Sagi, D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell 2004, 6, 447–458. [Google Scholar] [CrossRef]
- Wislez, M.; Fujimoto, N.; Izzo, J.G.; Hanna, A.E.; Cody, D.D.; Langley, R.R.; Tang, H.; Burdick, M.D.; Sato, M.; Minna, J.D.; et al. High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res. 2006, 66, 4198–4207. [Google Scholar] [CrossRef]
- Ancrile, B.B.; Counter, C.M. Oncogenic Ras-nduced Expression of Cytokines: A New Target of Anti-Cancer Therapeutics. Mol. Interventions 2008, 8, 22–27. [Google Scholar] [CrossRef]
- Maxwell, P.J.; Coulter, J.; Walker, S.M.; McKechnie, M.; Neisen, J.; McCabe, N.; Kennedy, R.D.; Salto-Tellez, M.; Albanese, C.; Waugh, D.J.J. Potentiation of Inflammatory CXCL8 Signalling Sustains Cell Survival in PTEN-deficient Prostate Carcinoma. Eur. Urol. 2013, 64, 177–188. [Google Scholar] [CrossRef]
- Lazennec, G.; Richmond, A. Chemokines and chemokine receptors: New insights into cancer-related inflammation. Trends Mol. Med. 2010, 16, 133–144. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef]
- Ammirante, M.; Luo, J.L.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010, 464, 302–305. [Google Scholar] [CrossRef]
- De Visser, K.E.; Jonkers, J. Towards understanding the role of cancer-associated inflammation in chemoresistance. Curr. Pharm. Des. 2009, 15, 1844–1853. [Google Scholar] [CrossRef]
- Matsushima, K.; Morishita, K. Molecular cloning of a human monocyte-derived neutrophil chemotactic factor (MDNCF) and the induction of MDNCF mRNA by interleukin 1 and tumor necrosis factor. J. Exp. Med. 1988, 167, 1883–1893. [Google Scholar] [CrossRef]
- Zeilhofer, H.U.; Schorr, W. Role of interleukin-8 in neutrophil signaling. Curr. Opin. Hematol. 2000, 7, 178–182. [Google Scholar] [CrossRef]
- Mazzucchelli, L.; Hauser, C.; Zgraggen, K.; Wagner, H.; Hess, M.; Laissue, J.A.; Mueller, C. Expression of interleukin-8 gene in inflammatory bowel disease is related to the histological grade of active inflammation. Am. J. Pathol. 1994, 144, 997–1007. [Google Scholar]
- Grimm, M.C.; Elsbury, S.K.; Pavli, P.; Doe, W.F. Interleukin 8: Cells of origin in inflammatory bowel disease. Gut 1996, 38, 90–98. [Google Scholar] [CrossRef]
- Brennan, F.M.; Zachariae, C.O.; Chantry, D.; Larsen, C.G.; Turner, M.; Maini, R.N.; Matsushima, K.; Feldmann, M. Detection of interleukin 8 biological activity in synovial fluids from patients with rheumatoid arthritis and production of interleukin 8 mRNA by isolated synovial cells. Eur. J. Immunol. 1990, 20, 2141–2144. [Google Scholar] [CrossRef]
- Hébert, C.A.; Baker, J.B. Interleukin-8: A review. Cancer Invest. 1993, 11, 743–750. [Google Scholar] [CrossRef]
- Ko, Y.C.; Mukaida, N.; Ishiyama, S.; Tokue, A.; Kawai, T.; Matsushima, K.; Kasahara, T. Elevated interleukin-8 levels in the urine of patients with urinary tract infections. Infect. Immun. 1993, 61, 1307–1314. [Google Scholar]
- Nasser, M.W.; Raghuwanshi, S.K.; Grant, D.J.; Jala, V.R.; Rajarathnam, K.; Richardson, R.M. Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J. Immunol. 2009, 183, 3425–3432. [Google Scholar] [CrossRef]
- Murphy, P.M. The molecular biology of leukocyte chemoattractant receptors. Annu. Rev. Immunol. 1994, 12, 593–633. [Google Scholar] [CrossRef]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef]
- Kobilka, B.K. G protein coupled receptor structure and activation. Biochim. Biophys. Acta 2007, 1768, 794–807. [Google Scholar] [CrossRef]
- Moser, B.; Loetscher, P. Lymphocyte traffic control by chemokines. Nat. Immunol. 2001, 2, 123–128. [Google Scholar] [CrossRef]
- Thelen, M. Dancing to the tune of chemokines. Nat. Immunol. 2001, 2, 129–134. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Chung, J.; Burger, M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am. J. Physiol. Lung. C 2001, 280, L1094–L1103. [Google Scholar]
- Zaslaver, A.; Feniger-Barish, R.; Ben-Baruch, A. Actin filaments are involved in the regulation of trafficking of two closely related chemokine receptors, CXCR1 and CXCR2. J. Immunol. 2001, 166, 1272–1284. [Google Scholar]
- Waugh, D.J.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef]
- Hoffmann, E.; Dittrich-Breiholz, O.; Holtmann, H.; Kracht, M. Multiple control of interleukin-8 gene expression. J. Leukocyte Biol. 2002, 72, 847–855. [Google Scholar]
- Shi, Q.; Xiong, Q.; Le, X.; Xie, K. Regulation of interleukin-8 expression by tumor-associated stress factors. J. Interf. Cytok. Res. 2001, 21, 553–566. [Google Scholar] [CrossRef]
- Imamura, R.; Konaka, K.; Matsumoto, N.; Hasegawa, M.; Fukui, M.; Mukaida, N.; Kinoshita, T.; Suda, T. Fas ligand induces cell-autonomous NF-kappaB activation and interleukin-8 production by a mechanism distinct from that of tumor necrosis factor-alpha. J. Biol. Chem. 2004, 279, 46415–46423. [Google Scholar] [CrossRef]
- Masckauchán, T.N.H.; Shawber, C.J.; Funahashi, Y.; Li, C.-M.; Kitajewski, J. Wnt/beta-catenin signaling induces proliferation, survival and interleukin-8 in human endothelial cells. Angiogenesis 2005, 8, 43–51. [Google Scholar] [CrossRef]
- Kim, K.S.; Rajagopal, V.; Gonsalves, C.; Johnson, C.; Kalra, V.K. A novel role of hypoxia-inducible factor in cobalt chloride-and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J. Immunol. 2006, 177, 7211–7224. [Google Scholar]
- Mukaida, N.; Okamoto, S.; Ishikawa, Y.; Matsushima, K. Molecular mechanism of interleukin-8 gene expression. J. Leukocyte Biol. 1994, 56, 554–558. [Google Scholar]
- Roebuck, K. Regulation of interleukin-8 gene expression. J. Interf. Cytok. Res. 1999, 19, 429–438. [Google Scholar] [CrossRef]
