Antimicrobial Peptides

Abstract
:1. Sources and History of Antimicrobial Peptides
2. Structure and Major Activities of AMPs
3. Major Categories of AMPs and Mechanisms of Action
3.1. Classification
3.1.2. Antibacterial Peptides
3.1.3. Antifungal Peptides
3.1.4. Antiparasitic Peptides
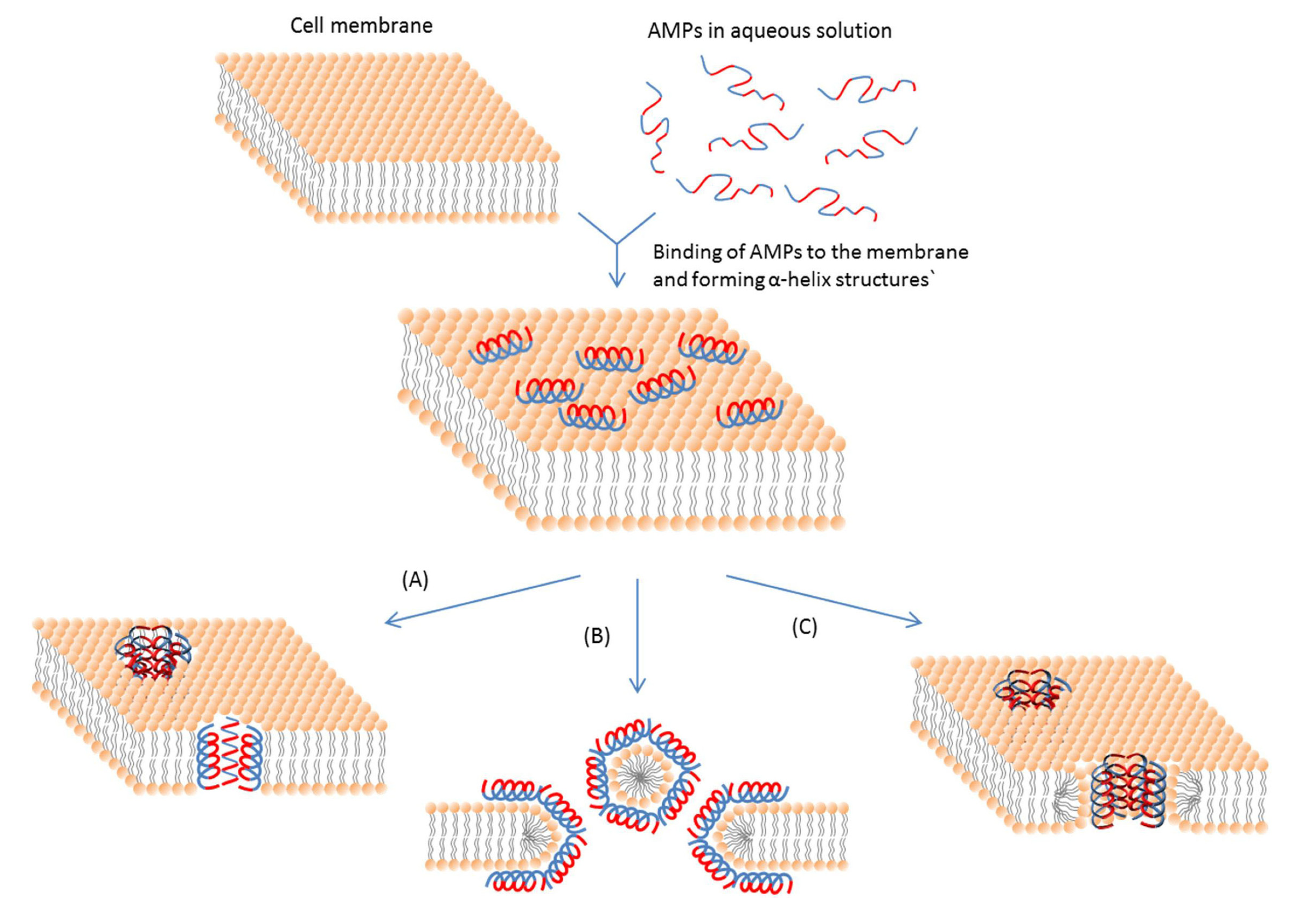
3.2. Mechanism of Action
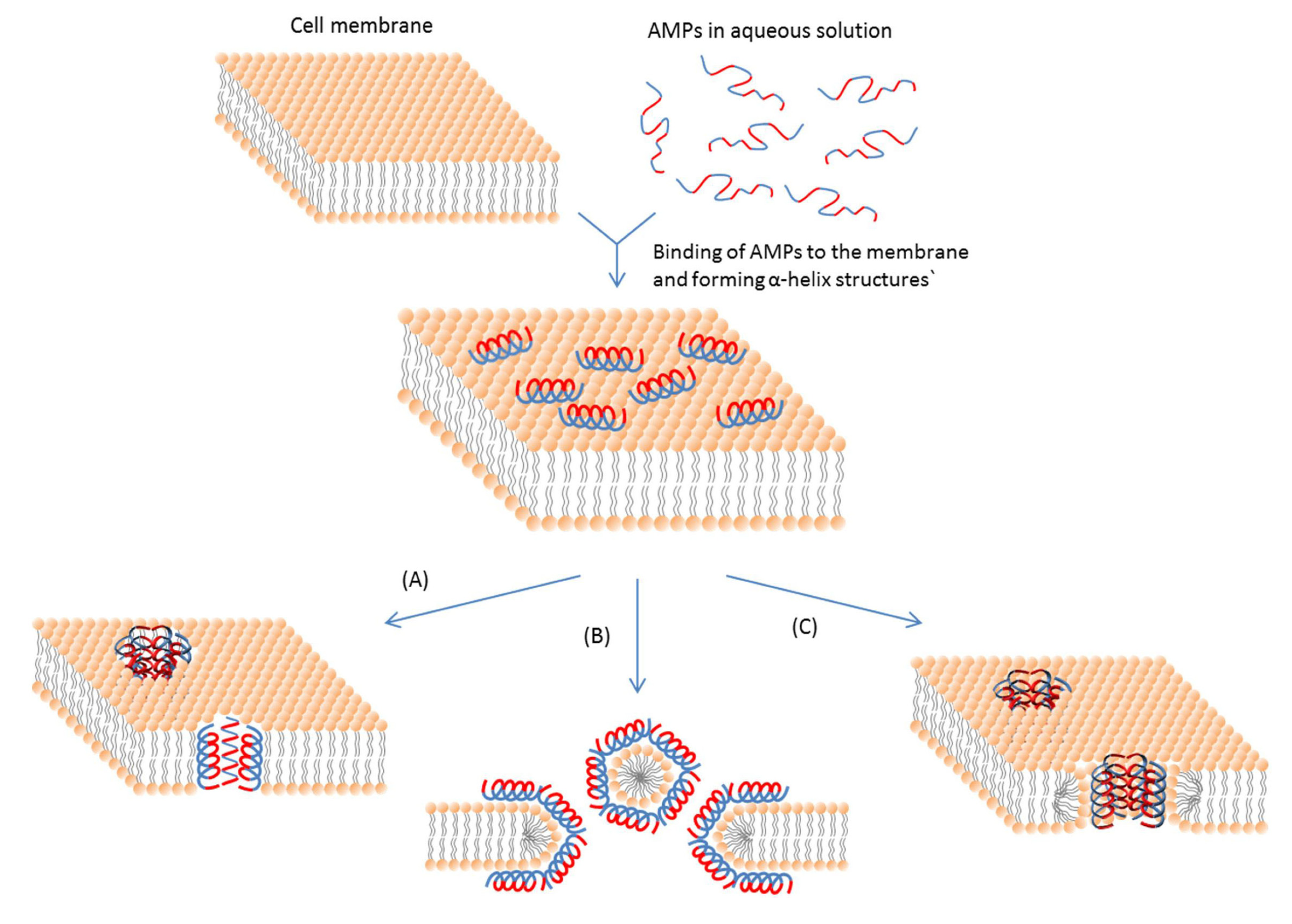
3.2.1. Membrane-Active AMPs

{kind=link}
{kind=link}
{kind=link}
| Interaction model | Mechanism | References |
|---|---|---|
| Carpet like (Detergent-like) | The peptide micelle touches the membrane first and coats a small area of the membrane. Then AMP molecules penetrate the lipid bilayer to let pore formation occur leaving holes behind. | [115,116,117] |
| Membrane thinning | AMPs insert themselves into only one side of the lipid bilayer. It can form a gap between lipid molecules at the chain region. This gap creates a force and pulls the neighboring lipid molecules to fill it. | [118,119,120] |
| Aggregate | AMPs stick to the membrane parallel to the surface. Then reorientation of AMPs occurs and they insert themselves into the membrane vertically to form sphere-like structures. | [115,121,122,123] |
| Toroidal pore | AMPs align perpendicularly into the bilayer structure with their hydrophobic regions associated with the center part of the lipid bilayer and their hydrophilic regions facing the pore. | [83,123] |
| Barrel-stave | Staves are formed first parallel to the cell membrane. Then barrels are formed and AMPs are inserted perpendicularly to the plane of the membrane bilayer. | [82,124,125] |
3.2.2. Intracellularly Active AMPs
4. Designing New Synthetic AMPs: Major Factors to Consider
4.1. Important Physiochemical Properties of AMPs
4.1.1. Length
4.1.2. Net Charge
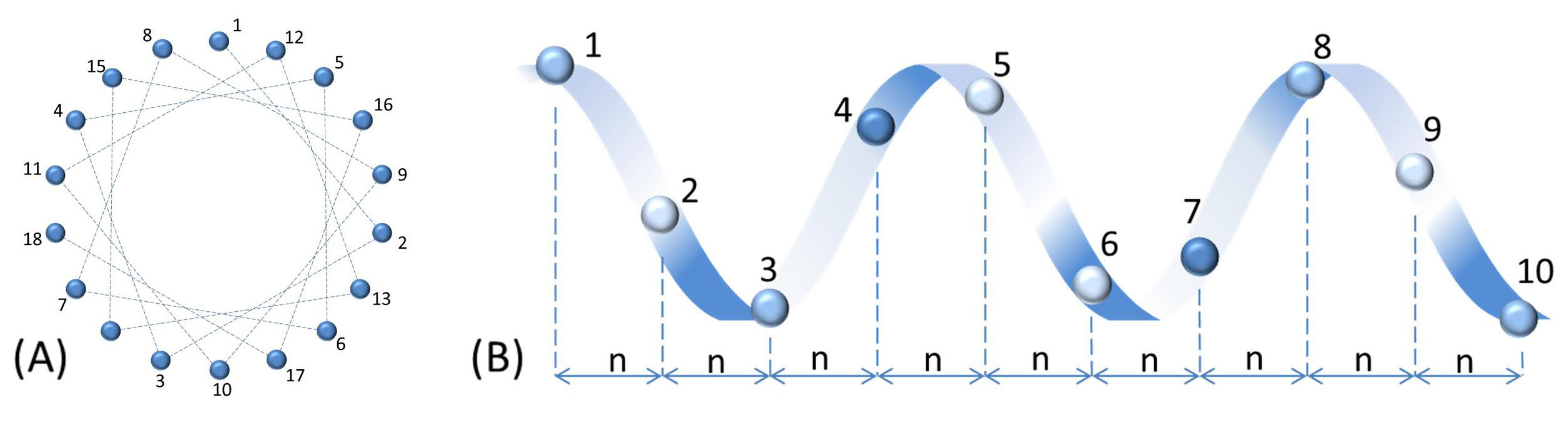
4.1.3. Helicity
4.1.4. Hydrophobicity
4.1.5. Amphipathicity
4.1.6. Solubility
4.2. The Relationship between Physiochemical Properties of AMPs
4.3. AMP Modifications
4.3.1. Modification of AMPs with Covalent Bonds
4.3.2. Modification of AMPs by Changing Amino Acid Content
4.3.3. Modification of AMPs by Amidation
4.3.4. Modification of AMPs with Unnatural Amino Acids
4.3.5. Modification of AMPs with Computer-Assisted Methods
4.4. New AMP Design by Homology Modeling
5. New Targets of AMPs: Biofilms, Persister Cells, and Drug Resistance Bacteria
5.1. Biofilm Control
5.2. Persister Control
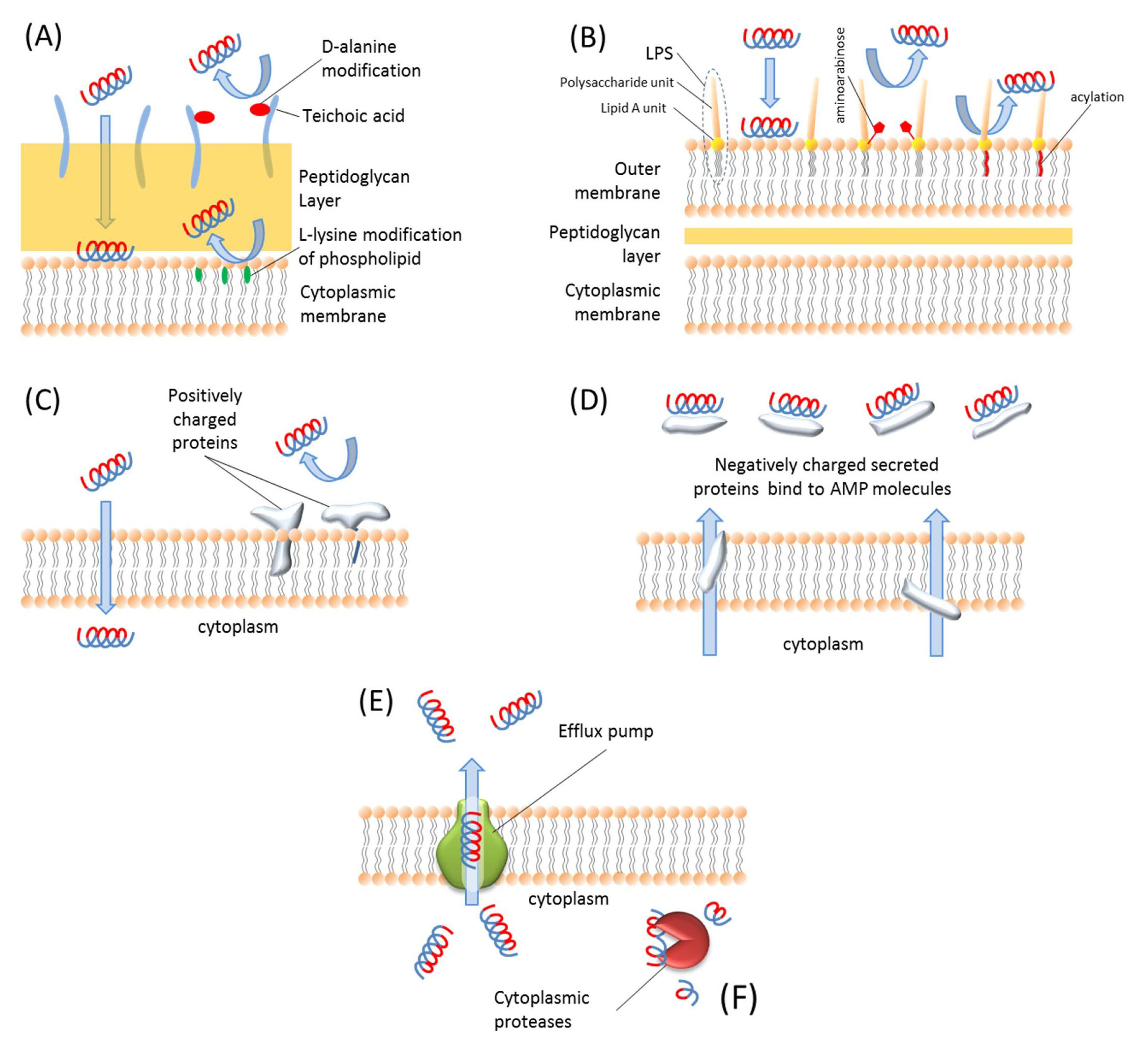
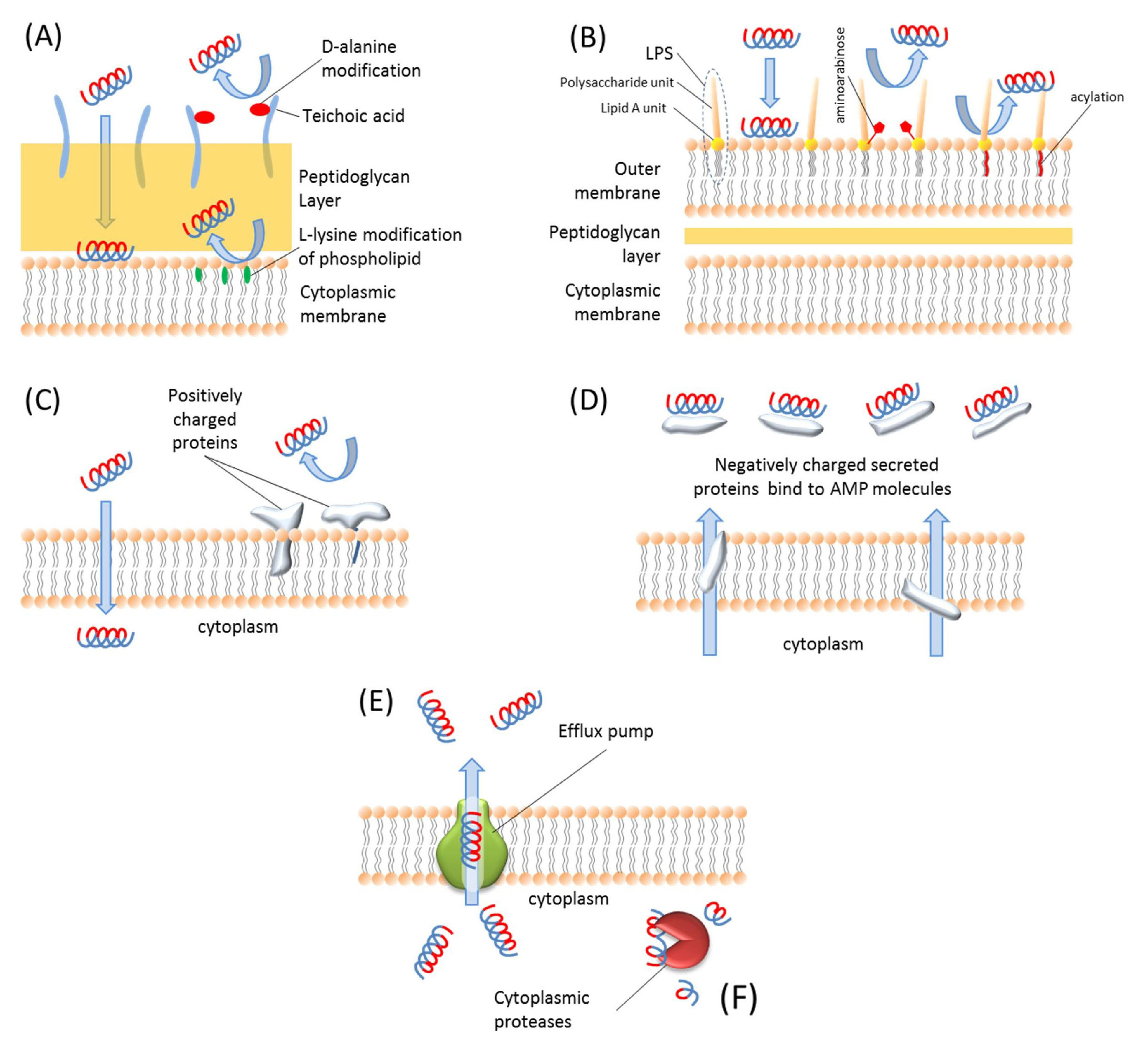
6. Resistance to Antimicrobial Peptides
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Brown, K.L.; Hancock, R.E. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef]
- Harris, F.; Dennison, S.R.; Phoenix, D.A. Anionic antimicrobial peptides from eukaryotic organisms. Curr. Protein Pept. Sci. 2009, 10, 585–606. [Google Scholar] [CrossRef]
- Groenink, J.; Walgreen-Weterings, E.; van’t Hof, W.; Veerman, E.C.; Nieuw Amerongen, A.V. Cationic amphipathic peptides, derived from bovine and human lactoferrins, with antimicrobial activity against oral pathogens. FEMS Microbiol. Lett. 1999, 179, 217–222. [Google Scholar]
- Bradshaw, J. Cationic antimicrobial peptides: Issues for potential clinical use. BioDrugs 2003, 17, 233–240. [Google Scholar] [CrossRef]
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-active host defense peptides—challenges and perspectives for the development of novel anticancer drugs. Chem. Phys. Lipids 2011, 164, 766–781. [Google Scholar] [CrossRef]
- Huang, Y.B.; Huang, J.F.; Chen, Y.X. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein Cell 2010, 1, 143–152. [Google Scholar]
- Dubos, R.J. Studies on a bactericidal agent extracted from a soil bacillus: I. Preparation of the agent. Its activity in vitro. J. Exp. Med. 1939, 70, 1–10. [Google Scholar] [CrossRef]
- Dubos, R.J. Studies on a bactericidal agent extracted from a soil bacillus: II. Protective effect of the bactericidal agent against experimental Pneumococcus infections in mice. J. Exp. Med. 1939, 70, 11–17. [Google Scholar] [CrossRef]
- Hotchkiss, R.D.; Dubos, R.J. Fractionation of the bactericidal agent from cultures of a soil Bacillus. J. Biol. Chem. 1940, 132, 791–792. [Google Scholar]
- Van Epps, H.L. Rene dubos: Unearthing antibiotics. J. Exp. Med. 2006, 203, 259. [Google Scholar] [CrossRef]
- Dubos, R.J.; Hotchkiss, R.D. The production of bactericidal substances by aerobic sporulating bacilli. J. Exp. Med. 1941, 73, 629–640. [Google Scholar] [CrossRef]
- Rammelkamp, C.H.; Weinstein, L. Toxic effects of tyrothricin, gramicidin and tyrocidine. J. Infect. Dis. 1942, 71, 166–173. [Google Scholar]
- Balls, A.K. A crystalline protein obtained from a lipoprotein of wheat flour. Cereal Chem. 1942, 19, 279–288. [Google Scholar]
- Ohtani, S.; Okada, T.; Yoshizumi, H.; Kagamiyama, H. Complete primary structures of two subunits of purothionin a, a lethal protein for brewer’s yeast from wheat flour. J. Biochem. 1977, 82, 753–767. [Google Scholar]
- Hirsch, J.G. Phagocytin: A bactericidal substance from polymorphonuclear leucocytes. J. Exp. Med. 1956, 103, 589–611. [Google Scholar] [CrossRef]
- Kiss, G.; Michl, H. Uber das giftsekret der gelbbauchunke, Bombinavariegata L. Toxicon 1962, 1, 33–34. [Google Scholar]
- Groves, M.L.; Peterson, R.F.; Kiddy, C.A. Poliomorphism in the red protein isolated from milk of individual cows. Nature 1965, 207, 1007–1008. [Google Scholar] [CrossRef]
- Zeya, H.I.; Spitznagel, J.K. Antibacterial and enzymic basic proteins from leukocyte lysosomes: Separation and identification. Science 1963, 142, 1085–1087. [Google Scholar]
- Zhao, X.; Wu, H.; Lu, H.; Li, G.; Huang, Q. Lamp: A database linking antimicrobial peptides. PLoS One 2013, 8, e66557. [Google Scholar] [CrossRef]
- Conlon, J.M.; Sonnevend, A. Antimicrobial peptides in frog skin secretions. Methods Mol. Biol. 2010, 618, 3–14. [Google Scholar]
- Radek, K.; Gallo, R. Antimicrobial peptides: Natural effectors of the innate immune system. Semin. Immunopathol. 2007, 29, 27–43. [Google Scholar] [CrossRef]
- Peters, B.M.; Shirtliff, M.E.; Jabra-Rizk, M.A. Antimicrobial peptides: Primeval molecules or future drugs? PLoS Pathog. 2010, 6, e1001067. [Google Scholar]
- Leippe, M. Antimicrobial and cytolytic polypeptides of amoeboid protozoa—Effector molecules of primitive phagocytes. Dev. Comp. Immunol. 1999, 23, 267–279. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Schauber, J.; Gallo, R.L. Antimicrobial peptides and the skin immune defense system. J. Allergy Clin. Immunol. 2008, 122, 261–266. [Google Scholar] [CrossRef]
