Radiosynthesis and Radiotracer Properties of a 7-(2-[18F]Fluoroethoxy)-6-methoxypyrrolidinylquinazoline for Imaging of Phosphodiesterase 10A with PET

 ,
,
Abstract
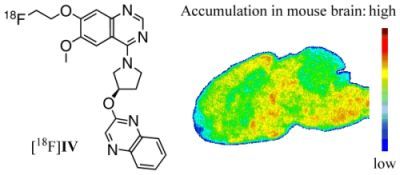
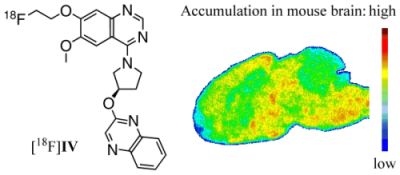
:
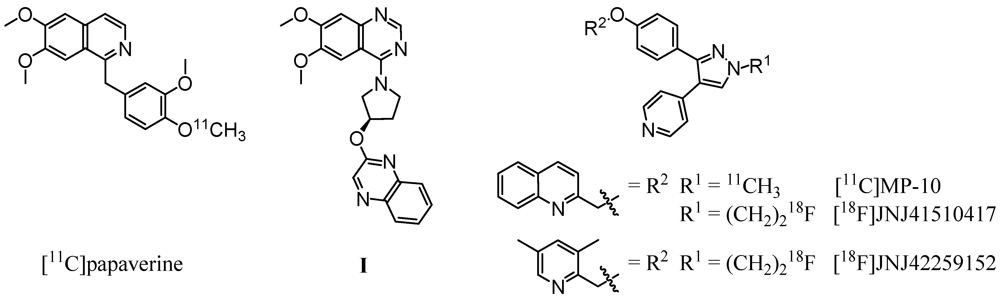
1. Introduction
2. Results and Discussion
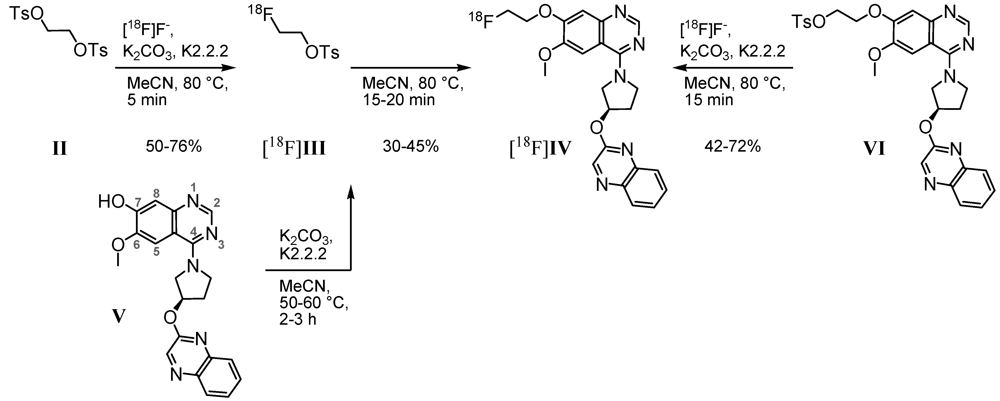
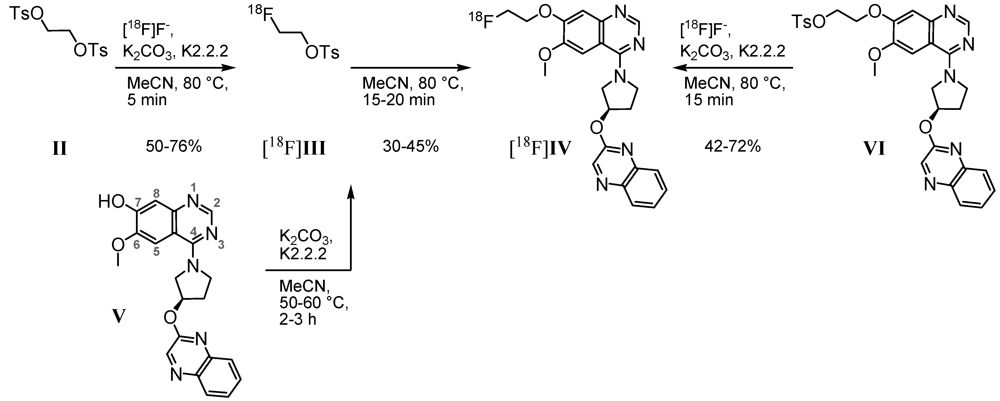
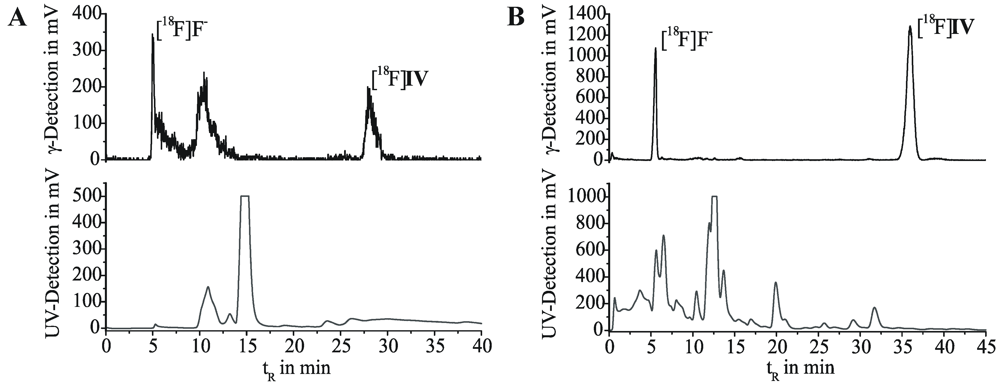
2.1. Radiosyntheses

2.2. Lipophilicity and Radioligand Stability in Vitro

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Specification | pH | Log D |
|---|---|---|---|
| Calculation | ACD/LogD | 7.2 | 2.27 |
| MarvinSketch | 7.0–7.4 | 3.52 ± 0.01 | |
| HPLC | Multospher, MeCN/NH4OAc | 7.0 | 2.63 ± 0.01 |
| Supelcosil, MeCN/NH4OAc | 7.0 | 2.63 ± 0.01 | |
| Shake flask | n-Octanol/phys. Phosphate | 7.2 | 2.73 ± 0.59 |
| n-Octanol/Phosphate | 7.2 | 2.64 ± 0.56 | |
| n-Octanol/Tris-HCl | 7.4 | 2.64 ± 0.55 | |
| Average | 7.2–7.4 | 2.67 ± 0.58 |
2.3. PDE10A Affinity and Selectivity
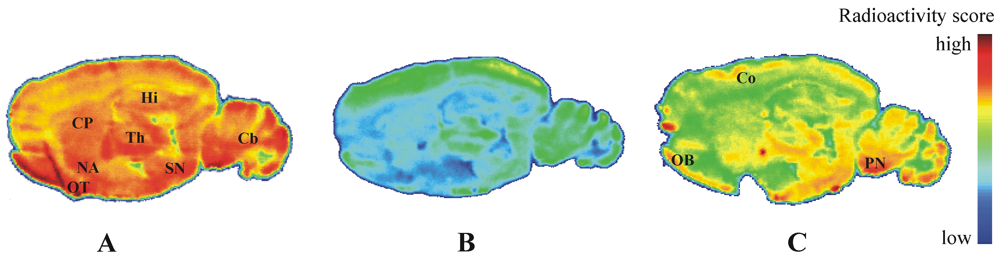
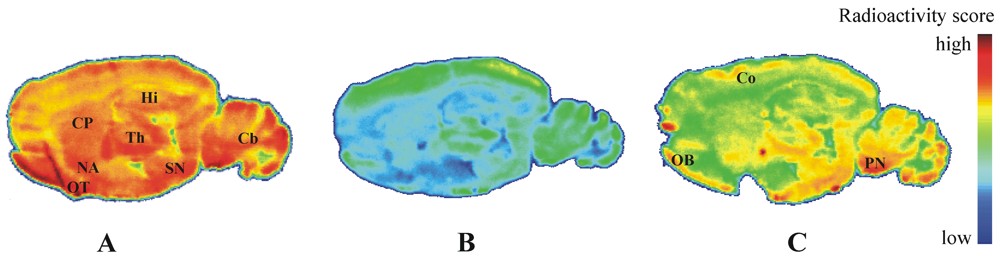
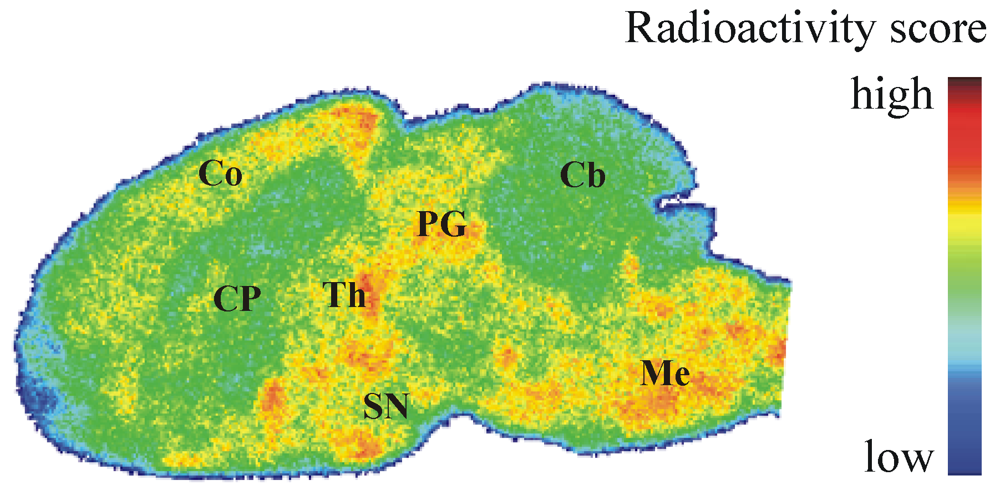
2.4. Autoradiography in Vitro
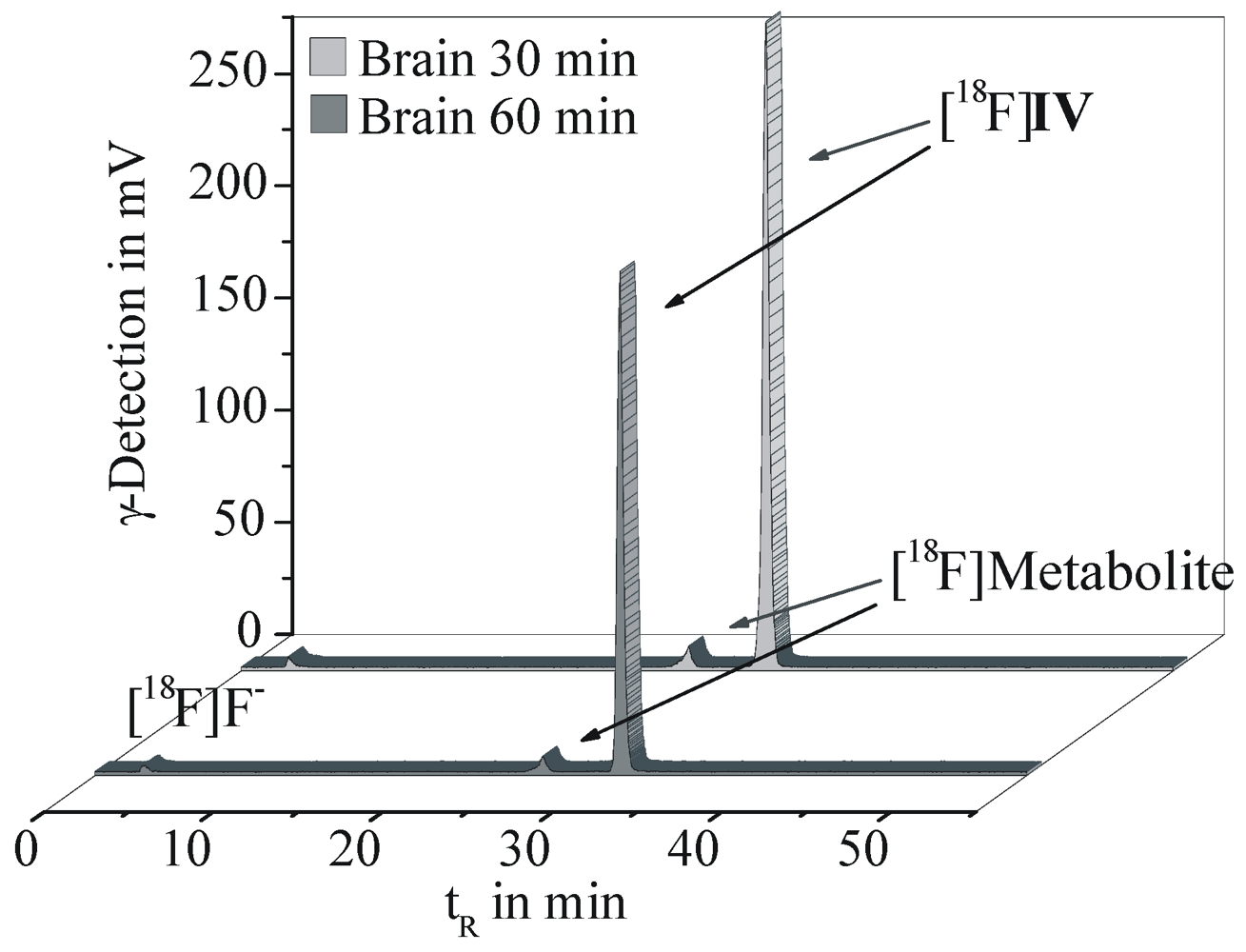
2.5. Radioligand Stability in Vivo
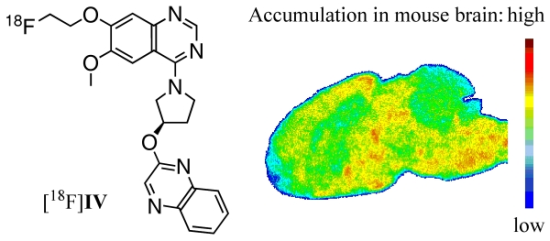
2.6. Biodistribution and Specificity in Vivo
| Control (% ID/g Wet Weight) | Blocking, 60 min p.i. | |||||
|---|---|---|---|---|---|---|
| Organ | 5 min p.i. (n = 4) | 30 min p.i. (n = 4) | 60 min p.i. (n = 6) | 120 min p.i. (n = 3) | MP-10 (n = 3) | I (n = 3) |
| Blood | 1.88 ± 0.08 | 1.69 ± 0.38 | 1.45 ± 0.20 | 1.05 ± 0.10 | 1.85 ± 0.50 | 1.72 ± 0.16 |
| Plasma | 3.97 ± 1.41 | 2.87 ± 0.54 | 2.59 ± 0.36 | 2.13 ± 0.80 | 3.34 ± 1.00 | 2.70 ± 0.24 |
| Brain | 1.99 ± 0.21 | 1.54 ± 0.30 | 1.08 ± 0.26 | 0.77 ± 0.10 | 1.30 ± 0.20 | 1.13 ± 0.17 |