- Siddiqui, R.A.; Akard, L.P.; Garcia, J.G.; Cui, Y.; English, D. Chemotactic migration triggers IL-8 generation in neutrophilic leukocytes. J. Immunol. 1999, 162, 1077–1083. [Google Scholar]
- Yu, Y.; Zeng, H.; Lyons, S.; Carlson, A.; Merlin, D.; Neish, A.S.; Gewirtz, A.T. TLR5-mediated activation of p38 MAPK regulates epithelial IL-8 expression via posttranscriptional mechanism. Am. J. Physiol. Gastr. L 2003, 285, G282–G290. [Google Scholar]
- Kuhns, D.; Gallin, J. Increased cell-associated IL-8 in human exudative and A23187-treated peripheral blood neutrophils. J. Immunol. 1995, 154, 6556–6562. [Google Scholar]
- Murphy, C.; McGurk, M.; Pettigrew, J.; Santinelli, A.; Mazzucchelli, R.; Johnston, P.G.; Montironi, R.; Waugh, D.J.J. Nonapical and cytoplasmic expression of interleukin-8, CXCR1, and CXCR2 correlates with cell proliferation and microvessel density in prostate cancer. Clin. Cancer Res. 2005, 11, 4117–4127. [Google Scholar] [CrossRef]
- Li, A.; Varney, M.L.; Singh, R.K. Expression of Interleukin 8 and Its Receptors in Human Colon Carcinoma Cells with Different Metastatic Potentials Expression of Interleukin 8 and Its Receptors in Human Colon Carcinoma Cells with Different Metastatic Potentials 1. Clin. Cancer Res. 2001, 3298–3304. [Google Scholar]
- Yuan, A.; Yang, P.C.; Yu, C.J.; Chen, W.J.; Lin, F.Y.; Kuo, S.H.; Luh, K.T. Interleukin-8 messenger ribonucleic acid expression correlates with tumor progression, tumor angiogenesis, patient survival, and timing of relapse in non-small-cell lung cancer. Am. J. Res. Crit. Care 2000, 162, 1957–1963. [Google Scholar] [CrossRef]
- Veltri, R.W.; Miller, M.C.; Zhao, G.; Ng, A.; Marley, G.M.; Wright, G.L., Jr.; Vessella, R.L.; Ralph, D. Interleukin-8 serum levels in patients with benign prostatic hyperplasia and prostate cancer. Urology 4295, 139–147. [Google Scholar]
- Seaton, A.; Scullin, P.; Maxwell, P.J.; Wilson, C.; Pettigrew, J.; Gallagher, R.; O’Sullivan, J.M.; Johnston, P.G.; Waugh, D.J.J. Interleukin-8 signaling promotes androgen-independent proliferation of prostate cancer cells via induction of androgen receptor expression and activation. Carcinogenesis 2008, 29, 1148–1156. [Google Scholar] [CrossRef]
- Araki, S.; Omori, Y.; Lyn, D.; Singh, R.K.; Meinbach, D.M.; Sandman, Y.; Lokeshwar, V.B.; Lokeshwar, B.L. Interleukin-8 is a molecular determinant of androgen independence and progression in prostate cancer. Cancer Res. 2007, 67, 6854–6862. [Google Scholar] [CrossRef]
- Lee, L.-F.; Louie, M.C.; Desai, S.J.; Yang, J.; Chen, H.-W.; Evans, C.P.; Kung, H.-J. Interleukin-8 confers androgen-independent growth and migration of LNCaP: Differential effects of tyrosine kinases Src and FAK. Oncogene 2004, 23, 2197–2205. [Google Scholar] [CrossRef]
- MacManus, C.F.; Pettigrew, J.; Seaton, A.; Wilson, C.; Maxwell, P.J.; Berlingeri, S.; Purcell, C.; McGurk, M.; Johnston, P.G.; Waugh, D.J.J. Interleukin-8 signaling promotes translational regulation of cyclin D in androgen-independent prostate cancer cells. Mol. Cancer Res. 2007, 5, 737–748. [Google Scholar] [CrossRef]
- Ning, Y.; Manegold, P.C.; Hong, Y.K.; Zhang, W.; Pohl, A.; Lurje, G.; Winder, T.; Yang, D.; LaBonte, M.J.; Wilson, P.M.; et al. Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int. J. Cancer 2011, 128, 2038–2049. [Google Scholar] [CrossRef]
- Luppi, F.; Longo, A.M.; de Boer, W.I.; Rabe, K.F.; Hiemstra, P.S. Interleukin-8 stimulates cell proliferation in non-small cell lung cancer through epidermal growth factor receptor transactivation. Lung Cancer 2007, 56, 25–33. [Google Scholar] [CrossRef]
- Gabellini, C.; Trisciuoglio, D.; Desideri, M.; Candiloro, A.; Ragazzoni, Y.; Orlandi, A.; Zupi, G.; del Bufalo, D. Functional activity of CXCL8 receptors, CXCR1 and CXCR2, on human malignant melanoma progression. Eur. J. Cancer 2009, 45, 2618–2627. [Google Scholar] [CrossRef]
- Strieter, R.M.; Belperio, J.A.; Phillips, R.J.; Keane, M.P. CXC chemokines in angiogenesis of cancer. Semin. Cancer Biol. 2004, 14, 195–200. [Google Scholar] [CrossRef]
- Kim, S.J.; Uehara, H.; Karashima, T.; Mccarty, M.; Shih, N.; Fidler, I.J. Expression of interleukin-8 correlates with angiogenesis, tumorigenicity, and metastasis of human prostate cancer cells implanted orthotopically in nude mice. Neoplasia 2001, 3, 33–42. [Google Scholar]
- Moore, B.B.; Arenberg, D.A.; Stoy, K.; Morgan, T.; Addison, C.L.; Morris, S.B.; Glass, M.; Wilke, C.; Xue, Y.Y.; Sitterding, S.; et al. Distinct CXC chemokines mediate tumorigenicity of prostate cancer cells. Am. J. Pathol. 1999, 154, 1503–1512. [Google Scholar] [CrossRef]
- Singh, S.; Varney, M.; Singh, R.K. Host CXCR2-dependent regulation of melanoma growth, angiogenesis, and experimental lung metastasis. Cancer Res. 2009, 69, 411–415. [Google Scholar] [CrossRef]
- Matsuo, Y.; Raimondo, M.; Woodward, T.A.; Wallace, M.B.; Gill, K.R.; Tong, Z.; Burdick, M.D.; Yang, Z.; Strieter, R.M.; Hoffman, R.M.; et al. CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer. Int. J. Cancer 2009, 125, 1027–1037. [Google Scholar] [CrossRef]
- Arenberg, D.A.; Kunkel, S.L.; Polverini, P.J.; Glass, M.; Burdick, M.D.; Strieter, R.M. Inhibition of interleukin-8 reduces tumorigenesis of human non-small cell lung cancer in SCID mice. J. Clin. Invest. 1996, 97, 2792–2802. [Google Scholar] [CrossRef]
- Reiland, J.; Furcht, L.T.; McCarthy, J.B. CXC-chemokines stimulate invasion and chemotaxis in prostate carcinoma cells through the CXCR2 receptor. Prostate 1999, 41, 78–88. [Google Scholar] [CrossRef]
- McFarlane, S.; Waugh, D. Personal communication, Centre for Cancer Research and Cell Biology, Queen’s University Belfast: Northern Ireland, 1 May 2013.