- Ma, Y.F.; Liu, C.B.; Liu, X.H.; Wu, J.; Yang, H.L.; Wang, Y.P.; Li, J.X.; Yu, H.N.; Lai, R. Peptidomics and genomics analysis of novel antimicrobial peptides from the frog, Rana nigrovittata. Genomics 2010, 95, 66–71. [Google Scholar]
- Hultmark, D.; Steiner, H.; Rasmuson, T.; Boman, H.G. Insect immunity. Purification and properties of three inducible bactericidal proteins from hemolymph of immunized pupae of Hyalophora cecropia. Eur. J. Biochem. 1980, 106, 7–16. [Google Scholar]
- Bals, R.; Wang, X.; Meegalla, R.L.; Wattler, S.; Weiner, D.J.; Nehls, M.C.; Wilson, J.M. Mouse beta-defensin 3 is an inducible antimicrobial peptide expressed in the epithelia of multiple organs. Infect. Immun. 1999, 67, 3542–3547. [Google Scholar]
- Ganz, T. The role of antimicrobial peptides in innate immunity. Integr. Comp. Biol. 2003, 43, 300–304. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Iwabuchi, K.; Matsuda, H.; Ogawa, H.; Nagaoka, I. Epithelial cell-derived human beta-defensin-2 acts as a chemotaxin for mast cells through a pertussis toxin-sensitive and phospholipase c-dependent pathway. Int. Immunol. 2002, 14, 421–426. [Google Scholar] [CrossRef]
- Hancock, R.E.; Scott, M.G. The role of antimicrobial peptides in animal defenses. Proc. Natl. Acad. Sci. USA 2000, 97, 8856–8861. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Biragyn, A.; Kwak, L.W.; Yang, D. Roles of antimicrobial peptides such as defensins in innate and adaptive immunity. Ann. Rheum. Dis. 2003, 62, ii17–ii21. [Google Scholar]
- Scott, M.G.; Rosenberger, C.M.; Gold, M.R.; Finlay, B.B.; Hancock, R.E. An alpha-helical cationic antimicrobial peptide selectively modulates macrophage responses to lipopolysaccharide and directly alters macrophage gene expression. J. Immunol. 2000, 165, 3358–3365. [Google Scholar]
- Nijnik, A.; Pistolic, J.; Filewod, N.C.; Hancock, R.E. Signaling pathways mediating chemokine induction in keratinocytes by cathelicidin ll-37 and flagellin. J. Innate Immun. 2012, 4, 377–386. [Google Scholar]
- Kindrachuk, J.; Jenssen, H.; Elliott, M.; Nijnik, A.; Magrangeas-Janot, L.; Pasupuleti, M.; Thorson, L.; Ma, S.; Easton, D.M.; Bains, M.; et al. Manipulation of innate immunity by a bacterial secreted peptide: Lantibiotic nisin z is selectively immunomodulatory. Innate Immun. 2013, 19, 315–327. [Google Scholar] [CrossRef]
- Birchler, T.; Seibl, R.; Buchner, K.; Loeliger, S.; Seger, R.; Hossle, J.P.; Aguzzi, A.; Lauener, R.P. Human toll-like receptor 2 mediates induction of the antimicrobial peptide human beta-defensin 2 in response to bacterial lipoprotein. Eur. J. Immunol. 2001, 31, 3131–3137. [Google Scholar]
- Larrick, J.W.; Hirata, M.; Balint, R.F.; Lee, J.; Zhong, J.; Wright, S.C. Human cap18: A novel antimicrobial lipopolysaccharide-binding protein. Infect. Immun. 1995, 63, 1291–1297. [Google Scholar]
- Brackett, D.J.; Lerner, M.R.; Lacquement, M.A.; He, R.; Pereira, H.A. A synthetic lipopolysaccharide-binding peptide based on the neutrophil-derived protein cap37 prevents endotoxin-induced responses in conscious rats. Infect. Immun. 1997, 65, 2803–2811. [Google Scholar]
- Zhang, G.H.; Mann, D.M.; Tsai, C.M. Neutralization of endotoxin in vitro and in vivo by a human lactoferrin-derived peptide. Infect. Immun. 1999, 67, 1353–1358. [Google Scholar]
- Loppnow, H.; Libby, P.; Freudenberg, M.; Krauss, J.H.; Weckesser, J.; Mayer, H. Cytokine induction by lipopolysaccharide (LPS) corresponds to lethal toxicity and is inhibited by nontoxic Rhodobacter capsulatus LPS. Infect. Immun. 1990, 58, 3743–3750. [Google Scholar]
- Powers, J.P.; Hancock, R.E. The relationship between peptide structure and antibacterial activity. Peptides 2003, 24, 1681–1691. [Google Scholar] [CrossRef]
- Bulet, P.; Stocklin, R.; Menin, L. Anti-microbial peptides: From invertebrates to vertebrates. Immunol. Rev. 2004, 198, 169–184. [Google Scholar] [CrossRef]
- McManus, A.M.; Dawson, N.F.; Wade, J.D.; Carrington, L.E.; Winzor, D.J.; Craik, D.J. Three-dimensional structure of rk-1: A novel alpha-defensin peptide. Biochemistry 2000, 39, 15757–15764. [Google Scholar]
- Uteng, M.; Hauge, H.H.; Markwick, P.R.; Fimland, G.; Mantzilas, D.; Nissen-Meyer, J.; Muhle-Goll, C. Three-dimensional structure in lipid micelles of the pediocin-like antimicrobial peptide sakacin p and a sakacin p variant that is structurally stabilized by an inserted c-terminal disulfide bridge. Biochemistry 2003, 42, 11417–11426. [Google Scholar]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar]
- Hsu, C.H.; Chen, C.; Jou, M.L.; Lee, A.Y.; Lin, Y.C.; Yu, Y.P.; Huang, W.T.; Wu, S.H. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res. 2005, 33, 4053–4064. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide antimicrobial agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef]
- Loeffler, J.M.; Nelson, D.; Fischetti, V.A. Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science 2001, 294, 2170–2172. [Google Scholar] [CrossRef]
- Naghmouchi, K.; le Lay, C.; Baah, J.; Drider, D. Antibiotic and antimicrobial peptide combinations: Synergistic inhibition of Pseudomonas fluorescens and antibiotic-resistant variants. Res. Microbiol. 2012, 163, 101–108. [Google Scholar] [CrossRef]
- Costa, F.; Carvalho, I.F.; Montelaro, R.C.; Gomes, P.; Martins, M.C. Covalent immobilization of antimicrobial peptides (AMPs) onto biomaterial surfaces. Acta Biomater. 2011, 7, 1431–1440. [Google Scholar] [CrossRef]
- Wade, J.D.; Lin, F.; Hossain, M.A.; Dawson, R.M. Chemical synthesis and biological evaluation of an antimicrobial peptide gonococcal growth inhibitor. Amino Acids 2012, 43, 2279–2283. [Google Scholar] [CrossRef]
- Piers, K.L.; Brown, M.H.; Hancock, R.E. Recombinant DNA procedures for producing small antimicrobial cationic peptides in bacteria. Gene 1993, 134, 7–13. [Google Scholar] [CrossRef]
- Ramos, R.; Moreira, S.; Rodrigues, A.; Gama, M.; Domingues, L. Recombinant expression and purification of the antimicrobial peptide magainin-2. Biotechnol. Prog. 2013, 29, 17–22. [Google Scholar] [CrossRef]
- Papo, N.; Oren, Z.; Pag, U.; Sahl, H.G.; Shai, Y. The consequence of sequence alteration of an amphipathic alpha-helical antimicrobial peptide and its diastereomers. J. Biol. Chem. 2002, 277, 33913–33921. [Google Scholar]