| Striatum | 2.32 ± 0.49 | 1.69 ± 0.44 | 1.14 ± 0.29 | 0.83 ± 0.10 | 1.38 ± 0.30 | 1.28 ± 0.27 |
| Heart | 4.01 ± 0.47 | 2.93 ± 0.71 | 2.16 ± 0.45 | 1.33 ± 0.20 | 2.89 ± 0.42 | 2.30 ± 0.22 |
| Lungs | 4.01 ± 0.51 | 3.51 ± 1.26 | 2.37 ± 0.62 | 1.46 ± 0.26 | 2.95 ± 0.86 | 2.62 ± 0.44 |
| Stomach | 6.14 ± 2.04 | 15.8 ± 6.09 | 12.6 ± 5.08 | 10.1 ± 5.96 | 14.6 ± 7.83 | 13.8 ± 1.65 |
| Intestine | 13.5 ± 4.15 | 43.2 ± 21.9 | 42.8 ± 33.5 | 49.3 ± 8.03 | 14.8 ± 4.16 | 44.3 ± 9.58 |
| Colon | 0.55 ± 0.05 | 0.88 ± 0.44 | 1.08 ± 0.43 | 3.49 ± 2.41 | 1.78 ± 0.78 | 1.07 ± 0.22 |
| Liver | 20.5 ± 1.87 | 16.4 ± 4.00 | 14.5 ± 4.54 | 7.05 ± 1.10 | 17.7 ± 3.87 | 12.9 ± 1.08 |
| Kidneys | 7.65 ± 0.54 | 5.34 ± 1.4 | 4.29 ± 0.93 | 2.13 ± 0.44 | 5.87 ± 1.56 | 4.01 ± 0.54 |
| Urine | 56.4 ± 105 | 18.5 ± 16.8 | 23.1 ± 9.31 | 30.9 ± 8.06 | 14.7 ± 7.17 | 18.0 ± 9.26 |
| Bladder | 1.85 ± 0.39 | 3.51 ± 1.39 | 2.60 ± 0.78 | 1.62 ± 0.28 | 2.93 ± 0.51 | 2.20 ± 0.49 |
| Spleen | 4.23 ± 0.51 | 2.85 ± 0.60 | 2.30 ± 0.25 | 1.21 ± 0.24 | 3.00 ± 0.79 | 2.48 ± 0.68 |
| Thymus | 3.10 ± 0.45 | 2.61 ± 0.50 | 2.08 ± 0.51 | 1.26 ± 0.07 | 2.55 ± 0.62 | 2.22 ± 0.08 |
| Pancreas | 5.19 ± 0.42 | 3.14 ± 0.70 | 2.77 ± 0.85 | 1.41 ± 0.07 | 3.19 ± 0.91 | 2.25 ± 0.26 |
| Adrenals | 25.4 ± 5.87 | 48.5 ± 16.2 | 28.2 ± 11.4 | 19.3 ± 13.2 | 36.0 ± 9.51 | 51.1 ± 17.9 |
| Gonads | 2.58 ± 0.46 | 3.94 ± 0.84 | 3.50 ± 1.22 | 1.64 ± 0.26 | 4.10 ± 1.41 | 3.30 ± 0.29 |
| Muscles | 1.75 ± 0.12 | 1.31 ± 0.32 | 1.22 ± 0.28 | 0.66 ± 0.01 | 1.29 ± 0.37 | 1.27 ± 0.25 |
| Skin | n.d. | n.d. | 1.45, 1.17 * | n.d. | 2.43 ± 0.44 | 2.26 ± 0.82 |
| Femurs | 2.00 ± 0.36 | 2.14 ± 1.13 | 2.95 ± 3.73 | 1.61 ± 0.35 | 1.73 ± 0.58 | 1.62 ± 0.72 |
| Femurs (fl.) | 1.11 ± 0.32 | 0.67 ± 0.24 | 0.58 ± 0.42 | 1.15 ± 0.18 | 0.92 ± 0.23 | 0.81 ± 0.13 |
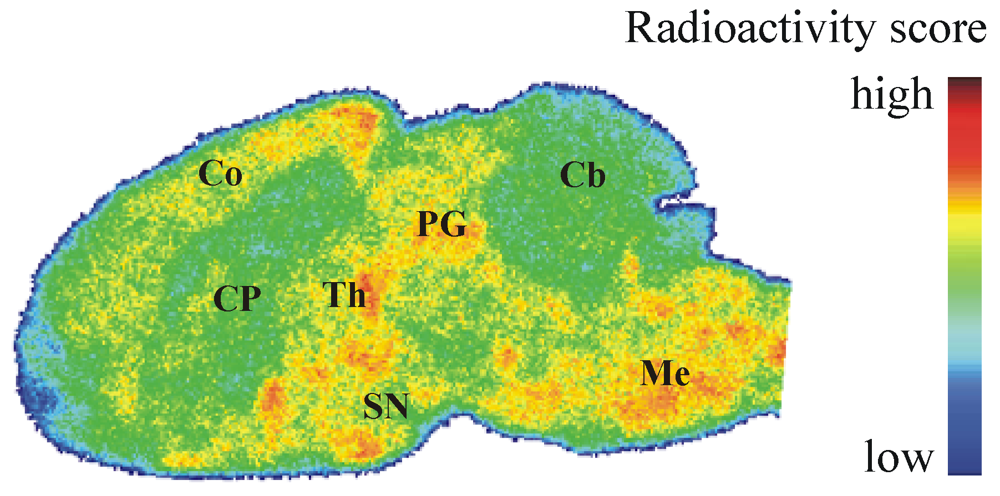
2.7. Autoradiography ex Vivo