- Wang, J.M.; Taraboletti, G.; Matsushima, K.; van Damme, J.; Mantovani, A. Induction of haptotactic migration of melanoma cells by neutrophil activating protein/interleukin-8. Biochem. Biophys. Res. Commun. 1990, 169, 165–170. [Google Scholar] [CrossRef]
- Wilson, A.J.; Byron, K.; Gibson, P.R. Interleukin-8 stimulates the migration of human colonic epithelial cells in vitro. Clin. Sci. (London) 1999, 97, 385–390. [Google Scholar] [CrossRef]
- Kuai, W.-X.; Wang, Q.; Yang, X.-Z.; Zhao, Y.; Yu, R.; Tang, X.-J. Interleukin-8 associates with adhesion, migration, invasion and chemosensitivity of human gastric cancer cells. World J. Gastroenterol. 2012, 18, 979–985. [Google Scholar] [CrossRef]
- Houghton, A.M. The paradox of tumor-associated neutrophils: Fueling tumor growth with cytotoxic substances. Cell Cycle 2010, 9, 1732–1737. [Google Scholar] [CrossRef]
- Tazzyman, S.; Lewis, C.E.; Murdoch, C. Neutrophils: Key mediators of tumour angiogenesis. Int. J. Exp. Pathol. 2009, 90, 222–231. [Google Scholar] [CrossRef]
- Jensen, T.O.; Schmidt, H.; Møller, H.J.; Donskov, F.; Høyer, M.; Sjoegren, P.; Christensen, I.J.; Steiniche, T. Intratumoral neutrophils and plasmacytoid dendritic cells indicate poor prognosis and are associated with pSTAT3 expression in AJCC stage I/II melanoma. Cancer 2012, 118, 2476–2485. [Google Scholar] [CrossRef]
- Rao, H.-L.; Chen, J.-W.; Li, M.; Xiao, Y.-B.; Fu, J.; Zeng, Y.-X.; Cai, M.-Y.; Xie, D. Increased intratumoral neutrophil in colorectal carcinomas correlates closely with malignant phenotype and predicts patients’ adverse prognosis. PLoS One 2012, 7, e30806. [Google Scholar]
- Jensen, H.K.; Donskov, F.; Marcussen, N.; Nordsmark, M.; Lundbeck, F.; von der Maase, H. Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J. Clin. Oncol. 2009, 27, 4709–4717. [Google Scholar] [CrossRef]
- Queen, M.M.; Ryan, R.E.; Holzer, R.G.; Keller-Peck, C.R.; Jorcyk, C.L. Breast cancer cells stimulate neutrophils to produce oncostatin M: Potential implications for tumor progression. Cancer Res. 2005, 65, 8896–8904. [Google Scholar] [CrossRef]
- Shang, K.; Bai, Y.-P.; Wang, C.; Wang, Z.; Gu, H.-Y.; Du, X.; Zhou, X.-Y.; Zheng, C.-L.; Chi, Y.-Y.; Mukaida, N.; et al. Crucial involvement of tumor-associated neutrophils in the regulation of chronic colitis-associated carcinogenesis in mice. PLoS One 2012, 7, e51848. [Google Scholar] [CrossRef]
- Tazzyman, S.; Barry, S.T.; Ashton, S.; Wood, P.; Blakey, D.; Lewis, C.E.; Murdoch, C. Inhibition of neutrophil infiltration into A549 lung tumors in vitro and in vivo using a CXCR2-specific antagonist is associated with reduced tumor growth. Int. J. Cancer 2011, 129, 847–858. [Google Scholar] [CrossRef]
- Farooq, S.M.; Stillie, R.; Svensson, M.; Svanborg, C.; Strieter, R.M.; Stadnyk, A.W. Therapeutic effect of blocking CXCR2 on neutrophil recruitment and dextran sodium sulfate-induced colitis. J. Pharmacol. Exp. Ther. 2009, 329, 123–129. [Google Scholar] [CrossRef]
- Jamieson, T.; Clarke, M.; Steele, C.W.; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.B.; Sansom, O.J. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Invest. 2012, 122, 3127–3144. [Google Scholar] [CrossRef]
- Maxwell, P.J.; Gallagher, R.; Seaton, A.; Wilson, C.; Scullin, P.; Pettigrew, J.; Stratford, I.J.; Williams, K.J.; Johnston, P.G.; Waugh, D.J.J. HIF-1 and NF-kappaB-mediated upregulation of CXCR1 and CXCR2 expression promotes cell survival in hypoxic prostate cancer cells. Oncogene 2007, 26, 7333–7345. [Google Scholar] [CrossRef]
- Wilson, C.; Purcell, C.; Seaton, A.; Oladipo, O.; Maxwell, P.J.; Sullivan, J.M.O.; Wilson, R.H.; Johnston, P.G.; Waugh, D.J.J. Chemotherapy-induced CXC-chemokine/CXC-chemokine receptor signaling in metastatic prostate cancer cells confers resistance to oxaliplatin through potentiation of nuclear factor-kappaB transcription and evasion of apoptosis. Pharmacology 2008, 327, 746–759. [Google Scholar]
- Wilson, C.; Wilson, T.; Johnston, P.G.; Longley, D.B.; Waugh, D.J.J. Interleukin-8 signaling attenuates TRAIL- and chemotherapy-induced apoptosis through transcriptional regulation of c-FLIP in prostate cancer cells. Mol. Cancer Ther. 2008, 7, 2649–2661. [Google Scholar] [CrossRef]
- Wilson, C.; Maxwell, P.J.; Longley, D.B.; Wilson, R.H.; Johnston, P.G.; Waugh, D.J.J. Constitutive and treatment-induced CXCL8-signalling selectively modulates the efficacy of anti-metabolite therapeutics in metastatic prostate cancer. PloS One 2012, 7, e36545. [Google Scholar]
- Ning, Y.; Labonte, M.J.; Zhang, W.; Bohanes, P.O.; Gerger, A.; Yang, D.; Benhaim, L.; Paez, D.; Rosenberg, D.O.; Nagulapalli Venkata, K.C.; et al. The CXCR2 antagonist, SCH-527123, shows antitumor activity and sensitizes cells to oxaliplatin in preclinical colon cancer models. Mol Cancer Ther. 2012, 11, 1353–1364. [Google Scholar] [CrossRef]
- Wang, Y.; Qu, Y.; Niu, X.L.; Sun, W.J.; Zhang, X.L.; Li, L.Z. Autocrine production of interleukin-8 confers cisplatin and paclitaxel resistance in ovarian cancer cells. Cytokine 2011, 56, 365–375. [Google Scholar] [CrossRef]