- Pacor, S.; Giangaspero, A.; Bacac, M.; Sava, G.; Tossi, A. Analysis of the cytotoxicity of synthetic antimicrobial peptides on mouse leucocytes: Implications for systemic use. J. Antimicrob. Chemother. 2002, 50, 339–348. [Google Scholar] [CrossRef]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar]
- Sieprawska-Lupa, M.; Mydel, P.; Krawczyk, K.; Wojcik, K.; Puklo, M.; Lupa, B.; Suder, P.; Silberring, J.; Reed, M.; Pohl, J.; et al. Degradation of human antimicrobial peptide ll-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother. 2004, 48, 4673–4679. [Google Scholar] [CrossRef]
- Svenson, J.; Stensen, W.; Brandsdal, B.O.; Haug, B.E.; Monrad, J.; Svendsen, J.S. Antimicrobial peptides with stability toward tryptic degradation. Biochemistry 2008, 47, 3777–3788. [Google Scholar]
- Eckert, R.; Qi, F.; Yarbrough, D.K.; He, J.; Anderson, M.H.; Shi, W. Adding selectivity to antimicrobial peptides: Rational design of a multidomain peptide against Pseudomonas spp. Antimicrob. Agents Chemother. 2006, 50, 1480–1488. [Google Scholar] [CrossRef]
- Bommarius, B.; Jenssen, H.; Elliott, M.; Kindrachuk, J.; Pasupuleti, M.; Gieren, H.; Jaeger, K.E.; Hancock, R.E.; Kalman, D. Cost-effective expression and purification of antimicrobial and host defense peptides in Escherichia coli. Peptides 2010, 31, 1957–1965. [Google Scholar]
- Duquesne, S.; Destoumieux-Garzon, D.; Zirah, S.; Knappe, T.A.; Goulard, C.; Peduzzi, J.; Marahiel, M.A.; Rebuffat, S. Post-translational modification and folding of a lasso-type gene-encoded antimicrobial peptide require two enzymes only in Escherichia coli. Adv. Exp. Med. Biol. 2009, 611, 35–36. [Google Scholar]
- Bagheri, M.; Beyermann, M.; Dathe, M. Immobilization reduces the activity of surface-bound cationic antimicrobial peptides with no influence upon the activity spectrum. Antimicrob. Agents Chemother. 2009, 53, 1132–1141. [Google Scholar] [CrossRef]
- Bader, M.W.; Sanowar, S.; Daley, M.E.; Schneider, A.R.; Cho, U.; Xu, W.; Klevit, R.E.; Le Moual, H.; Miller, S.I. Recognition of antimicrobial peptides by a bacterial sensor kinase. Cell 2005, 122, 461–472. [Google Scholar]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef]
- Phoenix, D.; Dennison, S.R.; Harris, F. Antimicrobial Peptides; Wiley-VCH: Weinheim, Germany, 2013; p. 231. [Google Scholar]
- Kirby, A.J. The lysozyme mechanism sorted—After 50 years. Nat. Struct. Biol. 2001, 8, 737–739. [Google Scholar] [CrossRef]
- Bastian, A.; Schafer, H. Human alpha-defensin 1 (hnp-1) inhibits adenoviral infection in vitro. Regul. Pept. 2001, 101, 157–161. [Google Scholar] [CrossRef]
- Horne, W.S.; Wiethoff, C.M.; Cui, C.; Wilcoxen, K.M.; Amorin, M.; Ghadiri, M.R.; Nemerow, G.R. Antiviral cyclic d,l-α-peptides: Targeting a general biochemical pathway in virus infections. Bioorg. Med. Chem. 2005, 13, 5145–5153. [Google Scholar]
- Robinson, W.E., Jr.; McDougall, B.; Tran, D.; Selsted, M.E. Anti-hiv-1 activity of indolicidin, an antimicrobial peptide from neutrophils. J. Leukoc. Biol. 1998, 63, 94–100. [Google Scholar]
- Sitaram, N.; Nagaraj, R. Interaction of antimicrobial peptides with biological and model membranes: Structural and charge requirements for activity. Biochim. Biophys. Acta 1999, 1462, 29–54. [Google Scholar]
- Belaid, A.; Aouni, M.; Khelifa, R.; Trabelsi, A.; Jemmali, M.; Hani, K. In vitro antiviral activity of dermaseptins against herpes simplex virus type 1. J. Med. Virol. 2002, 66, 229–234. [Google Scholar] [CrossRef]
- Yasin, B.; Wang, W.; Pang, M.; Cheshenko, N.; Hong, T.; Waring, A.J.; Herold, B.C.; Wagar, E.A.; Lehrer, R.I. Theta defensins protect cells from infection by herpes simplex virus by inhibiting viral adhesion and entry. J. Virol. 2004, 78, 5147–5156. [Google Scholar] [CrossRef]
- Tamamura, H.; Ishihara, T.; Otaka, A.; Murakami, T.; Ibuka, T.; Waki, M.; Matsumoto, A.; Yamamoto, N.; Fujii, N. Analysis of the interaction of an anti-hiv peptide, t22 ([tyr5, 12, lys7]-polyphemusin ii), with gp120 and cd4 by surface plasmon resonance. Biochim. Biophys. Acta 1996, 1298, 37–44. [Google Scholar]
- Song, B.H.; Lee, G.C.; Moon, M.S.; Cho, Y.H.; Lee, C.H. Human cytomegalovirus binding to heparan sulfate proteoglycans on the cell surface and/or entry stimulates the expression of human leukocyte antigen class I. J. Gen. Virol. 2001, 82, 2405–2413. [Google Scholar]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar]
- Laquerre, S.; Argnani, R.; Anderson, D.B.; Zucchini, S.; Manservigi, R.; Glorioso, J.C. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins b and c, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J. Virol. 1998, 72, 6119–6130. [Google Scholar]
- Andersen, J.H.; Jenssen, H.; Sandvik, K.; Gutteberg, T.J. Anti-hsv activity of lactoferrin and lactoferricin is dependent on the presence of heparan sulphate at the cell surface. J. Med. Virol. 2004, 74, 262–271. [Google Scholar]
- Jenssen, H.; Andersen, J.H.; Uhlin-Hansen, L.; Gutteberg, T.J.; Rekdal, O. Anti-hsv activity of lactoferricin analogues is only partly related to their affinity for heparan sulfate. Antiviral Res. 2004, 61, 101–109. [Google Scholar]
- Liu, Y.; Gong, W.; Huang, C.C.; Herr, W.; Cheng, X. Crystal structure of the conserved core of the herpes simplex virus transcriptional regulatory protein vp16. Genes Dev. 1999, 13, 1692–1703. [Google Scholar]
- Sinha, S.; Cheshenko, N.; Lehrer, R.I.; Herold, B.C. Np-1, a rabbit alpha-defensin, prevents the entry and intercellular spread of herpes simplex virus type 2. Antimicrob. Agents Chemother. 2003, 47, 494–500. [Google Scholar]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef]
- Zhang, L.; Rozek, A.; Hancock, R.E. Interaction of cationic antimicrobial peptides with model membranes. J. Biol. Chem. 2001, 276, 35714–35722. [Google Scholar]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin ii: Buforin ii kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef]
- Otvos, L.; O, I.; Rogers, M.E.; Consolvo, P.J.; Condie, B.A.; Lovas, S.; Bulet, P.; Blaszczyk-Thurin, M. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 2000, 39, 14150–14159. [Google Scholar]
- Kragol, G.; Lovas, S.; Varadi, G.; Condie, B.A.; Hoffmann, R.; Otvos, L. The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry 2001, 40, 3016–3026. [Google Scholar]
- Brumfitt, W.; Salton, M.R.; Hamilton-Miller, J.M. Nisin, alone and combined with peptidoglycan-modulating antibiotics: Activity against methicillin-resistant Staphylococcus aureus and vancomycin-resistant enterococci. J. Antimicrob. Chemother. 2002, 50, 731–734. [Google Scholar] [CrossRef]
