Targeting Platelet Thrombin Receptor Signaling to Prevent Thrombosis

{kind=link}
{kind=link}
Abstract
:1. Introduction
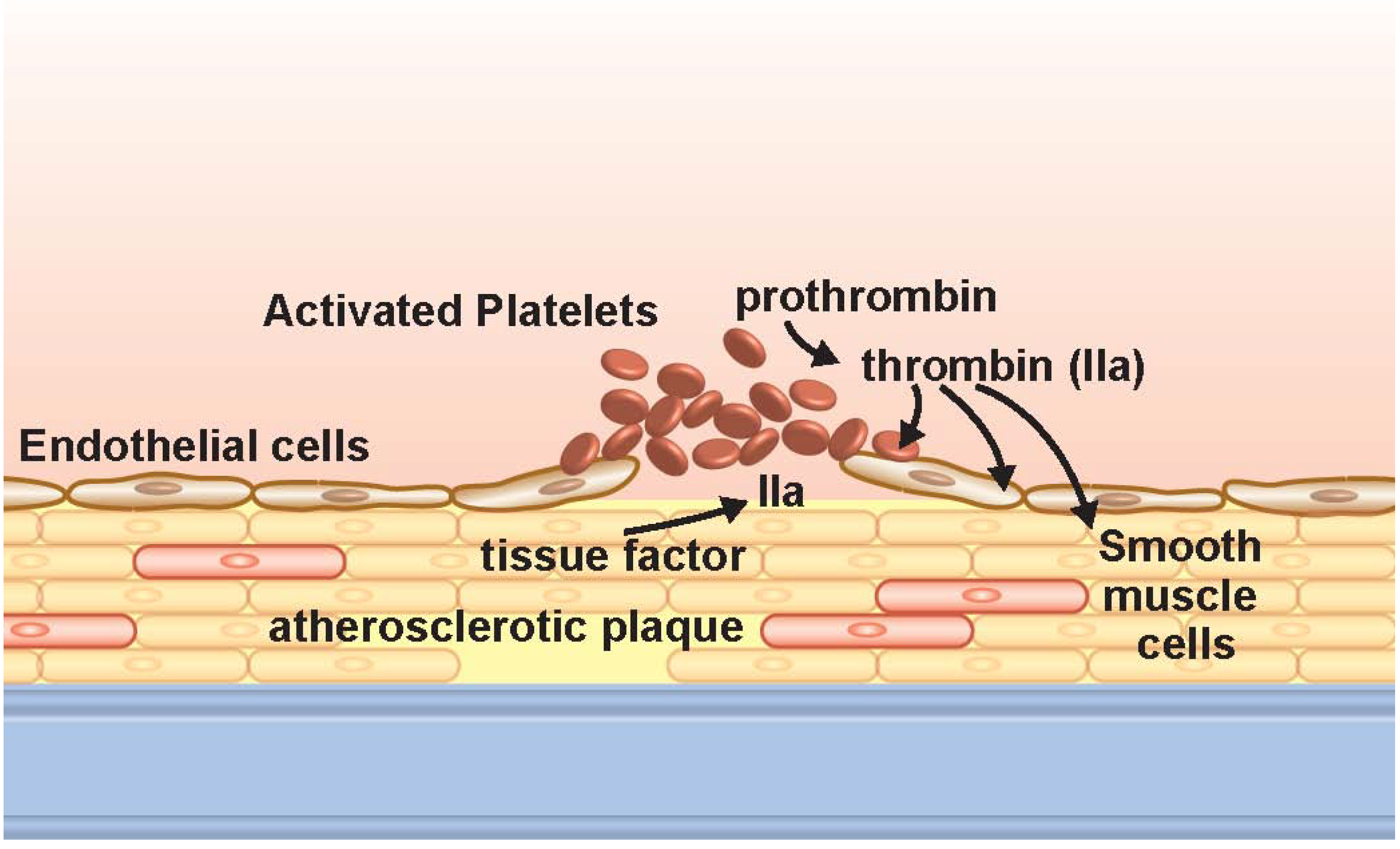
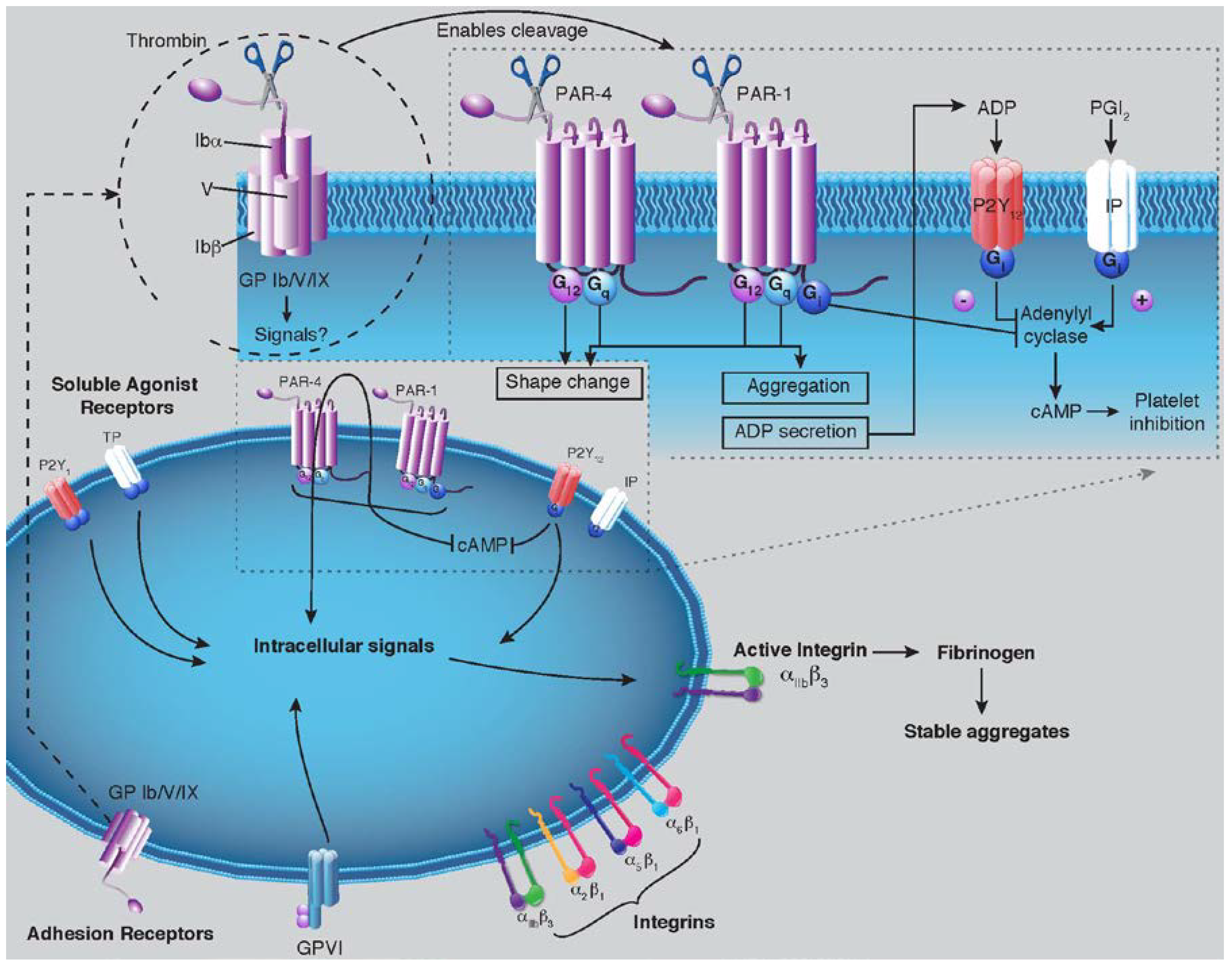