- Shi, Q.; Le, X.; Abbruzzese, J.L.; Wang, B.; Mujaida, N.; Matsushima, K.; Huang, S.; Xiong, Q.; Xie, K. Cooperation between transcription factor AP-1 and NF-kappaB in the induction of interleukin-8 in human pancreatic adenocarcinoma cells by hypoxia. J. Interf. Cytok. Res. 1999, 19, 1363–1371. [Google Scholar] [CrossRef]
- Xie, K. Interleukin-8 and human cancer biology. Cytokine Growth F. R. 2001, 12, 375–391. [Google Scholar] [CrossRef]
- Hashimoto, S.; Matsumoto, K.; Gon, Y.; Maruoka, S.; Takeshita, I.; Hayashi, S.; Koura, T.; Kujime, K.; Horie, T. p38 Mitogen-activated protein kinase regulates IL-8 expression in human pulmonary vascular endothelial cells. Eur. Respir. J. 1999, 13, 1357–1364. [Google Scholar]
- Li, H.; Nord, E.P. CD40 ligation stimulates MCP-1 and IL-8 production, TRAF6 recruitment, and MAPK activation in proximal tubule cells. Am. J. Physiol. Renal. 2002, 282, F1020–F1033. [Google Scholar]
- Ridley, S.H.; Sarsfield, S.J.; Lee, J.C.; Bigg, H.F.; Cawston, T.E.; Taylor, D.J.; DeWitt, D.L.; Saklatvala, J. Actions of IL-1 are selectively controlled by p38 mitogen-activated protein kinase: Regulation of prostaglandin H synthase-2, metalloproteinases, and IL-6 at different levels. J. Immunol. 1997, 158, 3165–3173. [Google Scholar]
- Kuldo, J.M.; Westra, J.; Asgeirsdottir, S.A.; Kok, R.J.; Oosterhuis, K.; Rots, M.G.; Schouten, J.P.; Limburg, P.C.; Molema, G. Differential effects of NF-{kappa}B and p38 MAPK inhibitors and combinations thereof on TNF-{alpha}- and IL-1{beta}-induced proinflammatory status of endothelial cells in vitro. Am. J. Physiol. Cell Ph. 2005, 289, C1229–C1239. [Google Scholar] [CrossRef]
- Bonavia, R.; Inda, M.M.; Vandenberg, S.; Cheng, S.Y.; Nagane, M.; Hadwiger, P.; Tan, P.; Sah, D.W.; Cavenee, W.K.; Furnari, F.B. EGFRvIII promotes glioma angiogenesis and growth through the NF-kappaB, interleukin-8 pathway. Oncogene 2012, 31, 4054–4066. [Google Scholar] [CrossRef]
- Lomas, D.A.; Lipson, D.A.; Miller, B.E.; Willits, L.; Keene, O.; Barnacle, H.; Barnes, N.C.; Tal-Singer, R.; Investigators, L.S. An oral inhibitor of p38 MAP kinase reduces plasma fibrinogen in patients with chronic obstructive pulmonary disease. J. Clin. Pharmacol. 2012, 52, 416–424. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Wang, L.; Wang, X.; Xia, J. Regulatory mechanisms of interleukin-8 production induced by tumour necrosis factor-alpha in human hepatocellular carcinoma cells. J. Cell Mol. Med. 2012, 16, 496–506. [Google Scholar] [CrossRef]
- Fernandes, A.F.; Bian, Q.; Jiang, J.K.; Thomas, C.J.; Taylor, A.; Pereira, P.; Shang, F. Proteasome inactivation promotes p38 mitogen-activated protein kinase-dependent phosphatidylinositol 3-kinase activation and increases interleukin-8 production in retinal pigment epithelial cells. Mol. Biol. Cell 2009, 20, 3690–3699. [Google Scholar] [CrossRef]
- Osawa, Y.; Nagaki, M.; Banno, Y.; Brenner, D.A.; Asano, T.; Nozawa, Y.; Moriwaki, H.; Nakashima, S. Tumor necrosis factor alpha-induced interleukin-8 production via NF-kappaB and phosphatidylinositol 3-kinase/Akt pathways inhibits cell apoptosis in human hepatocytes. Infect. Immun. 2002, 70, 6294–6301. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Zhang, M.; Jin, M.; Bai, C.; Wang, X. Potential mechanism of interleukin-8 production from lung cancer cells: An involvement of EGF-EGFR-PI3K-Akt-Erk pathway. J. Cell Physiol. 2012, 227, 35–43. [Google Scholar] [CrossRef]
- Bagrodia, S.; Smeal, T.; Abraham, R.T. Mechanisms of intrinsic and acquired resistance to kinase-targeted therapies. Pigm. Cell Melanoma R 2012, 25, 819–831. [Google Scholar] [CrossRef]
- Britschgi, A.; Andraos, R.; Brinkhaus, H.; Klebba, I.; Romanet, V.; Muller, U.; Murakami, M.; Radimerski, T.; Bentires-Alj, M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: A rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell 2012, 22, 796–811. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Z.; Wang, L.; Zhang, X. NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell Mol. Immunol. 2009, 6, 327–334. [Google Scholar] [CrossRef]
- Palayoor, S.T.; Youmell, M.Y.; Calderwood, S.K.; Coleman, C.N.; Price, B.D. Constitutive activation of IkappaB kinase alpha and NF-kappaB in prostate cancer cells is inhibited by ibuprofen. Oncogene 1999, 18, 7389–7394. [Google Scholar] [CrossRef]
- Gasparian, A.V; Yao, Y.J.; Kowalczyk, D.; Lyakh, L.A.; Karseladze, A.; Slaga, T.J.; Budunova, I.V. The role of IKK in constitutive activation of NF-kappaB transcription factor in prostate carcinoma cells. J. Cell Sci. 2002, 115, 141–151. [Google Scholar]
- Suh, J.; Payvandi, F.; Edelstein, L.C.; Amenta, P.S.; Zong, W.X.; Gelinas, C.; Rabson, A.B. Mechanisms of constitutive NF-kappaB activation in human prostate cancer cells. Prostate 2002, 52, 183–200. [Google Scholar] [CrossRef]
- Tafani, M.; Pucci, B.; Russo, A.; Schito, L.; Pellegrini, L.; Perrone, G.A.; Villanova, L.; Salvatori, L.; Ravenna, L.; Petrangeli, E.; Russo, M.A. Modulators of HIF1alpha and NFkB in Cancer Treatment: Is it a Rational Approach for Controlling Malignant Progression? Front. Pharmacol. 2013, 4, 13. [Google Scholar]