- De Lucca, A.J.; Bland, J.M.; Jacks, T.J.; Grimm, C.; Walsh, T.J. Fungicidal and binding properties of the natural peptides cecropin b and dermaseptin. Med. Mycol. 1998, 36, 291–298. [Google Scholar]
- De Lucca, A.J.; Walsh, T.J. Antifungal peptides: Novel therapeutic compounds against emerging pathogens. Antimicrob. Agents Chemother. 1999, 43, 1–11. [Google Scholar]
- Lee, Y.T.; Kim, D.H.; Suh, J.Y.; Chung, J.H.; Lee, B.L.; Lee, Y.; Choi, S. Structural characteristics of tenecin 3, an insect antifungal protein. Biochem. Mol. Biol. Int. 1999, 47, 369–376. [Google Scholar]
- Yokoyama, S.; Iida, Y.; Kawasaki, Y.; Minami, Y.; Watanabe, K.; Yagi, F. The chitin-binding capability of cy-amp1 from cycad is essential to antifungal activity. J. Pept. Sci. 2009, 15, 492–497. [Google Scholar]
- Pushpanathan, M.; Rajendhran, J.; Jayashree, S.; Sundarakrishnan, B.; Jayachandran, S.; Gunasekaran, P. Identification of a novel antifungal peptide with chitin-binding property from marine metagenome. Protein Pept. Lett. 2012, 19, 1289–1296. [Google Scholar] [CrossRef]
- Fujimura, M.; Ideguchi, M.; Minami, Y.; Watanabe, K.; Tadera, K. Purification, characterization, and sequencing of novel antimicrobial peptides, Tu-AMP 1 and Tu-AMP 2, from bulbs of tulip (Tulipa gesneriana L.). Biosci. Biotechnol. Biochem. 2004, 68, 571–577. [Google Scholar]
- Lehrer, R.I.; Szklarek, D.; Ganz, T.; Selsted, M.E. Correlation of binding of rabbit granulocyte peptides to Candida albicans with candidacidal activity. Infect. Immun. 1985, 49, 207–211. [Google Scholar]
- Terras, F.R.; Schoofs, H.M.; De Bolle, M.F.; Van Leuven, F.; Rees, S.B.; Vanderleyden, J.; Cammue, B.P.; Broekaert, W.F. Analysis of two novel classes of plant antifungal proteins from radish (Raphanus sativus L.) seeds. J. Biol. Chem. 1992, 267, 15301–15309. [Google Scholar]
- Van der Weerden, N.L.; Hancock, R.E.; Anderson, M.A. Permeabilization of fungal hyphae by the plant defensin nad1 occurs through a cell wall-dependent process. J. Biol. Chem. 2010, 285, 37513–37520. [Google Scholar] [CrossRef]
- Moerman, L.; Bosteels, S.; Noppe, W.; Willems, J.; Clynen, E.; Schoofs, L.; Thevissen, K.; Tytgat, J.; Van Eldere, J.; van der Walt, J.; et al. Antibacterial and antifungal properties of α-helical, cationic peptides in the venom of scorpions from southern Africa. Eur. J. Biochem. 2002, 269, 4799–4810. [Google Scholar] [CrossRef]
- Jiang, Z.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 2008, 90, 369–383. [Google Scholar] [CrossRef]
- Lee, D.G.; Hahm, K.S.; Shin, S.Y. Structure and fungicidal activity of a synthetic antimicrobial peptide, p18, and its truncated peptides. Biotechnol. Lett. 2004, 26, 337–341. [Google Scholar]
- Lee, D.G.; Kim, H.K.; Kim, S.A.; Park, Y.; Park, S.C.; Jang, S.H.; Hahm, K.S. Fungicidal effect of indolicidin and its interaction with phospholipid membranes. Biochem. Bioph. Res. Co. 2003, 305, 305–310. [Google Scholar]
- Barbault, F.; Landon, C.; Guenneugues, M.; Meyer, J.P.; Schott, V.; Dimarcq, J.L.; Vovelle, F. Solution structure of alo-3: A new knottin-type antifungal peptide from the insect Acrocinus longimanus. Biochemistry 2003, 42, 14434–14442. [Google Scholar]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef]
- Alberola, J.; Rodriguez, A.; Francino, O.; Roura, X.; Rivas, L.; Andreu, D. Safety and efficacy of antimicrobial peptides against naturally acquired leishmaniasis. Antimicrob. Agents Chemother. 2004, 48, 641–643. [Google Scholar] [CrossRef]
- Park, Y.; Jang, S.H.; Lee, D.G.; Hahm, K.S. Antinematodal effect of antimicrobial peptide, pmap-23, isolated from porcine myeloid against Caenorhabditis elegans. J. Pept. Sci. 2004, 10, 304–311. [Google Scholar] [CrossRef]
- Brogden, K.A.; Ackermann, M.; Huttner, K.M. Small, anionic, and charge-neutralizing propeptide fragments of zymogens are antimicrobial. Antimicrob. Agents Chemother. 1997, 41, 1615–1617. [Google Scholar]
- Lai, R.; Liu, H.; Lee, W.H.; Zhang, Y. An anionic antimicrobial peptide from toad Bombina maxima. Biochem. Bioph. Res. Co. 2002, 295, 796–799. [Google Scholar]
- Steffen, H.; Rieg, S.; Wiedemann, I.; Kalbacher, H.; Deeg, M.; Sahl, H.G.; Peschel, A.; Gotz, F.; Garbe, C.; Schittek, B. Naturally processed dermcidin-derived peptides do not permeabilize bacterial membranes and kill microorganisms irrespective of their charge. Antimicrob. Agents Chemother. 2006, 50, 2608–2620. [Google Scholar]
- Selsted, M.E.; Novotny, M.J.; Morris, W.L.; Tang, Y.Q.; Smith, W.; Cullor, J.S. Indolicidin, a novel bactericidal tridecapeptide amide from neutrophils. J. Biol. Chem. 1992, 267, 4292–4295. [Google Scholar]
- Subbalakshmi, C.; Sitaram, N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol. Lett. 1998, 160, 91–96. [Google Scholar] [CrossRef]
- Krajewski, K.; Marchand, C.; Long, Y.Q.; Pommier, Y.; Roller, P.P. Synthesis and hiv-1 integrase inhibitory activity of dimeric and tetrameric analogs of indolicidin. Bioorg. Med. Chem. Lett. 2004, 14, 5595–5598. [Google Scholar]
- Lee, D.G.; Kim, P.I.; Park, Y.K.; Woo, E.R.; Choi, J.S.; Choi, C.H.; Hahm, K.S. Design of novel peptide analogs with potent fungicidal activity, based on pmap-23 antimicrobial peptide isolated from porcine myeloid. Biochem. Bioph. Res. Co. 2002, 293, 231–238. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Rock, C.O. Transcriptional regulation in bacterial membrane lipid synthesis. J. Lipid Res. 2009, 50, S115–S119. [Google Scholar] [CrossRef]
- He, K.; Ludtke, S.J.; Worcester, D.L.; Huang, H.W. Neutron scattering in the plane of membranes: Structure of alamethicin pores. Biophys. J. 1996, 70, 2659–2666. [Google Scholar] [CrossRef]
- Madani, F.; Lindberg, S.; Langel, U.; Futaki, S.; Graslund, A. Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar]
- Bechinger, B. Detergent-like properties of magainin antibiotic peptides: A 31p solid-state nmr spectroscopy study. Biochim. Biophys. Acta 2005, 1712, 101–108. [Google Scholar]
- Bolintineanu, D.S.; Kaznessis, Y.N. Computational studies of protegrin antimicrobial peptides: A review. Peptides 2011, 32, 188–201. [Google Scholar] [CrossRef]
- Mecke, A.; Lee, D.K.; Ramamoorthy, A.; Orr, B.G.; Holl, M.M.B. Membrane thinning due to antimicrobial peptide binding: An atomic force microscopy study of msi-78 in lipid bilayers. Biophys. J. 2005, 89, 4043–4050. [Google Scholar]