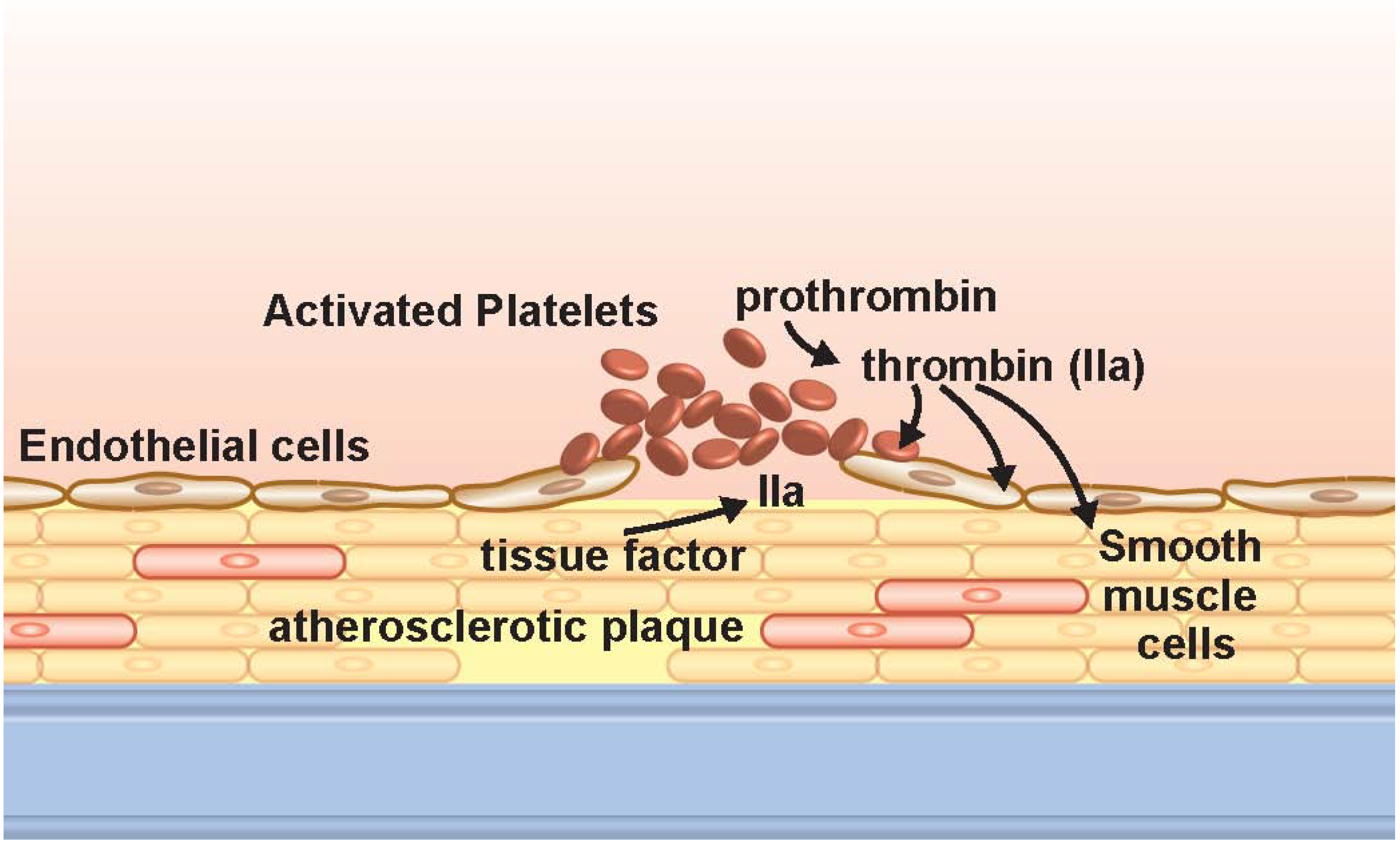
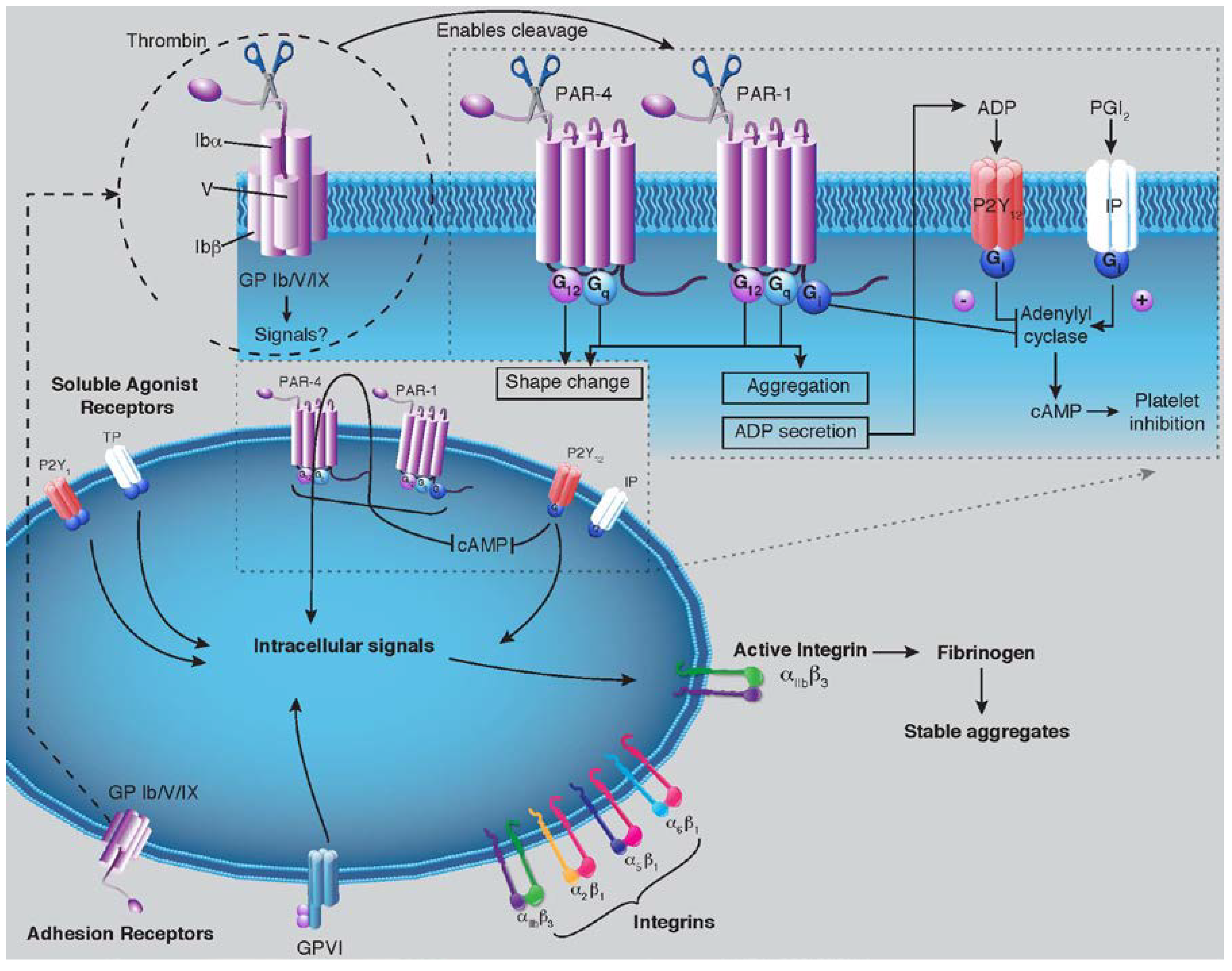
2. Discussion
2.1. Platelet Biology and Thrombin
2.2. PAR-1 Structure and Mechanisms
2.3. Clinical Trials Involving PAR-1
2.3.1. Vorapaxar (SCH 530348)
2.3.2. Atopaxar (E5555)
2.4. Pooled Analysis of Current Trials and Major Bleeding
2.5. Future Pharmacological Agents
2.6. PAR-1 Inhibitors and P2Y12 Antagonism
2.7. Comparison with New Oral Anticoagulants
3. Conclusions
Acknowledgements
Conflict of Interest
References
- ISIS-2 (Second International Study of Infarct Survival) Collaborative Group. Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS-2. Lancet 1988, 2, 349–360.