- Lee, D.F.; Hung, M.C. Advances in targeting IKK and IKK-related kinases for cancer therapy. Clin. Cancer Res. 2008, 14, 5656–5662. [Google Scholar] [CrossRef]
- Boccadoro, M.; Morgan, G.; Cavenagh, J. Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy. Cancer Cell 2005, 5, 18. [Google Scholar] [CrossRef]
- Williams, S.A.; McConkey, D.J. The proteasome inhibitor bortezomib stabilizes a novel active form of p53 in human LNCaP-Pro5 prostate cancer cells. Cancer Res. 2003, 63, 7338–7344. [Google Scholar]
- Kamat, A.M.; Karashima, T.; Davis, D.W.; Lashinger, L.; Bar-Eli, M.; Millikan, R.; Shen, Y.; Dinney, C.P.; McConkey, D.J. The proteasome inhibitor bortezomib synergizes with gemcitabine to block the growth of human 253JB-V bladder tumors in vivo. Mol. Cancer Ther. 2004, 3, 279–290. [Google Scholar]
- Tsapakidis, K.; Vlachostergios, P.J.; Voutsadakis, I.A.; Befani, C.D.; Patrikidou, A.; Hatzidaki, E.; Daliani, D.D.; Moutzouris, G.; Liakos, P.; Papandreou, C.N. Bortezomib reverses the proliferative and antiapoptotic effect of neuropeptides on prostate cancer cells. Int. J. Urol. 2012, 19, 565–574. [Google Scholar] [CrossRef]
- Plummer, S.M.; Holloway, K.A.; Manson, M.M.; Munks, R.J.; Kaptein, A.; Farrow, S.; Howells, L. Inhibition of cyclo-oxygenase 2 expression in colon cells by the chemopreventive agent curcumin involves inhibition of NF-kappaB activation via the NIK/IKK signalling complex. Oncogene 1999, 18, 6013–6020. [Google Scholar] [CrossRef]
- Chen, Y.R.; Tan, T.H. Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene 1998, 17, 173–178. [Google Scholar]
- Bierhaus, A.; Zhang, Y.; Quehenberger, P.; Luther, T.; Haase, M.; Muller, M.; Mackman, N.; Ziegler, R.; Nawroth, P.P. The dietary pigment curcumin reduces endothelial tissue factor gene expression by inhibiting binding of AP-1 to the DNA and activation of NF-kappa B. Thromb. Haemostasis 1997, 77, 772–782. [Google Scholar]
- Shishodia, S.; Singh, T.; Chaturvedi, M.M. Modulation of transcription factors by curcumin. Adv. Exp. Med. Biol. 2007, 595, 127–148. [Google Scholar] [CrossRef]
- Zhou, H.; Beevers, C.S.; Huang, S. The targets of curcumin. Curr. Drug Targets 2011, 12, 332–347. [Google Scholar] [CrossRef]
- Strimpakos, A.S.; Sharma, R.A. Curcumin: Preventive and therapeutic properties in laboratory studies and clinical trials. Antioxid. Redox Sign. 2008, 10, 511–545. [Google Scholar] [CrossRef]
- Hidaka, H.; Ishiko, T.; Furuhashi, T.; Kamohara, H.; Suzuki, S.; Miyazaki, M.; Ikeda, O.; Mita, S.; Setoguchi, T.; Ogawa, M. Curcumin inhibits interleukin 8 production and enhances interleukin 8 receptor expression on the cell surface: Impact on human pancreatic carcinoma cell growth by autocrine regulation. Cancer 2002, 95, 1206–1214. [Google Scholar] [CrossRef]
- Kwon, O.J.; Au, B.T.; Collins, P.D.; Baraniuk, J.N.; Adcock, I.M.; Chung, K.F.; Barnes, P.J. Inhibition of interleukin-8 expression by dexamethasone in human cultured airway epithelial cells. Immunology 1994, 81, 389–394. [Google Scholar]
- Chang, M.M.; Juarez, M.; Hyde, D.M.; Wu, R. Mechanism of dexamethasone-mediated interleukin-8 gene suppression in cultured airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L107–L115. [Google Scholar]
- Eddleston, J.; Herschbach, J.; Wagelie-Steffen, A.L.; Christiansen, S.C.; Zuraw, B.L. The anti-inflammatory effect of glucocorticoids is mediated by glucocorticoid-induced leucine zipper in epithelial cells. J. Allergy Clin. Immun. 2007, 119, 115–122. [Google Scholar] [CrossRef]
- Scheinman, R.I.; Gualberto, A.; Jewell, C.M.; Cidlowski, J.A.; Baldwin, A.S., Jr. Characterization of mechanisms involved in transrepression of NF-kappa B by activated glucocorticoid receptors. Mol. Cell. Biol. 1995, 15, 943–953. [Google Scholar]
- Dauletbaev, N.; Eklove, D.; Mawji, N.; Iskandar, M.; di Marco, S.; Gallouzi, I.E.; Lands, L.C. Down-regulation of cytokine-induced interleukin-8 requires inhibition of p38 mitogen-activated protein kinase (MAPK) via MAPK phosphatase 1-dependent and -independent mechanisms. J. Biol. Chem. 2011, 286, 15998–16007. [Google Scholar]
- Chen, J.J.W.; Yao, P.-L.; Yuan, A.; Hong, T.-M.; Shun, C.-T.; Kuo, M.-L.; Lee, Y.-C.; Yang, P.-C. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: Its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin. Cancer Res. 2003, 9, 729–737. [Google Scholar]
- Venkitaraman, R.; Thomas, K.; Huddart, R.A.; Horwich, A.; Dearnaley, D.P.; Parker, C.C. Efficacy of low-dose dexamethasone in castration-refractory prostate cancer. BJU Int. 2008, 101, 440–443. [Google Scholar]
- Yano, A.; Fujii, Y.; Iwai, A.; Kageyama, Y.; Kihara, K. Glucocorticoids suppress tumor angiogenesis and in vivo growth of prostate cancer cells. Clin. Cancer Res. 2006, 12, 3003–3009. [Google Scholar] [CrossRef]
- Wilson, C.; Scullin, P.; Worthington, J.; Seaton, A.; Maxwell, P.; O’Rourke, D.; Johnston, P.G.; McKeown, S.R.; Wilson, R.H.; O’Sullivan, J.M.; et al. Dexamethasone potentiates the antiangiogenic activity of docetaxel in castration-resistant prostate cancer. Br. J. Cancer 2008, 99, 2054–2064. [Google Scholar] [CrossRef]