- Ludtke, S.; He, K.; Huang, H. Membrane thinning caused by magainin 2. Biochemistry 1995, 34, 16764–16769. [Google Scholar] [CrossRef]
- Chen, F.Y.; Lee, M.T.; Huang, H.W. Evidence for membrane thinning effect as the mechanism for peptide-induced pore formation. Biophys. J. 2003, 84, 3751–3758. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry 1996, 35, 11361–11368. [Google Scholar] [CrossRef]
- Matsuzaki, K. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta Biomembr. 1998, 1376, 391–400. [Google Scholar]
- Wu, M.; Maier, E.; Benz, R.; Hancock, R.E. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 1999, 38, 7235–7242. [Google Scholar]
- Ehrenstein, G.; Lecar, H. Electrically gated ionic channels in lipid bilayers. Q. Rev. Biophys. 1977, 10, 1–34. [Google Scholar] [CrossRef]
- Shimazaki, K.; Tazume, T.; Uji, K.; Tanaka, M.; Kumura, H.; Mikawa, K.; Shimo-Oka, T. Properties of a heparin-binding peptide derived from bovine lactoferrin. J. Dairy. Sci. 1998, 81, 2841–2849. [Google Scholar]
- Cudic, M.; Otvos, L. Intracellular targets of antibacterial peptides. Curr. Drug Targets 2002, 3, 101–106. [Google Scholar] [CrossRef]
- Otvos, L. Antibacterial peptides and proteins with multiple cellular targets. J. Pept. Sci. 2005, 11, 697–706. [Google Scholar] [CrossRef]
- Mookherjee, N.; Lippert, D.N.; Hamill, P.; Falsafi, R.; Nijnik, A.; Kindrachuk, J.; Pistolic, J.; Gardy, J.; Miri, P.; Naseer, M.; et al. Intracellular receptor for human host defense peptide ll-37 in monocytes. J. Immunol. 2009, 183, 2688–2696. [Google Scholar] [CrossRef]
- Chen, L.; Harrison, S.D. Cell-penetrating peptides in drug development: Enabling intracellular targets. Biochem. Soc. Trans. 2007, 35, 821–825. [Google Scholar]
- Marchand, C.; Krajewski, K.; Lee, H.F.; Antony, S.; Johnson, A.A.; Amin, R.; Roller, P.; Kvaratskhelia, M.; Pommier, Y. Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Res. 2006, 34, 5157–5165. [Google Scholar]
- Nicolas, P. Multifunctional host defense peptides: Intracellular-targeting antimicrobial peptides. FEBS J. 2009, 276, 6483–6496. [Google Scholar] [CrossRef]
- Hilpert, K.; McLeod, B.; Yu, J.; Elliott, M.R.; Rautenbach, M.; Ruden, S.; Burck, J.; Muhle-Goll, C.; Ulrich, A.S.; Keller, S.; et al. Short cationic antimicrobial peptides interact with ATP. Antimicrob. Agents Chemother. 2010, 54, 4480–4483. [Google Scholar]
- Boman, H.G.; Agerberth, B.; Boman, A. Mechanisms of action on Escherichia coli of cecropin p1 and pr-39, two antibacterial peptides from pig intestine. Infect. Immun. 1993, 61, 2978–2984. [Google Scholar]
- Xiong, Y.Q.; Yeaman, M.R.; Bayer, A.S. In vitro antibacterial activities of platelet microbicidal protein and neutrophil defensin against Staphylococcus aureus are influenced by antibiotics differing in mechanism of action. Antimicrob. Agents Chemother. 1999, 43, 1111–1117. [Google Scholar]
- Castle, M.; Nazarian, A.; Yi, S.S.; Tempst, P. Lethal effects of apidaecin on Escherichia coli involve sequential molecular interactions with diverse targets. J. Biol. Chem. 1999, 274, 32555–32564. [Google Scholar]
- Nishikata, M.; Kanehira, T.; Oh, H.; Tani, H.; Tazaki, M.; Kuboki, Y. Salivary histatin as an inhibitor of a protease produced by the oral bacterium Bacteroides gingivalis. Biochem. Bioph. Res. Co. 1991, 174, 625–630. [Google Scholar] [CrossRef]
- Couto, M.A.; Harwig, S.S.; Lehrer, R.I. Selective inhibition of microbial serine proteases by enap-2, an antimicrobial peptide from equine neutrophils. Infect. Immun. 1993, 61, 2991–2994. [Google Scholar]
- Keppi, E.; Pugsley, A.P.; Lambert, J.; Wicker, C.; Dimarcq, J.L.; Hoffmann, J.A.; Hoffmann, D. Mode of action of diptericin-a, a bactericidal peptide induced in the hemolymph of Phormia terranovae larvae. Arch. Insect Biochem. 1989, 10, 229–239. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kubo, T.; Natori, S. Purification and characterization of a diptericin homologue from Sarcophaga peregrina (flesh fly). Biochem. J. 1992, 287, 573–578. [Google Scholar]
- Scheit, K.H.; Reddy, E.S.; Bhargava, P.M. Seminaplasmin is a potent inhibitor of E. coli RNA polymerase in vivo. Nature 1979, 279, 728–731. [Google Scholar] [CrossRef]
- Chitnis, S.N.; Prasad, K.S.; Bhargava, P.M. Isolation and characterization of autolysis-defective mutants of Escherichia coli that are resistant to the lytic activity of seminalplasmin. J. Gen. Microbiol. 1990, 136, 463–469. [Google Scholar]
- Chitnis, S.N.; Prasad, K.S.; Bhargava, P.M. Bacteriolytic activity of seminalplasmin. J. Gen. Microbiol. 1987, 133, 1265–1271. [Google Scholar]
- Jones, A.T. Macropinocytosis: Searching for an endocytic identity and role in the uptake of cell penetrating peptides. J. Cell Mol. Med. 2007, 11, 670–684. [Google Scholar] [CrossRef]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef]
- Kobayashi, S.; Takeshima, K.; Park, C.B.; Kim, S.C.; Matsuzaki, K. Interactions of the novel antimicrobial peptide buforin 2 with lipid bilayers: Proline as a translocation promoting factor. Biochemistry 2000, 39, 8648–8654. [Google Scholar]
- Park, C.B.; Yi, K.S.; Matsuzaki, K.; Kim, M.S.; Kim, S.C. Structure-activity analysis of buforin ii, a histone h2a-derived antimicrobial peptide: The proline hinge is responsible for the cell-penetrating ability of buforin ii. Proc. Natl. Acad. Sci. USA 2000, 97, 8245–8250. [Google Scholar]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, alpha-helical antimicrobial peptides. Biopolymers 2000, 55, 4–30. [Google Scholar]
- Westerhoff, H.V.; Juretic, D.; Hendler, R.W.; Zasloff, M. Magainins and the disruption of membrane-linked free-energy transduction. Proc. Natl. Acad. Sci. USA 1989, 86, 6597–6601. [Google Scholar]
- Subbalakshmi, C.; Nagaraj, R.; Sitaram, N. Biological activities of c-terminal 15-residue synthetic fragment of melittin: Design of an analog with improved antibacterial activity. FEBS Lett. 1999, 448, 62–66. [Google Scholar] [CrossRef]
- Park, Y.; Park, S.C.; Park, H.K.; Shin, S.Y.; Kim, Y.; Hahm, K.S. Structure-activity relationship of hp (2–20) analog peptide: Enhanced antimicrobial activity by n-terminal random coil region deletion. Biopolymers 2007, 88, 199–207. [Google Scholar] [CrossRef]
- Oren, Z.; Shai, Y. A class of highly potent antibacterial peptides derived from pardaxin, a pore-forming peptide isolated from moses sole fish Pardachirus marmoratus. Eur. J. Biochem. 1996, 237, 303–310. [Google Scholar]
- Pace, C.N.; Scholtz, J.M. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 1998, 75, 422–427. [Google Scholar] [CrossRef]