3. Experimental Section
3.1. General
3.2. Radiochemistry
3.2.1. Two-Step Radiosynthesis of (R)-7-(2-[18F]Fluoroethoxy)-6-methoxy-4-(3-(quinoxalin-2-yloxy)pyrrolidin-1-yl)quinazoline ([18F]IV)
3.2.2. One-Step Radiosynthesis of [18F]IV
3.3. Determination of Lipophilicity of [18/19F]IV
3.3.1. Determination of Log D7.2–7.4 Values by Shake-Flask Method
3.3.2. Determination of log D7.0 Values by RP-HPLC Retention
3.4. In Vitro Characterization
3.4.1. PDE10A Affinity
3.4.2. Data Analysis
3.4.3. In Vitro Autoradiographic Study
3.5. Biodistribution and Metabolism Studies in Mice
3.5.1. Metabolic Stability
3.5.2. Biodistribution and Regional Brain Uptake Studies
3.5.3. Ex Vivo Autoradiographic Study
4. Conclusions
Acknowledgments
References
- Beavo, J.A. Cyclic nucleotide phosphodiesterases: Functional implications of multiple isoforms. Physiol. Rev. 1995, 75, 725–748. [Google Scholar]
- Francis, S.H.; Turko, I.V.; Corbin, J.D. Cyclic nucleotide phosphodiesterases: Relating structure and function. Prog. Nucleic Acid Res. Mol. Biol. 2001, 65, 1–52. [Google Scholar]
- Keravis, T.; Lugnier, C. Cyclic nucleotide phosphodiesterases (PDE) and peptide motifs. Curr. Pharm. Des. 2010, 16, 1114–1125. [Google Scholar] [CrossRef]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef]
- Surapisitchat, J.; Beavo, J.A. Phosphodiesterase families. In Transduction Mechanisms in Cellular Signaling: Cell Signaling Collection, 1st; Dennis, E.A., Bradshaw, R.A., Eds.; Academic Press, Elsevier Inc.: San Diego, CA, USA, 2011; pp. 375–380. [Google Scholar]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Fujishige, K.; Kotera, J.; Omori, K. Striatum- and testis-specific phosphodiesterase PDE10A. Isolation and characterization of a rat PDE10A. Eur. J. Biochem. 1999, 266, 1118–1127. [Google Scholar] [CrossRef]