- Yusuf, S.; Zhao, F.; Mehta, S.R.; Chrolavicius, S.; Tognoni, G.; Fox, K.K. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N. Engl. J. Med. 2001, 345, 494–502. [Google Scholar] [CrossRef]
- Wallentin, L.; Becker, R.C.; Budaj, A.; Cannon, C.P.; Emanuelsson, H.; Held, C.; Horrow, J.; Husted, S.; James, S.; Katus, H.; et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2009, 361, 1045–1057. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Braunwald, E.; McCabe, C.H.; Montalescot, G.; Ruzyllo, W.; Gottlieb, S.; Neumann, F.J.; Ardissino, D.; De Servi, S.; Murphy, S.A.; et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N. Engl. J. Med. 2007, 357, 2001–2015. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.; Adams, R.; Carnethon, M.; De Simone, G.; Ferguson, T.B.; Flegal, K.; Ford, E.; Furie, K.; Go, A.; Greenlund, K.; et al. Heart disease and stroke statistics—2009 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009, 119, 480–486. [Google Scholar] [CrossRef]
- Lincoff, A.M.; Kleiman, N.S.; Kereiakes, D.J.; Feit, F.; Bittl, J.A.; Jackman, J.D.; Sarembock, I.J.; Cohen, D.J.; Spriggs, D.; Ebrahimi, R.; et al. Long-term efficacy of bivalirudin and provisional glycoprotein IIb/IIIa blockade vs. heparin and planned glycoprotein IIb/IIIa blockade during percutaneous coronary revascularization: REPLACE-2 randomized trial. JAMA 2004, 292, 696–703. [Google Scholar] [CrossRef]
- Leger, A.J.; Covic, L.; Kuliopulos, A. Protease-activated receptors in cardiovascular diseases. Circulation 2006, 114, 1070–1077. [Google Scholar] [CrossRef]
- Brummel, K.E.; Paradis, S.G.; Butenas, S.; Mann, K.G. Thrombin functions during tissue factor-induced blood coagulation. Blood 2002, 100, 148–152. [Google Scholar] [CrossRef]
- Camerer, E.; Duong, D.N.; Hamilton, J.R.; Coughlin, S.R. Combined deficiency of protease-activated receptor-4 and fibrinogen recapitulates the hemostatic defect but not the embryonic lethality of prothrombin deficiency. Blood 2004, 103, 152–154. [Google Scholar] [CrossRef]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Invest. 2005, 115, 3378–3384. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Spronk, H.M.; ten Cate, H. The hemostatic system as a modulator of atherosclerosis. N. Engl. J. Med. 2011, 364, 1746–1760. [Google Scholar] [CrossRef]
- Vu, T.K.; Hung, D.T.; Wheaton, V.I.; Coughlin, S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 1991, 64, 1057–1068. [Google Scholar] [CrossRef]
- Coughlin, S.R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 2005, 3, 1800–1814. [Google Scholar] [CrossRef]
- Ossovskaya, V.S.; Bunnett, N.W. Protease-activated receptors: contribution to physiology and disease. Physiol. Rev. 2004, 84, 579–621. [Google Scholar] [CrossRef]
- Kahn, M.L.; Nakanishi-Matsui, M.; Shapiro, M.J.; Ishihara, H.; Coughlin, S.R. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Invest. 1999, 103, 879–887. [Google Scholar] [CrossRef]
- Cornelissen, I.; Palmer, D.; David, T.; Wilsbacher, L.; Concengco, C.; Conley, P.; Pandey, A.; Coughlin, S.R. Roles and interactions among protease-activated receptors and P2ry12 in hemostasis and thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 18605–18610. [Google Scholar] [CrossRef]