- Lee, D.G.; Kim, H.N.; Park, Y.K.; Kim, H.K.; Choi, B.H.; Choi, C.H.; Hahm, K.S. Design of novel analogue peptides with potent antibiotic activity based on the antimicrobial peptide, hp (2–20), derived from n-terminus of Helicobacter pylori ribosomal protein L1. Biochim. Biophys. Acta 2002, 1598, 185–194. [Google Scholar]
- Kustanovich, I.; Shalev, D.E.; Mikhlin, M.; Gaidukov, L.; Mor, A. Structural requirements for potent versus selective cytotoxicity for antimicrobial dermaseptin s4 derivatives. J. Biol. Chem. 2002, 277, 16941–16951. [Google Scholar]
- Zelezetsky, I.; Pacor, S.; Pag, U.; Papo, N.; Shai, Y.; Sahl, H.G.; Tossi, A. Controlled alteration of the shape and conformational stability of alpha-helical cell-lytic peptides: Effect on mode of action and cell specificity. Biochem. J. 2005, 390, 177–188. [Google Scholar]
- Dathe, M.; Wieprecht, T.; Nikolenko, H.; Handel, L.; Maloy, W.L.; MacDonald, D.L.; Beyermann, M.; Bienert, M. Hydrophobicity, hydrophobic moment and angle subtended by charged residues modulate antibacterial and haemolytic activity of amphipathic helical peptides. FEBS Lett. 1997, 403, 208–212. [Google Scholar] [CrossRef]
- Fernandez-Vidal, M.; Jayasinghe, S.; Ladokhin, A.S.; White, S.H. Folding amphipathic helices into membranes: Amphiphilicity trumps hydrophobicity. J. Mol. Biol. 2007, 370, 459–470. [Google Scholar] [CrossRef]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic alpha helical antimicrobial peptides. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef]
- Ryan, L.; Lamarre, B.; Diu, T.; Ravi, J.; Judge, P.J.; Temple, A.; Carr, M.; Cerasoli, E.; Su, B.; Jenkinson, H.F. Anti-antimicrobial peptides: Folding-mediated host defense antagonists. J. Biol. Chem. 2013, 288, 20162–20172. [Google Scholar] [CrossRef]
- Tew, G.N.; Liu, D.; Chen, B.; Doerksen, R.J.; Kaplan, J.; Carroll, P.J.; Klein, M.L.; de Grado, W.F. De novo design of biomimetic antimicrobial polymers. Proc. Natl. Acad. Sci. USA 2002, 99, 5110–5114. [Google Scholar]
- Goumon, Y.; Strub, J.M.; Moniatte, M.; Nullans, G.; Poteur, L.; Hubert, P.; van Dorsselaer, A.; Aunis, D.; Metz-Boutigue, M.H. The c-terminal bisphosphorylated proenkephalin-a-(209–237)-peptide from adrenal medullary chromaffin granules possesses antibacterial activity. Eur. J. Biochem. 1996, 235, 516–525. [Google Scholar]
- Kreil, G. d-amino acids in animal peptides. Annu. Rev. Biochem. 1997, 66, 337–345. [Google Scholar] [CrossRef]
- Kamatani, Y.; Minakata, H.; Nomoto, K.; Kim, K.H.; Yongsiri, A.; Takeuchi, H. Isolation of achatin-I, a neuroactive tetrapeptide having a d-phenylalanine residue, from Achatina ganglia, and its effects on Achatina giant neurones. Comp. Biochem. Physiol. C 1991, 98, 97–103. [Google Scholar]
- Hancock, R.E.; Chapple, D.S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999, 43, 1317–1323. [Google Scholar]
- Rifflet, A.; Gavalda, S.; Tene, N.; Orivel, J.; Leprince, J.; Guilhaudis, L.; Genin, E.; Vetillard, A.; Treilhou, M. Identification and characterization of a novel antimicrobial peptide from the venom of the ant Tetramorium bicarinatum. Peptides 2012, 38, 363–370. [Google Scholar] [CrossRef]
- Oman, T.J.; Boettcher, J.M.; Wang, H.; Okalibe, X.N.; van der Donk, W.A. Sublancin is not a lantibiotic but an s-linked glycopeptide. Nat. Chem. Biol. 2011, 7, 78–80. [Google Scholar]
- Mangoni, M.E.; Aumelas, A.; Charnet, P.; Roumestand, C.; Chiche, L.; Despaux, E.; Grassy, G.; Calas, B.; Chavanieu, A. Change in membrane permeability induced by protegrin 1: Implication of disulphide bridges for pore formation. FEBS Lett. 1996, 383, 93–98. [Google Scholar] [CrossRef]
- Shinnar, A.E.; Butler, K.L.; Park, H.J. Cathelicidin family of antimicrobial peptides: Proteolytic processing and protease resistance. Bioorg. Chem. 2003, 31, 425–436. [Google Scholar] [CrossRef]
- Yasin, B.; Pang, M.; Turner, J.S.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I.; Wagar, E.A. Evaluation of the inactivation of infectious herpes simplex virus by host-defense peptides. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 187–194. [Google Scholar]
- Rozek, A.; Powers, J.P.; Friedrich, C.L.; Hancock, R.E. Structure-based design of an indolicidin peptide analogue with increased protease stability. Biochemistry 2003, 42, 14130–14138. [Google Scholar]
- Osapay, K.; Tran, D.; Ladokhin, A.S.; White, S.H.; Henschen, A.H.; Selsted, M.E. Formation and characterization of a single trp-trp cross-link in indolicidin that confers protease stability without altering antimicrobial activity. J. Biol. Chem. 2000, 275, 12017–12022. [Google Scholar]
- Houston, M.E., Jr.; Kondejewski, L.H.; Karunaratne, D.N.; Gough, M.; Fidai, S.; Hodges, R.S.; Hancock, R.E. Influence of preformed alpha-helix and alpha-helix induction on the activity of cationic antimicrobial peptides. J. Pept. Res. 1998, 52, 81–88. [Google Scholar]
- Zhang, L.; Benz, R.; Hancock, R.E. Influence of proline residues on the antibacterial and synergistic activities of alpha-helical peptides. Biochemistry 1999, 38, 8102–8111. [Google Scholar] [CrossRef]
- Nell, M.J.; Tjabringa, G.S.; Wafelman, A.R.; Verrijk, R.; Hiemstra, P.S.; Drijfhout, J.W.; Grote, J.J. Development of novel ll-37 derived antimicrobial peptides with LPS and LTA neutralizing and antimicrobial activities for therapeutic application. Peptides 2006, 27, 649–660. [Google Scholar]
- Goblyos, A.; Schimmel, K.J.; Valentijn, A.R.; Fathers, L.M.; Cordfunke, R.A.; Chan, H.L.; Oostendorp, J.; Nibbering, P.H.; Drijfhout, J.W.; Hiemstra, P.S.; et al. Development of a nose cream containing the synthetic antimicrobial peptide p60.4ac for eradication of methicillin-resistant Staphylococcus aureus carriage. J. Pharm. Sci. 2013, 102, 3539–3544. [Google Scholar]
- Bessalle, R.; Kapitkovsky, A.; Gorea, A.; Shalit, I.; Fridkin, M. All-d-magainin: Chirality, antimicrobial activity and proteolytic resistance. FEBS Lett. 1990, 274, 151–155. [Google Scholar]
- Giuliani, A.; Rinaldi, A.C. Beyond natural antimicrobial peptides: Multimeric peptides and other peptidomimetic approaches. Cell Mol. Life Sci. 2011, 68, 2255–2266. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, S.C.; Yoon, M.Y.; Hahm, K.S.; Park, Y. C-terminal amidation of pmap-23: Translocation to the inner membrane of gram-negative bacteria. Amino Acids 2011, 40, 183–195. [Google Scholar]