- Lakics, V.; Karran, E.H.; Boess, F.G. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010, 59, 367–374. [Google Scholar] [CrossRef]
- Seeger, T.F.; Bartlett, B.; Coskran, T.M.; Culp, J.S.; James, L.C.; Krull, D.L.; Lanfear, J.; Ryan, A.M.; Schmidt, C.J.; Strick, C.A.; et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res. 2003, 985, 113–126. [Google Scholar] [CrossRef]
- Coskran, T.M.; Morton, D.; Menniti, F.S.; Adamowicz, W.O.; Kleiman, R.J.; Ryan, A.M.; Strick, C.A.; Schmidt, C.J.; Stephenson, D.T. Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J. Histochem. Cytochem. 2006, 54, 1205–1213. [Google Scholar] [CrossRef]
- Xie, Z.; Adamowicz, W.O.; Eldred, W.D.; Jakowski, A.B.; Kleiman, R.J.; Morton, D.G.; Stephenson, D.T.; Strick, C.A.; Williams, R.D.; Menniti, F.S. Cellular and subcellular localization of PDE10A, a striatum-enriched phosphodiesterase. Neuroscience 2006, 139, 597–607. [Google Scholar] [CrossRef]
- Siuciak, J.A.; Chapin, D.S.; Harms, J.F.; Lebel, L.A.; McCarthy, S.A.; Chambers, L.; Shrikhande, A.; Wong, S.; Menniti, F.S.; Schmidt, C.J. Inhibition of the striatum-enriched phosphodiesterase PDE10A: A novel approach to the treatment of psychosis. Neuropharmacology 2006, 51, 386–396. [Google Scholar] [CrossRef]
- Grauer, S.M.; Pulito, V.L.; Navarra, R.L.; Kelly, M.P.; Kelley, C.; Graf, R.; Langen, B.; Logue, S.; Brennan, J.; Jiang, L.; et al. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J. Pharmacol. Exp. Ther. 2009, 331, 574–590. [Google Scholar] [CrossRef]
- Siuciak, J.A.; McCarthy, S.A.; Chapin, D.S.; Martin, A.N.; Harms, J.F.; Schmidt, C.J. Behavioral characterization of mice deficient in the phosphodiesterase-10A (PDE10A) enzyme on a C57/Bl6N congenic background. Neuropharmacology 2008, 54, 417–427. [Google Scholar] [CrossRef]