- Kato, Y.; Kita, Y.; Hirasawa-Taniyama, Y.; Nishio, M.; Mihara, K.; Ito, K.; Yamanaka, T.; Seki, J.; Miyata, S.; Mutoh, S. Inhibition of arterial thrombosis by a protease-activated receptor 1 antagonist, FR171113, in the guinea pig. Eur. J. Pharmacol. 2003, 473, 163–169. [Google Scholar] [CrossRef]
- Derian, C.K.; Damiano, B.P.; Addo, M.F.; Darrow, A.L.; D'Andrea, M.R.; Nedelman, M.; Zhang, H.C.; Maryanoff, B.E.; Andrade-Gordon, P. Blockade of the thrombin receptor protease-activated receptor-1 with a small-molecule antagonist prevents thrombus formation and vascular occlusion in nonhuman primates. J. Pharmacol. Exp. Ther. 2003, 304, 855–861. [Google Scholar] [CrossRef]
- Chintala, M.; Shimizu, K.; Ogawa, M.; Yamaguchi, H.; Doi, M.; Jensen, P. Basic and translational research on proteinase-activated receptors: Antagonism of the proteinase-activated receptor 1 for thrombin, a novel approach to antiplatelet therapy for atherothrombotic disease. J. Pharmacol. Sci. 2008, 108, 433–438. [Google Scholar] [CrossRef]
- Kosoglou, T.; Kraft, W.K.; Kumar, B.; Statkevich, P.; Xuan, F.; Ma, L.; Jennings, L.K.; Schiller, J.E.; Langdon, R.B.; et al. Pharmacokinetics and pharmacodynamics of the novel PAR-1 antagonist vorapaxar in patients with end-stage renal disease. Eur. J. Clin. Pharmacol. 2012, 68, 1049–1056. [Google Scholar] [CrossRef]
- Zhang, C.; Srinivasan, Y.; Arlow, D.H.; Fung, J.J.; Palmer, D.; Zheng, Y.; Green, H.F.; Pandey, A.; Dror, R.O.; Shaw, D.E.; et al. High-resolution crystal structure of human protease-activated receptor 1. Nature 2012, 492, 387–392. [Google Scholar] [CrossRef]
- Becker, R.C.; Moliterno, D.J.; Jennings, L.K.; Pieper, K.S.; Pei, J.; Niederman, A.; Ziada, K.M.; Berman, G.; Strony, J.; Joseph, D.; et al. Safety and tolerability of SCH 530348 in patients undergoing non-urgent percutaneous coronary intervention: A randomised, double-blind, placebo-controlled phase II study. Lancet 2009, 373, 919–928. [Google Scholar] [CrossRef]
- Goto, S.; Yamaguchi, T.; Ikeda, Y.; Kato, K.; Yamaguchi, H.; Jensen, P. Safety and exploratory efficacy of the novel thrombin receptor (PAR-1) antagonist SCH530348 for non-ST-segment elevation acute coronary syndrome. J. Atheroscler. Thromb. 2010, 17, 156–164. [Google Scholar] [CrossRef]
- Tricoci, P.; Huang, Z.; Held, C.; Moliterno, D.J.; Armstrong, P.W.; van de Werf, F.; White, H.D.; Aylward, P.E.; Wallentin, L.; Chen, E.; et al. Thrombin-receptor antagonist vorapaxar in acute coronary syndromes. N. Engl. J. Med. 2012, 366, 20–33. [Google Scholar] [CrossRef]
- Morrow, D.A.; Braunwald, E.; Bonaca, M.P.; Ameriso, S.F.; Dalby, A.J.; Fish, M.P.; Fox, K.A.; Lipka, L.J.; Liu, X.; Nicolau, J.C.; et al. Vorapaxar in the secondary prevention of atherothrombotic events. N. Engl. J. Med. 2012, 366, 1404–1413. [Google Scholar] [CrossRef]
- Bonaca, M.P.; Scirica, B.M.; Creager, M.A.; Olin, J.W.; Bounameaux, H.; Dellborg, M.; Lamp, J.M.; Murphy, S.A.; Braunwald, E.; Morrow, D.A. Vorapaxar in Patients with Peripheral Artery Disease: Results from TRA2{degrees}P-TIMI 50. Circulation 2013, 127, 1522–1529. [Google Scholar] [CrossRef]
- Takeuchi, M. Pharmacodynamics and safety of a novel Protease Activated Receptor-1 antagonist E5555 for healthy volunteers. In Proceedings of ESC Congress, Vienna, Austria, 1–5 September 2007.