- Berthold, N.; Czihal, P.; Fritsche, S.; Sauer, U.; Schiffer, G.; Knappe, D.; Alber, G.; Hoffmann, R. Novel apidaecin 1b analogs with superior serum stabilities for treatment of infections by gram-negative pathogens. Antimicrob. Agents Chemother. 2013, 57, 402–409. [Google Scholar]
- Gupta, M.; Chauhan, V.S. De novo design of α,β-didehydrophenylalanine containing peptides: From models to applications. Biopolymers 2011, 95, 161–173. [Google Scholar] [CrossRef]
- Mathur, P.; Jagannathan, N.R.; Chauhan, V.S. Alpha, beta-dehydrophenylalanine containing cecropin-melittin hybrid peptides: Conformation and activity. J. Pept. Sci. 2007, 13, 253–262. [Google Scholar]
- Torino, D.; Mollica, A.; Pinnen, F.; Feliciani, F.; Lucente, G.; Fabrizi, G.; Portalone, G.; Davis, P.; Lai, J.; Ma, S.W.; et al. Synthesis and evaluation of new endomorphin-2 analogues containing (z)-alpha,beta-didehydrophenylalanine (delta(z)phe) residues. J. Med. Chem. 2010, 53, 4550–4554. [Google Scholar]
- Avrahami, D.; Shai, Y. Bestowing antifungal and antibacterial activities by lipophilic acid conjugation to d,l-amino acid-containing antimicrobial peptides: A plausible mode of action. Biochemistry 2003, 42, 14946–14956. [Google Scholar] [CrossRef]
- Schneider, G.; Schrodl, W.; Wallukat, G.; Muller, J.; Nissen, E.; Ronspeck, W.; Wrede, P.; Kunze, R. Peptide design by artificial neural networks and computer-based evolutionary search. Proc. Natl. Acad. Sci. USA 1998, 95, 12179–12184. [Google Scholar]
- Fjell, C.D.; Jenssen, H.; Hilpert, K.; Cheung, W.A.; Pante, N.; Hancock, R.E.; Cherkasov, A. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 2009, 52, 2006–2015. [Google Scholar]
- Belda, I.; Llora, X.; Giralt, E. Evolutionary algorithms and de novo peptide design. Soft. Comput. 2006, 10, 295–304. [Google Scholar] [CrossRef]
- Loose, C.; Jensen, K.; Rigoutsos, I.; Stephanopoulos, G. A linguistic model for the rational design of antimicrobial peptides. Nature 2006, 443, 867–869. [Google Scholar]
- Jenssen, H.; Fjell, C.D.; Cherkasov, A.; Hancock, R.E. QSAR modeling and computer-aided design of antimicrobial peptides. J. Pept. Sci. 2008, 14, 110–114. [Google Scholar]
- Cherkasov, A.; Hilpert, K.; Jenssen, H.; Fjell, C.D.; Waldbrook, M.; Mullaly, S.C.; Volkmer, R.; Hancock, R.E. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 2009, 4, 65–74. [Google Scholar]
- Fjell, C.D.; Jenssen, H.; Cheung, W.A.; Hancock, R.E.; Cherkasov, A. Optimization of antibacterial peptides by genetic algorithms and cheminformatics. Chem. Biol. Drug Des. 2011, 77, 48–56. [Google Scholar]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 124–124. [Google Scholar]
- Tossi, A.; Tarantino, C.; Romeo, D. Design of synthetic antimicrobial peptides based on sequence analogy and amphipathicity. Eur. J. Biochem. 1997, 250, 549–558. [Google Scholar]
- Mor, A.; Nicolas, P. The nh2-terminal alpha-helical domain 1-18 of dermaseptin is responsible for antimicrobial activity. J. Biol. Chem. 1994, 269, 1934–1939. [Google Scholar]
- Storici, P.; Scocchi, M.; Tossi, A.; Gennaro, R.; Zanetti, M. Chemical synthesis and biological activity of a novel antibacterial peptide deduced from a pig myeloid cDNA. FEBS Lett. 1994, 337, 303–307. [Google Scholar]
- Romani, A.A.; Baroni, M.C.; Taddei, S.; Ghidini, F.; Sansoni, P.; Cavirani, S.; Cabassi, C.S. In vitro activity of novel in silico-developed antimicrobial peptides against a panel of bacterial pathogens. J. Pept. Sci. 2013, 19, 554–565. [Google Scholar]
- Cruz, J.; Ortiz, C.C.; Guzman, F.; Cardenas, C.; Fernandez-Lafuente, R.; Torres, R.G. Design and activity of novel lactoferrampin analogues against O157:H7 enterohemorrhagic Escherichia coli. Biopolymers 2013. [Google Scholar] [CrossRef]
- Darouiche, R.O. Treatment of infections associated with surgical implants. N. Engl. J. Med. 2004, 350, 1422–1429. [Google Scholar] [CrossRef]
- Lewis, K. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 2001, 45, 999–1007. [Google Scholar] [CrossRef]
- Harro, J.M.; Peters, B.M.; O’May, G.A.; Archer, N.; Kerns, P.; Prabhakara, R.; Shirtliff, M.E. Vaccine development in Staphylococcus aureus: Taking the biofilm phenotype into consideration. FEMS Immunol. Med. Microbiol. 2010, 59, 306–323. [Google Scholar]
- Sutherland, I. Biofilm exopolysaccharides: A strong and sticky framework. Microbiology 2001, 147, 3–9. [Google Scholar]
- Mah, T.F.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Dunne, W.M.; Mason, E.O.; Kaplan, S.L. Diffusion of rifampin and vancomycin through a Staphylococcus epidermidis biofilm. Antimicrob. Agents Chemother. 1993, 37, 2522–2526. [Google Scholar] [CrossRef]
- Keren, I.; Kaldalu, N.; Spoering, A.; Wang, Y.P.; Lewis, K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004, 234, 187–187. [Google Scholar]
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138. [Google Scholar]
- Yao, Y.; Sturdevant, D.E.; Otto, M. Genomewide analysis of gene expression in Staphylococcus epidermidis biofilms: Insights into the pathophysiology of S. epidermidis biofilms and the role of phenol-soluble modulins in formation of biofilms. J. Infect. Dis. 2005, 191, 289–298. [Google Scholar]
- Otto, M. Bacterial evasion of antimicrobial peptides by biofilm formation. Curr. Top. Microbiol. Immunol. 2006, 306, 251–258. [Google Scholar] [CrossRef]
- Singh, P.K.; Parsek, M.R.; Greenberg, E.P.; Welsh, M.J. A component of innate immunity prevents bacterial biofilm development. Nature 2002, 417, 552–555. [Google Scholar]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.; Rehm, B.H.; Hancock, R.E. Human host defense peptide ll-37 prevents bacterial biofilm formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar]
- De la Fuente-Nunez, C.; Korolik, V.; Bains, M.; Nguyen, U.; Breidenstein, E.B.; Horsman, S.; Lewenza, S.; Burrows, L.; Hancock, R.E. Inhibition of bacterial biofilm formation and swarming motility by a small synthetic cationic peptide. Antimicrob. Agents Chemother. 2012, 56, 2696–2704. [Google Scholar]
- Gao, G.; Lange, D.; Hilpert, K.; Kindrachuk, J.; Zou, Y.; Cheng, J.T.; Kazemzadeh-Narbat, M.; Yu, K.; Wang, R.; Straus, S.K.; et al. The biocompatibility and biofilm resistance of implant coatings based on hydrophilic polymer brushes conjugated with antimicrobial peptides. Biomaterials 2011, 32, 3899–3909. [Google Scholar] [CrossRef]
- Lucke, M.; Schmidmaier, G.; Sadoni, S.; Wildemann, B.; Schiller, R.; Haas, N.P.; Raschke, M. Gentamicin coating of metallic implants reduces implant-related osteomyelitis in rats. Bone 2003, 32, 521–531. [Google Scholar] [CrossRef]