- Lopez-Armada, M.J.; Sanchez-Pernaute, O.; Largo, R.; Diez-Ortego, I.; Palacios, I.; Egido, J.; Herrero-Beaumont, G. Modulation of cell recruitment by anti-inflammatory agents in antigen-induced arthritis. Ann. Rheum. Dis. 2002, 61, 1027–1030. [Google Scholar] [CrossRef]
- Gustafson-Svard, C.; Lilja, I.; Hallbook, O.; Sjodahl, R. Cyclo-oxygenase and colon cancer: Clues to the aspirin effect? Ann. Med. 1997, 29, 247–252. [Google Scholar] [CrossRef]
- Bosetti, C.; Rosato, V.; Gallus, S.; Cuzick, J.; la Vecchia, C. Aspirin and cancer risk: A quantitative review to 2011. Ann. Oncol. 2012, 23, 1403–1415. [Google Scholar] [CrossRef]
- Ouyang, N.; Ji, P.; Williams, J.L. A novel NSAID derivative, phospho-ibuprofen, prevents AOM-induced colon cancer in rats. Int. J. Oncol. 2013, 42, 643–650. [Google Scholar]
- Ji, H.; Greening, D.W.; Kapp, E.A.; Moritz, R.L.; Simpson, R.J. Secretome-based proteomics reveals sulindac-modulated proteins released from colon cancer cells. Proteom. Clin. Appl. 2009, 3, 433–451. [Google Scholar] [CrossRef]
- Housby, J.N.; Cahill, C.M.; Chu, B.; Prevelige, R.; Bickford, K.; Stevenson, M.A.; Calderwood, S.K. Non-steroidal anti-inflammatory drugs inhibit the expression of cytokines and induce HSP70 in human monocytes. Cytokine 1999, 11, 347–358. [Google Scholar] [CrossRef]
- Bekes, E.M.; Schweighofer, B.; Kupriyanova, T.A.; Zajac, E.; Ardi, V.C.; Quigley, J.P.; Deryugina, E.I. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am. J. Pathol. 2011, 179, 1455–1470. [Google Scholar] [CrossRef]
- Bizzarri, C.; Beccari, A.R.; Bertini, R.; Cavicchia, M.R.; Giorgini, S.; Allegretti, M. ELR+ CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol. Therapeut. 2006, 112, 139–149. [Google Scholar] [CrossRef]
- Mahler, D.A.; Huang, S.; Tabrizi, M.; Bell, G.M. Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in COPD: A pilot study. Chest 2004, 126, 926–934. [Google Scholar] [CrossRef]
- Huang, S.; Mills, L.; Mian, B.; Tellez, C.; McCarty, M.; Yang, X.-D.; Gudas, J.M.; Bar-Eli, M. Fully humanized neutralizing antibodies to interleukin-8 (ABX-IL8) inhibit angiogenesis, tumor growth, and metastasis of human melanoma. Am. J. Pathol. 2002, 161, 125–134. [Google Scholar] [CrossRef]
- Skov, L.; Beurskens, F.J.; Zachariae, C.O.C.; Reitamo, S.; Teeling, J.; Satijn, D.; Knudsen, K.M.; Boot, E.P.J.; Hudson, D.; Baadsgaard, O.; et al. IL-8 as antibody therapeutic target in inflammatory diseases: Reduction of clinical activity in palmoplantar pustulosis. J. Immunol. 2008, 181, 669–679. [Google Scholar]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef]
- Saintigny, P.; Massarelli, E.; Lin, S.; Ahn, Y.-H.; Chen, Y.; Goswami, S.; Erez, B.; O’Reilly, M.S.; Liu, D.; Lee, J.J.; et al. CXCR2 expression in tumor cells is a poor prognostic factor and promotes invasion and metastasis in lung adenocarcinoma. Cancer Res. 2013, 73, 571–582. [Google Scholar] [CrossRef]
- Zhu, Y.M.; Bagstaff, S.M.; Woll, P.J. Production and upregulation of granulocyte chemotactic protein-2/CXCL6 by IL-1beta and hypoxia in small cell lung cancer. Br. J. Cancer 2006, 94, 1936–1941. [Google Scholar] [CrossRef]
- Hussain, F.; Freissmuth, M.; Volkel, D.; Thiele, M.; Douillard, P.; Antoine, G.; Thurner, P.; Ehrlich, H.; Schwarz, H.-P.; Scheiflinger, F.; et al. Human Anti-Macrophage Migration Inhibitory Factor (MIF) Antibodies Inhibit Growth of Human Prostate Cancer Cells In Vitro and In Vivo. Mol. Cancer Ther. 2013, 12, 1223–1234. [Google Scholar] [CrossRef]
- Zhu, Y.M.; Webster, S.J.; Flower, D.; Woll, P.J. Interleukin-8/CXCL8 is a growth factor for human lung cancer cells. Br. J. Cancer 2004, 91, 1970–1976. [Google Scholar] [CrossRef]
- Sánchez, J.; Moldobaeva, A.; McClintock, J.; Jenkins, J.; Wagner, E. The role of CXCR2 in systemic neovascularization of the mouse lung. J. Appl. Physiol. 2007, 103, 594–599. [Google Scholar] [CrossRef]
- Allegretti, M.; Bertini, R.; Cesta, M.C.; Bizzarri, C.; di Bitondo, R.; di Cioccio, V.; Galliera, E.; Berdini, V.; Topai, A.; Zampella, G.; et al. 2-Arylpropionic CXC chemokine receptor 1 (CXCR1) ligands as novel noncompetitive CXCL8 inhibitors. J. Med. Chem. 2005, 48, 4312–4331. [Google Scholar] [CrossRef]
- Moriconi, A.; Cesta, M.C.; Cervellera, M.N.; Aramini, A.; Coniglio, S.; Colagioia, S.; Beccari, A.R.; Bizzarri, C.; Cavicchia, M.R.; Locati, M.; et al. Design of noncompetitive interleukin-8 inhibitors acting on CXCR1 and CXCR2. J. Med. Chem. 2007, 50, 3984–4002. [Google Scholar] [CrossRef]
- Ginestier, C.; Liu, S.; Diebel, M.E.; Korkaya, H.; Luo, M.; Brown, M.; Wicinski, J.; Cabaud, O.; Charafe-Jauffret, E.; Birnbaum, D.; Guan, J.-L.; Dontu, G.; Wicha, M.S. CXCR1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. J. Clin. Invest. 2010, 120, 485–497. [Google Scholar] [CrossRef]
- Hitting Cancer at Its Core: At San Antonio Breast Cancer Symposium Eyes Focused on Reparixin, a Drug Fruit of Dompé’s Research, with Action Aimed at Breast Cancer Stem Cells. Available online: www.pharmaceutical-tech.com/press/pressrelease_archives.asp?PID=1242/ (accessed online 3 July 2013).