- Serebruany, V.; Sabaeva, E.; Booze, C.; Atar, O.D.; Eisert, C.; Hanley, D. Distribution of dipyridamole in blood components among post-stroke patients treated with extended release formulation. Thromb. Haemost. 2009, 102, 538–543. [Google Scholar]
- Goto, S.; Ogawa, H.; Takeuchi, M.; Flather, M.D.; Bhatt, D.L. Double-blind, placebo-controlled Phase II studies of the protease-activated receptor 1 antagonist E5555 (atopaxar) in Japanese patients with acute coronary syndrome or high-risk coronary artery disease. Eur. Heart J. 2010, 31, 2601–2613. [Google Scholar] [CrossRef]
- O'Donoghue, M.L.; Bhatt, D.L.; Wiviott, S.D.; Goodman, S.G.; Fitzgerald, D.J.; Angiolillo, D.J.; Goto, S.; Montalescot, G.; Zeymer, U.; Aylward, P.E.; et al. Safety and tolerability of atopaxar in the treatment of patients with acute coronary syndromes: The lessons from antagonizing the cellular effects of Thrombin-Acute Coronary Syndromes Trial. Circulation 2011, 123, 1843–1853. [Google Scholar] [CrossRef]
- Wiviott, S.D.; Flather, M.D.; O'Donoghue, M.L.; Goto, S.; Fitzgerald, D.J.; Cura, F.; Aylward, P.; Guetta, V.; Dudek, D.; Contant, C.F.; et al. Randomized trial of atopaxar in the treatment of patients with coronary artery disease: The lessons from antagonizing the cellular effect of Thrombin-Coronary Artery Disease Trial. Circulation 2011, 123, 1854–1863. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ghose, A.; Sharma, A.; Guha, G.; Mukherjee, D.; Frankel, R. Comparing newer oral anti-platelets prasugrel and ticagrelor in reduction of ischemic events-evidence from a network meta-analysis. J. Thromb. Thrombolysis 2012. [Google Scholar] [CrossRef]
- Diener, H.C.; Bogousslavsky, J.; Brass, L.M.; Cimminiello, C.; Csiba, L.; Kaste, M.; Leys, D.; Matias-Guiu, J.; Rupprecht, H.J.; investigators, M. Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): randomised, double-blind, placebo-controlled trial. Lancet 2004, 364, 331–337. [Google Scholar] [CrossRef]
- Sacco, R.L.; Diener, H.C.; Yusuf, S.; Cotton, D.; Ounpuu, S.; Lawton, W.A.; Palesch, Y.; Martin, R.H.; Albers, G.W.; Bath, P.; et al. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N. Engl. J. Med. 2008, 359, 1238–1251. [Google Scholar] [CrossRef]
- Falcone, G.J.; Brouwers, H.B.; Rosand, J. Risk of intracranial hemorrhage with protease-activated receptor-1 antagonists. Stroke 2012, 43, 3158–3159. [Google Scholar] [CrossRef]
- Siller-Matula, J.M.; Krumphuber, J.; Jilma, B. Pharmacokinetic, pharmacodynamic and clinical profile of novel antiplatelet drugs targeting vascular diseases. Br. J. Pharmacol. 2010, 159, 502–517. [Google Scholar] [CrossRef]