- Siuciak, J.A.; McCarthy, S.A.; Chapin, D.S.; Fujiwara, R.A.; James, L.C.; Williams, R.D.; Stock, J.L.; McNeish, J.D.; Strick, C.A.; Menniti, F.S.; et al. Genetic deletion of the striatum-enriched phosphodiesterase PDE10A: Evidence for altered striatal function. Neuropharmacology 2006, 51, 374–385. [Google Scholar] [CrossRef]
- O'Connor, V.; Genin, A.; Davis, S.; Karishma, K.K.; Doyère, V.; de Zeeuw, C.I.; Sanger, G.; Hunt, S.P.; Richter-Levin, G.; Mallet, J.; et al. Differential amplification of intron-containing transcripts reveals long term potentiation-associated up-regulation of specific Pde10A phosphodiesterase splice variants. J. Biol. Chem. 2004, 279, 15841–15849. [Google Scholar]
- Hu, H.; McCaw, E.A.; Hebb, A.L.O.; Gomez, G.T.; Denovan-Wright, E.M. Mutant huntingtin affects the rate of transcription of striatum-specific isoforms of phosphodiesterase 10A. Eur. J. Neurosci. 2004, 20, 3351–3363. [Google Scholar] [CrossRef]
- Siuciak, J.A. The role of phosphodiesterases in schizophrenia: Therapeutic implications. CNS Drugs 2008, 22, 983–993. [Google Scholar] [CrossRef]
- Siuciak, J.A.; Strick, C.A. Phosphodiesterase 10A inhibitors as a novel therapeutic approach for schizophrenia. Expert Opin. Drug Discov. 2007, 2, 1001–1009. [Google Scholar] [CrossRef]
- Kehler, J.; Ritzén, A.; Greve, D.R. The potential therapeutic use of phosphodiesterase 10 inhibitors. Expert Opin. Ther. Pat. 2007, 17, 147–158. [Google Scholar] [CrossRef]
- Kehler, J.; Kilburn, J.P. Patented PDE10A inhibitors: Novel compounds since 2007. Expert Opin. Ther. Pat. 2009, 19, 1715–1725. [Google Scholar] [CrossRef]
- Kehler, J.; Nielsen, J. PDE10A inhibitors: Novel therapeutic drugs for schizophrenia. Curr. Pharm. Des. 2011, 17, 137–150. [Google Scholar] [CrossRef]
- Tu, Z.; Xu, J.; Jones, L.A.; Li, S.; Mach, R.H. Carbon-11 labeled papaverine as a PET tracer for imaging PDE10A: Radiosynthesis, in vitro and in vivo evaluation. Nucl. Med. Biol. 2010, 37, 509–516. [Google Scholar] [CrossRef]