- Price, J.S.; Tencer, A.F.; Arm, D.M.; Bohach, G.A. Controlled release of antibiotics from coated orthopedic implants. J. Biomed. Mater. Res. 1996, 30, 281–286. [Google Scholar] [CrossRef]
- Gollwitzer, H.; Ibrahim, K.; Meyer, H.; Mittelmeier, W.; Busch, R.; Stemberger, A. Antibacterial poly(d,l-lactic acid) coating of medical implants using a biodegradable drug delivery technology. J. Antimicrob. Chemoth. 2003, 51, 585–591. [Google Scholar]
- Yoshinari, M.; Kato, T.; Matsuzaka, K.; Hayakawa, T.; Shiba, K. Prevention of biofilm formation on titanium surfaces modified with conjugated molecules comprised of antimicrobial and titanium-binding peptides. Biofouling 2010, 26, 103–110. [Google Scholar] [CrossRef]
- Helmerhorst, E.J.; Hodgson, R.; van’t Hof, W.; Veerman, E.C.; Allison, C.; Nieuw Amerongen, A.V. The effects of histatin-derived basic antimicrobial peptides on oral biofilms. J. Dent. Res. 1999, 78, 1245–1250. [Google Scholar] [CrossRef]
- Wei, G.X.; Campagna, A.N.; Bobek, L.A. Effect of muc7 peptides on the growth of bacteria and on Streptococcus mutans biofilm. J. Antimicrob. Chemother. 2006, 57, 1100–1109. [Google Scholar]
- Chennupati, S.K.; Chiu, A.G.; Tamashiro, E.; Banks, C.A.; Cohen, M.B.; Bleier, B.S.; Kofonow, J.M.; Tam, E.; Cohen, N.A. Effects of an ll-37-derived antimicrobial peptide in an animal model of biofilm Pseudomonas sinusitis. Am. J. Rhinol. Allergy 2009, 23, 46–51. [Google Scholar] [CrossRef]
- Shigeta, M.; Tanaka, G.; Komatsuzawa, H.; Sugai, M.; Suginaka, H.; Usui, T. Permeation of antimicrobial agents through Pseudomonas aeruginosa biofilms: A simple method. Chemotherapy 1997, 43, 340–345. [Google Scholar] [CrossRef]
- Liu, Z.G.; Young, A.W.; Hu, P.; Rice, A.J.; Zhou, C.H.; Zhan, Y.K.; Kallenbach, N.R. Tuning the membrane selectivity of antimicrobial peptides by using multivalent design. ChemBioChem 2007, 8, 2063–2065. [Google Scholar]
- Hou, S.Y.; Zhou, C.H.; Liu, Z.G.; Young, A.W.; Shi, Z.H.; Ren, D.C.; Kallenbach, N.R. Antimicrobial dendrimer active against Escherichia coli biofilms. Bioorg. Med. Chem. Lett. 2009, 19, 5478–5481. [Google Scholar]
- Hou, S.; Liu, Z.; Young, A.W.; Mark, S.L.; Kallenbach, N.R.; Ren, D. Effects of trp- and arg-containing antimicrobial-peptide structure on inhibition of Escherichia coli planktonic growth and biofilm formation. Appl. Environ. Microbiol. 2010, 76, 1967–1974. [Google Scholar]
- Okuda, K.I.; Zendo, T.; Sugimoto, S.; Iwase, T.; Tajima, A.; Yamada, S.; Sonomoto, K.; Mizunoe, Y. Effects of bacteriocins on methicillin-resistant Staphylococcus aureus biofilm. Antimicrob. Agents Chemother. 2013, 57, 5572–5579. [Google Scholar]
- Hirt, H.; Gorr, S.U. Antimicrobial peptide gl13k is effective in reducing biofilms of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 4903–4910. [Google Scholar] [CrossRef]
- Gopal, R.; Lee, J.H.; Kim, Y.G.; Kim, M.S.; Seo, C.H.; Park, Y. Anti-microbial, anti-biofilm activities and cell selectivity of the nrc-16 peptide derived from witch flounder, Glyptocephalus cynoglossus. Mar. Drugs 2013, 11, 1836–1852. [Google Scholar] [CrossRef]
- Leitch, E.C.; Willcox, M.D. Lactoferrin increases the susceptibility of S. epidermidis biofilms to lysozyme and vancomycin. Curr. Eye Res. 1999, 19, 12–19. [Google Scholar] [CrossRef]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Zhou, C.; Kallenbach, N.R.; Ren, D. Control of bacterial persister cells by trp/arg-containing antimicrobial peptides. Appl. Environ. Microbiol. 2011, 77, 4878–4885. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef]
- Lewis, L.A.; Choudhury, B.; Balthazar, J.T.; Martin, L.E.; Ram, S.; Rice, P.A.; Stephens, D.S.; Carlson, R.; Shafer, W.M. Phosphoethanolamine substitution of lipid a and resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect. Immun. 2009, 77, 1112–1120. [Google Scholar] [CrossRef]
- Gunn, J.S. Bacterial modification of LPS and resistance to antimicrobial peptides. J. Endotoxin Res. 2001, 7, 57–62. [Google Scholar]
- Guo, L.; Lim, K.B.; Poduje, C.M.; Daniel, M.; Gunn, J.S.; Hackett, M.; Miller, S.I. Lipid a acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 1998, 95, 189–198. [Google Scholar] [CrossRef]
- Guina, T.; Yi, E.C.; Wang, H.; Hackett, M.; Miller, S.I. A phop-regulated outer membrane protease of Salmonella enterica serovar typhimurium promotes resistance to alpha-helical antimicrobial peptides. J. Bacteriol. 2000, 182, 4077–4086. [Google Scholar]
- Shafer, W.M.; Qu, X.; Waring, A.J.; Lehrer, R.I. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc. Natl. Acad. Sci. USA 1998, 95, 1829–1833. [Google Scholar]
- Del Castillo, F.J.; del Castillo, I.; Moreno, F. Construction and characterization of mutations at codon 751 of the Escherichia coli gyrB gene that confer resistance to the antimicrobial peptide microcin b17 and alter the activity of DNA gyrase. J. Bacteriol. 2001, 183, 2137–2140. [Google Scholar] [CrossRef]
- Friedrich, C.; Scott, M.G.; Karunaratne, N.; Yan, H.; Hancock, R.E. Salt-resistant alpha-helical cationic antimicrobial peptides. Antimicrob. Agents Chemother. 1999, 43, 1542–1548. [Google Scholar]
- Yeaman, M.R.; Bayer, A.S.; Koo, S.P.; Foss, W.; Sullam, P.M. Platelet microbicidal proteins and neutrophil defensin disrupt the Staphylococcus aureus cytoplasmic membrane by distinct mechanisms of action. J. Clin. Invest. 1998, 101, 178–187. [Google Scholar]
- Vuong, C.; Voyich, J.M.; Fischer, E.R.; Braughton, K.R.; Whitney, A.R.; DeLeo, F.R.; Otto, M. Polysaccharide intercellular adhesin (pia) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol. 2004, 6, 269–275. [Google Scholar]
- Miller, S.I.; Kukral, A.M.; Mekalanos, J.J. A two-component regulatory system (phop phoq) controls Salmonella typhimurium virulence. Proc. Natl. Acad. Sci. USA 1989, 86, 5054–5058. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bahar, A.A.; Ren, D. Antimicrobial Peptides. Pharmaceuticals 2013, 6, 1543-1575. https://doi.org/10.3390/ph6121543
Bahar AA, Ren D. Antimicrobial Peptides. Pharmaceuticals. 2013; 6(12):1543-1575. https://doi.org/10.3390/ph6121543
Chicago/Turabian StyleBahar, Ali Adem, and Dacheng Ren. 2013. "Antimicrobial Peptides" Pharmaceuticals 6, no. 12: 1543-1575. https://doi.org/10.3390/ph6121543
APA StyleBahar, A. A., & Ren, D. (2013). Antimicrobial Peptides. Pharmaceuticals, 6(12), 1543-1575. https://doi.org/10.3390/ph6121543