- Dompe Signs Up First Patient for Reparixin Phase III Trial. Available online: www.clinicalprofessionals.co.uk/blog/dompe-signs-up-first-patient-for-reparixin-phase-iii-trial/ (accessed on 3 July 2013).
- Citro, A.; Cantarelli, E.; Maffi, P. CXCR1/2 inhibition enhances pancreatic islet survival after transplantation. J. Clin. Invest. 2012, 122, 3647–3651. [Google Scholar] [CrossRef]
- Available online: www.clinicaltrials.gov (accessed on 3 July 2013). Study number NCT00248040.
- Available online: www.clinicaltrials.gov (accessed on 3 July 2013). Study number NCT00224406.
- Barsante, M.M.; Cunha, T.M.; Allegretti, M.; Cattani, F.; Policani, F.; Bizzarri, C.; Tafuri, W.L.; Poole, S.; Cunha, F.Q.; Bertini, R.; et al. Blockade of the chemokine receptor CXCR2 ameliorates adjuvant-induced arthritis in rats. Br. J. Pharmacol. 2008, 153, 992–1002. [Google Scholar]
- Russo, R.C.; Guabiraba, R.; Garcia, C.C.; Barcelos, L.S.; Roffê, E.; Souza, A.L.S.; Amaral, F.A.; Cisalpino, D.; Cassali, G.D.; et al. Role of the chemokine receptor CXCR2 in bleomycin-induced pulmonary inflammation and fibrosis. Am. J. Resp. Cell Mol. 2009, 40, 410–421. [Google Scholar] [CrossRef]
- Salchow, K.; Bond, M.E.; Evans, S.C.; Press, N.J.; Charlton, S.J.; Hunt, P.A.; Bradley, M.E. A common intracellular allosteric binding site for antagonists of the CXCR2 receptor. Br. J. Pharmacol. 2010, 159, 1429–1439. [Google Scholar] [CrossRef]
- Gonsiorek, W.; Fan, X.; Hesk, D.; Fossetta, J. Pharmacological characterization of Sch527123, a potent allosteric CXCR1/CXCR2 antagonist. J. Pharmacol. Exp. Ther. 2007, 322, 477–485. [Google Scholar] [CrossRef]
- Varney, M.L.; Singh, S.; Li, A.; Mayer-Ezell, R.; Bond, R.; Singh, R.K. Small molecule antagonists for CXCR2 and CXCR1 inhibit human colon cancer liver metastases. Cancer Lett. 2011, 300, 180–188. [Google Scholar] [CrossRef]
- Holz, O.; Khalilieh, S.; Ludwig-Sengpiel, A.; Watz, H.; Stryszak, P.; Soni, P.; Tsai, M.; Sadeh, J.; Magnussen, H. SCH527123, a novel CXCR2 antagonist, inhibits ozone-induced neutrophilia in healthy subjects. Eur. Respir. J. 2010, 35, 564–570. [Google Scholar] [CrossRef]
- Available online: https://www.ersnetsecure.org/public/prg_congres.abstract?ww_i_presentation=46823 (accessed on 3 July 2013).
- Nair, P.; Gaga, M.; Zervas, E.; Alagha, K.; Hargreave, F.E.; O’Byrne, P.M.; Stryszak, P.; Gann, L.; Sadeh, J.; Chanez, P. Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: A randomized, placebo-controlled clinical trial. Clin. Exp. Allergy 2012, 42, 1097–1103. [Google Scholar] [CrossRef]
- White, J.R.; Lee, J.M.; Young, P.R.; Hertzberg, R.P.; Jurewicz, A.J.; Chaikin, M.A.; Widdowson, K.; Foley, J.J.; Martin, L.D.; Griswold, D.E.; et al. Identification of a potent, selective non-peptide CXCR2 antagonist that inhibits interleukin-8-induced neutrophil migration. J. Biol. Chem. 1998, 273, 10095–10098. [Google Scholar] [CrossRef]
- Pease, J.; Horuk, R. Chemokine receptor antagonists. J. Med. Chem. 2012, 55, 9363–9392. [Google Scholar] [CrossRef]
- Bento, A.F.; Leite, D.F.P.; Claudino, R.F.; Hara, D.B.; Leal, P.C.; Calixto, J.B. The selective nonpeptide CXCR2 antagonist SB225002 ameliorates acute experimental colitis in mice. J. Leukocyte Biol. 2008, 84, 1213–1221. [Google Scholar] [CrossRef]
- Jin, Q.; Nie, H.; McCleland, B.W.; Widdowson, K.L.; Palovich, M.R.; Elliott, J.D.; Goodman, R.M.; Burman, M.; Sarau, H.M.; Ward, K.W.; et al. Discovery of potent and orally bioavailable N,N′-diarylurea antagonists for the CXCR2 chemokine receptor. Bioorg. Med. Chem. Lett. 2004, 14, 4375–4378. [Google Scholar] [CrossRef]
- Lazaar, A.L.; Sweeney, L.E.; MacDonald, A.J.; Alexis, N.E.; Chen, C.; Tal-Singer, R. SB-656933, a novel CXCR2 selective antagonist, inhibits ex vivo neutrophil activation and ozone-induced airway inflammation in humans. Br. J. Clin. Pharmacol. 2011, 72, 282–293. [Google Scholar] [CrossRef]
- Moss, R.B.; Mistry, S.J.; Konstan, M.W.; Pilewski, J.M.; Kerem, E.; Tal-Singer, R.; Lazaar, A.L. CF2110399 Investigators. Safety and early treatment effects of the CXCR2 antagonist SB-656933 in patients with cystic fibrosis. J. Cyst. Fibros. 2013, 12, 241–248. [Google Scholar] [CrossRef]
- Available online: www.clinicaltrials.gov (accessed on 3 July 2013). Study number NCT00748410.