- Tu, Z.; Fan, J.; Li, S.; Jones, L.A.; Cui, J.; Padakanti, P.K.; Xu, J.; Zeng, D.; Shoghi, K.I.; Perlmutter, J.S.; Mach, R.H. Radiosynthesis and in vivo evaluation of [11C]MP-10 as a PET probe for imaging PDE10A in rodent and non-human primate brain. Bioorg. Med. Chem. 2011, 19, 1666–1673. [Google Scholar]
- Plisson, C.; Salinas, C.; Weinzimmer, D.; Labaree, D.; Lin, S.-F.; Ding, Y.-S.; Jakobsen, S.; Smith, P.W.; Eiji, K.; Carson, R.E.; et al. Radiosynthesis and in vivo evaluation of [11C]MP-10 as a positron emission tomography radioligand for phosphodiesterase 10A. Nucl. Med. Biol. 2011, 38, 875–884. [Google Scholar] [CrossRef]
- Verhoest, P.R.; Chapin, D.S.; Corman, M.; Fonseca, K.; Harms, J.F.; Hou, X.; Marr, E.S.; Menniti, F.S.; Nelson, F.; O'Connor, R.; et al. Discovery of a novel class of phosphodiesterase 10A inhibitors and identification of clinical candidate 2-[4-(1-methyl-4-pyridin-4-yl-1H-pyrazol-3-yl)-phenoxymethyl]-quinoline (PF-2545920) for the treatment of schizophrenia. J. Med. Chem. 2009, 52, 5188–5196. [Google Scholar]
- Celen, S.; Angelis, M.D.; Chitneni, S.K.; Alcazar, J.; Koole, M.; Dedeurwaerdere, S.; Steckler, T.; Schmidt, M.; Laere, K.V.; Verbruggen, A.; et al. Synthesis and preliminary biological evaluation of [18F]JNJ41510417 as a radioligand for positron emission tomography imaging of phosphodiesterase-10A in the brain. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, S236. [Google Scholar]
- Celen, S.; Koole, M.; Angelis, M.D.; Sannen, I.; Chitneni, S.K.; Alcazar, J.; Dedeurwaerdere, S.; Moechars, D.; Schmidt, M.; Verbruggen, A.; et al. Preclinical evaluation of 18F-JNJ41510417 as a radioligand for PET imaging of phosphodiesterase-10A in the brain. J. Nucl. Med. 2010, 51, 1584–1591. [Google Scholar]