- Zhang, P.; Gruber, A.; Kasuda, S.; Kimmelstiel, C.; O'Callaghan, K.; Cox, D.H.; Bohm, A.; Baleja, J.D.; Covic, L.; Kuliopulos, A. Suppression of arterial thrombosis without affecting hemostatic parameters with a cell-penetrating PAR1 pepducin. Circulation 2012, 126, 83–91. [Google Scholar] [CrossRef]
- Connolly, S.J.; Ezekowitz, M.D.; Yusuf, S.; Eikelboom, J.; Oldgren, J.; Parekh, A.; Pogue, J.; Reilly, P.A.; Themeles, E.; Varrone, J.; Wang, S.; et al. Dabigatran versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2009, 361, 1139–1151. [Google Scholar] [CrossRef]
- Granger, C.B.; Alexander, J.H.; McMurray, J.J.; Lopes, R.D.; Hylek, E.M.; Hanna, M.; Al-Khalidi, H.R.; Ansell, J.; Atar, D.; Avezum, A.; et al. Apixaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2011, 365, 981–992. [Google Scholar] [CrossRef]
- Patel, M.R.; Mahaffey, K.W.; Garg, J.; Pan, G.; Singer, D.E.; Hacke, W.; Breithardt, G.; Halperin, J.L.; Hankey, G.J.; Piccini, J.P.; et al. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N. Engl. J. Med. 2011, 365, 883–891. [Google Scholar] [CrossRef]
- Schulman, S.; Kearon, C.; Kakkar, A.K.; Schellong, S.; Eriksson, H.; Baanstra, D.; Kvamme, A.M.; Friedman, J.; Mismetti, P.; Goldhaber, S.Z.; et al. Extended use of dabigatran, warfarin, or placebo in venous thromboembolism. N. Engl. J. Med. 2013, 368, 709–718. [Google Scholar] [CrossRef]
- EINSTEIN Investigators; Bauersachs, R.; Berkowitz, S.D.; Brenner, B.; Buller, H.R.; Decousus, H.; Gallus, A.S.; Lensing, A.W.; Misselwitz, F.; Prins, M.H.; et al. Oal rivaroxaban for symptomatic venous thromboembolism. N. Engl. J. Med. 2010, 363, 2499–2510. [Google Scholar] [CrossRef]
- Mega, J.L.; Braunwald, E.; Wiviott, S.D.; Bassand, J.P.; Bhatt, D.L.; Bode, C.; Burton, P.; Cohen, M.; Cook-Bruns, N.; Fox, K.A.; et al. Rivaroxaban in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 366, 9–19. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wallace, E.L.; Smyth, S.S. Targeting Platelet Thrombin Receptor Signaling to Prevent Thrombosis. Pharmaceuticals 2013, 6, 915-928. https://doi.org/10.3390/ph6080915
Wallace EL, Smyth SS. Targeting Platelet Thrombin Receptor Signaling to Prevent Thrombosis. Pharmaceuticals. 2013; 6(8):915-928. https://doi.org/10.3390/ph6080915
Chicago/Turabian StyleWallace, Eric L., and Susan S. Smyth. 2013. "Targeting Platelet Thrombin Receptor Signaling to Prevent Thrombosis" Pharmaceuticals 6, no. 8: 915-928. https://doi.org/10.3390/ph6080915
APA StyleWallace, E. L., & Smyth, S. S. (2013). Targeting Platelet Thrombin Receptor Signaling to Prevent Thrombosis. Pharmaceuticals, 6(8), 915-928. https://doi.org/10.3390/ph6080915