- Chapman, R.W.; Phillips, J.E; Hipkin, R.W.; Curran, A.K.; Lundell, D.; Fine, J.S. CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol. Therapeut. 2009, 121, 55–68. [Google Scholar] [CrossRef]
- De Boer, W.I.; Yao, H.; Rahman, I. Future therapeutic treatment of COPD: Struggle between oxidants and cytokines. Int. J. Chron. Obstruct. Pulmon. Dis. 2007, 2, 205–228. [Google Scholar]
- Virtala, R.; Ekman, A-K.; Jansson, L.; Westin, U.; Cardell, L.O. Airway inflammation evaluated in a human nasal lipopolysaccharide challenge model by investigating the effect of a CXCR2 inhibitor. Clin. Exp. Allergy. 2012, 42, 590–596. [Google Scholar] [CrossRef]
- Hunt, F.; Austin, C.; Austin, R.; Bonnert, R.; Cage, P.; Christie, J.; Christie, M.; Dixon, C.; Hill, S.; Jewell, R.; Martin, I.; Robinson, D.; Willis, P. SAR studies on thiazolo[4,5-d]pyrimidine based CXCR2 antagonists involving a novel tandem displacement reaction. Bioorg. Med. Chem. Lett. 2007, 17, 2731–2734. [Google Scholar] [CrossRef]
- The safety, tolerability and pharmacokinetics of AZD5069, a novel CXCR2 antagonist, in healthy Japanese volunteers. Available online: www.richmondpharmacology.com/downloads/Publications/ERS JSAD-JMAD poster_57.pdf (accessed on 3 July 2013).
- Available online: www.clinicaltrials.gov/ (accessed on 3 July 2013). Study number NCT01233232.
- Available online: www.clinicaltrials.gov/ (accessed on 3 July 2013). Study number NCT01255592.
- Available online: www.clinicaltrials.gov/ (accessed on 3 July 2013). Study number NCT01704495.
- O’Callaghan, K.; Kuliopulos, A.; Covic, L. Turning receptors on and off with intracellular pepducins: New insights into G-protein-coupled receptor drug development. J. Biol. Chem. 2012, 287, 12787–12796. [Google Scholar] [CrossRef]
- Kaneider, N.C.; Agarwal, A.; Leger, A.J.; Kuliopulos, A. Reversing systemic inflammatory response syndrome with chemokine receptor pepducins. Nat. Med. 2005, 11, 661–665. [Google Scholar] [CrossRef]
- Dimond, P.; Carlson, K.; Bouvier, M.; Gerard, C.; Xu, L.; Covic, L.; Agarwal, A.; Ernst, O.P.; Janz, J.M.; Schwartz, T.W.; et al. G protein-coupled receptor modulation with pepducins: Moving closer to the clinic. Ann. N. Y. Acad. Sci. 2011, 1226, 34–49. [Google Scholar] [CrossRef]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef]
- Mendonça, L.S.; Moreira, J.N.; De Lima, M.C.P.; Simões, S. Co-encapsulation of anti-BCR-ABL siRNA and imatinib mesylate in transferrin receptor-targeted sterically stabilized liposomes for chronic myeloid leukemia treatment. Biotechnol. Bioeng. 2010, 107, 884–893. [Google Scholar] [CrossRef]
- Di Paolo, D.; Brignole, C.; Pastorino, F.; Carosio, R.; Zorzoli, A.; Rossi, M.; Loi, M.; Pagnan, G.; Emionite, L.; Cilli, M.; et al. Neuroblastoma-targeted nanoparticles entrapping siRNA specifically knockdown ALK. Mol. Ther. 2011, 19, 1131–1140. [Google Scholar] [CrossRef]
- Merritt, W.M.; Lin, Y.G.; Spannuth, W.A.; Fletcher, M.S.; Kamat, A.A.; Han, L.Y.; Landen, C.N.; Jennings, N.; de Geest, K.; Langley, R.R.; et al. Effect of interleukin-8 gene silencing with liposome-encapsulated small interfering RNA on ovarian cancer cell growth. J. Natl. Cancer I. 2008, 100, 359–372. [Google Scholar] [CrossRef]
- Trotman, L.C.; Niki, M.; Dotan, Z.A.; Koutcher, J.A.; di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; van Dyke, T.; et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 2003, 1, E59. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Campbell, L.M.; Maxwell, P.J.; Waugh, D.J.J. Rationale and Means to Target Pro-Inflammatory Interleukin-8 (CXCL8) Signaling in Cancer. Pharmaceuticals 2013, 6, 929-959. https://doi.org/10.3390/ph6080929
Campbell LM, Maxwell PJ, Waugh DJJ. Rationale and Means to Target Pro-Inflammatory Interleukin-8 (CXCL8) Signaling in Cancer. Pharmaceuticals. 2013; 6(8):929-959. https://doi.org/10.3390/ph6080929
Chicago/Turabian StyleCampbell, Laura M., Pamela J. Maxwell, and David J.J. Waugh. 2013. "Rationale and Means to Target Pro-Inflammatory Interleukin-8 (CXCL8) Signaling in Cancer" Pharmaceuticals 6, no. 8: 929-959. https://doi.org/10.3390/ph6080929
APA StyleCampbell, L. M., Maxwell, P. J., & Waugh, D. J. J. (2013). Rationale and Means to Target Pro-Inflammatory Interleukin-8 (CXCL8) Signaling in Cancer. Pharmaceuticals, 6(8), 929-959. https://doi.org/10.3390/ph6080929