- Andrés, J.-I.; de Angelis, M.; Alcázar, J.; Iturrino, L.; Langlois, X.; Dedeurwaerdere, S.; Lenaerts, I.; Vanhoof, G.; Celen, S.; Bormans, G. Synthesis, in vivo occupancy, and radiolabeling of potent phosphodiesterase subtype-10 inhibitors as candidates for positron emission tomography imaging. J. Med. Chem. 2011, 54, 5820–5835. [Google Scholar] [CrossRef]
- Celen, S.; de Angelis, M.; Koole, M.; Alcazar, J.; Sannen, I.; Cornelis, J.; Dedeurwaerdere, S.; Schmidt, M.; van Laere, K.; Verbruggen, A.; et al. [18F]JNJ42259152 as potential radioligand for positron emission tomography imaging of phosphodiesterase-10A in the brain. J. Labelled Comp. Radiopharm. 2011, 54, S82. [Google Scholar]
- Chappie, T.A.; Humphrey, J.M.; Allen, M.P.; Estep, K.G.; Fox, C.B.; Lebel, L.A.; Liras, S.; Marr, E.S.; Menniti, F.S.; Pandit, J.; et al. Discovery of a Series of 6,7-Dimethoxy-4-pyrrolidylquinazoline PDE10A inhibitors. J. Med. Chem. 2007, 50, 182–185. [Google Scholar]
- Nieber, K.; Erdmann, S.; Briel, D.; Schwan, G.; Kubicova, L.; Barbar Asskar, G.; Sträter, N.; Zahn, M.; Brust, P.; Funke, U.; et al. Neue Halogenalkoxychinazoline, deren Herstellung und. Verwendung. Patent Appl. 00401P0051DE, 20 October 2010. [Google Scholar]
- Block, D.; Coenen, H.H.; Stöcklin, G. The N.C.A. nucleophilic 18F-fluorination of 1,N-disubstituted alkanes as fluoroalkylation agents. J. Labelled Comp. Radiopharm. 1987, 24, 1029–1042. [Google Scholar] [CrossRef]
- Block, D.; Coenen, H.H.; Stöcklin, G. N.C.A. 18F-fluoroalkylation of H-acidic compounds. J. Labelled Comp. Radiopharm. 1988, 25, 201–216. [Google Scholar] [CrossRef]
- Waterhouse, R.N. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol. Imaging Biol. 2003, 5, 376–389. [Google Scholar] [CrossRef]
- OECD (Organisation for Economic Co-operation and Development), Partition coefficient (n-octanol/water), high performance liquid chromatography (HPLC) method. In OECD Guideline for Testing of Chemicals; Paris, France, 2004; p. 11.
- Motulsky, H.J.; Christopoulos, A. Fitting Models to Biological Data Using Linear and Nonlinear Regression. A Practical Guide to Curve Fitting, 2nd ed; GraphPad Software, Inc.: San Diego, CA, USA, 2003; p. 351. [Google Scholar]
- Pike, V.W. PET radiotracers: Crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef]
- Reinhardt, R.R.; Bondy, C.A. Differential cellular pattern of gene expression for two distinct cGMP-inhibited cyclic nucleotide phosphodiesterases in developing and mature rat brain. Neuroscience 1996, 72, 567–578. [Google Scholar] [CrossRef]
- Seliskar, M.; Rozman, D. Mammalian cytochromes P450—Importance of tissue specificity. Biochim. Biophys. Acta 1770, 458–466. [Google Scholar]
- Block, D.; Klatte, B.; Knöchel, A.; Beckmann, R.; Holm, U. N.C.A. [18F]-labelling of aliphatic compounds in high yields via aminopolyether-supported nucleophilic substitution. J. Labelled Comp. Radiopharm. 1986, 23, 467–477. [Google Scholar] [CrossRef]
- Xing, D.; Chen, P.; Keil, R.; Kilts, C.D.; Shi, B.; Camp, V.M.; Malveaux, G.; Ely, T.; Owens, M.J.; Votaw, J.; et al. Synthesis, biodistribution, and primate imaging of fluorine-18 labeled 2b-carbo-1'-fluoro-2-propoxy-3b-(4-chlorophenyl)tropanes: Ligands for the imaging of dopamine transporters by positron emission tomography. J. Med. Chem. 2000, 43, 639–648. [Google Scholar] [CrossRef]
- Höfgen, N.; Stange, H.; Schindler, R.; Lankau, H.J.; Grunwald, C.; Langen, B.; Egerland, U.; Tremmel, P.; Pangalos, M.N.; Marquis, K.L.; et al. Discovery of imidazo[1,5-a]pyrido[3,2-e]pyrazines as a new class of phosphodiesterase 10A inhibitiors. J. Med. Chem. 2010, 53, 4399–4411. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Funke, U.; Deuther-Conrad, W.; Schwan, G.; Maisonial, A.; Scheunemann, M.; Fischer, S.; Hiller, A.; Briel, D.; Brust, P. Radiosynthesis and Radiotracer Properties of a 7-(2-[18F]Fluoroethoxy)-6-methoxypyrrolidinylquinazoline for Imaging of Phosphodiesterase 10A with PET. Pharmaceuticals 2012, 5, 169-188. https://doi.org/10.3390/ph5020169
Funke U, Deuther-Conrad W, Schwan G, Maisonial A, Scheunemann M, Fischer S, Hiller A, Briel D, Brust P. Radiosynthesis and Radiotracer Properties of a 7-(2-[18F]Fluoroethoxy)-6-methoxypyrrolidinylquinazoline for Imaging of Phosphodiesterase 10A with PET. Pharmaceuticals. 2012; 5(2):169-188. https://doi.org/10.3390/ph5020169
Chicago/Turabian StyleFunke, Uta, Winnie Deuther-Conrad, Gregor Schwan, Aurélie Maisonial, Matthias Scheunemann, Steffen Fischer, Achim Hiller, Detlef Briel, and Peter Brust. 2012. "Radiosynthesis and Radiotracer Properties of a 7-(2-[18F]Fluoroethoxy)-6-methoxypyrrolidinylquinazoline for Imaging of Phosphodiesterase 10A with PET" Pharmaceuticals 5, no. 2: 169-188. https://doi.org/10.3390/ph5020169
APA StyleFunke, U., Deuther-Conrad, W., Schwan, G., Maisonial, A., Scheunemann, M., Fischer, S., Hiller, A., Briel, D., & Brust, P. (2012). Radiosynthesis and Radiotracer Properties of a 7-(2-[18F]Fluoroethoxy)-6-methoxypyrrolidinylquinazoline for Imaging of Phosphodiesterase 10A with PET. Pharmaceuticals, 5(2), 169-188. https://doi.org/10.3390/ph5020169