Design, Synthesis and Hydrolytic Behavior of Mutual Prodrugs of NSAIDs with Gabapentin Using Glycol Spacers

Abstract
:1. Introduction
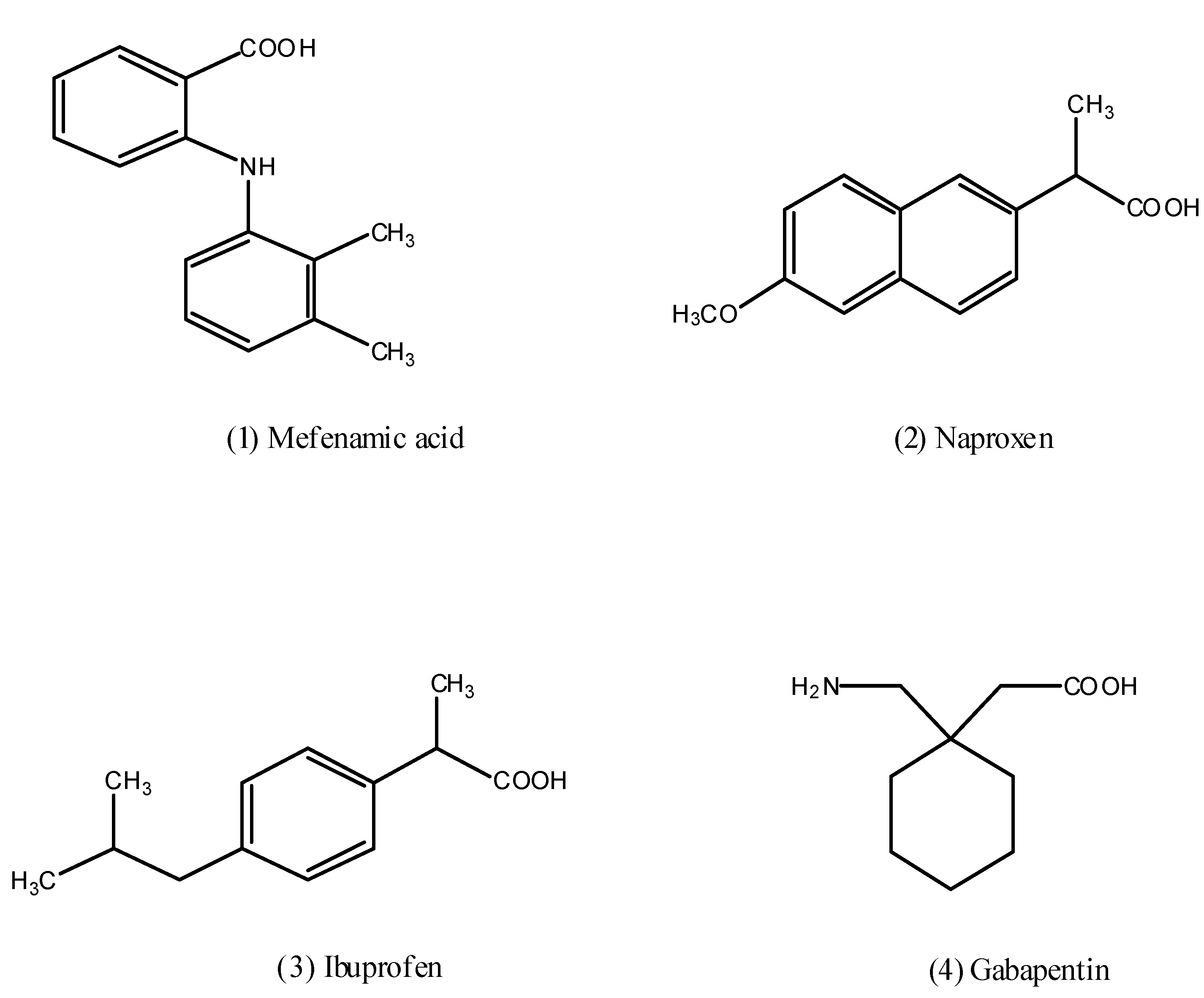
2. Results and Discussion
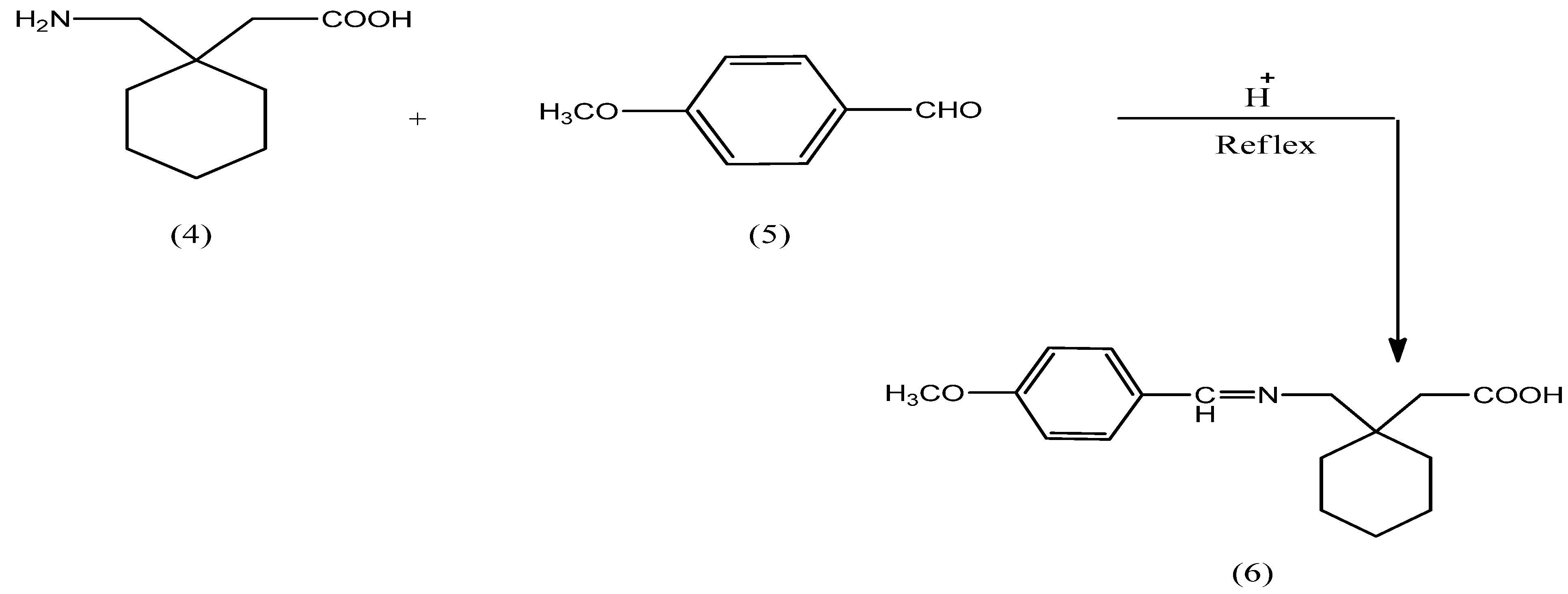
2.1. Chemistry
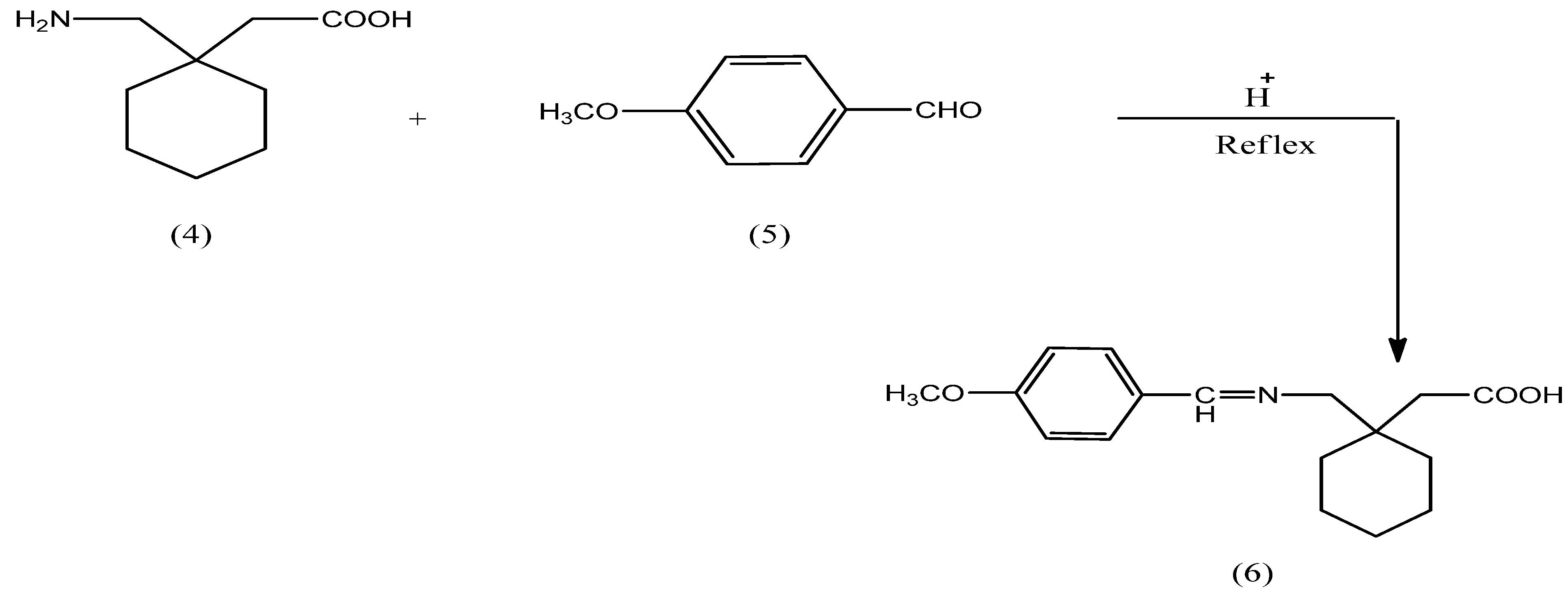
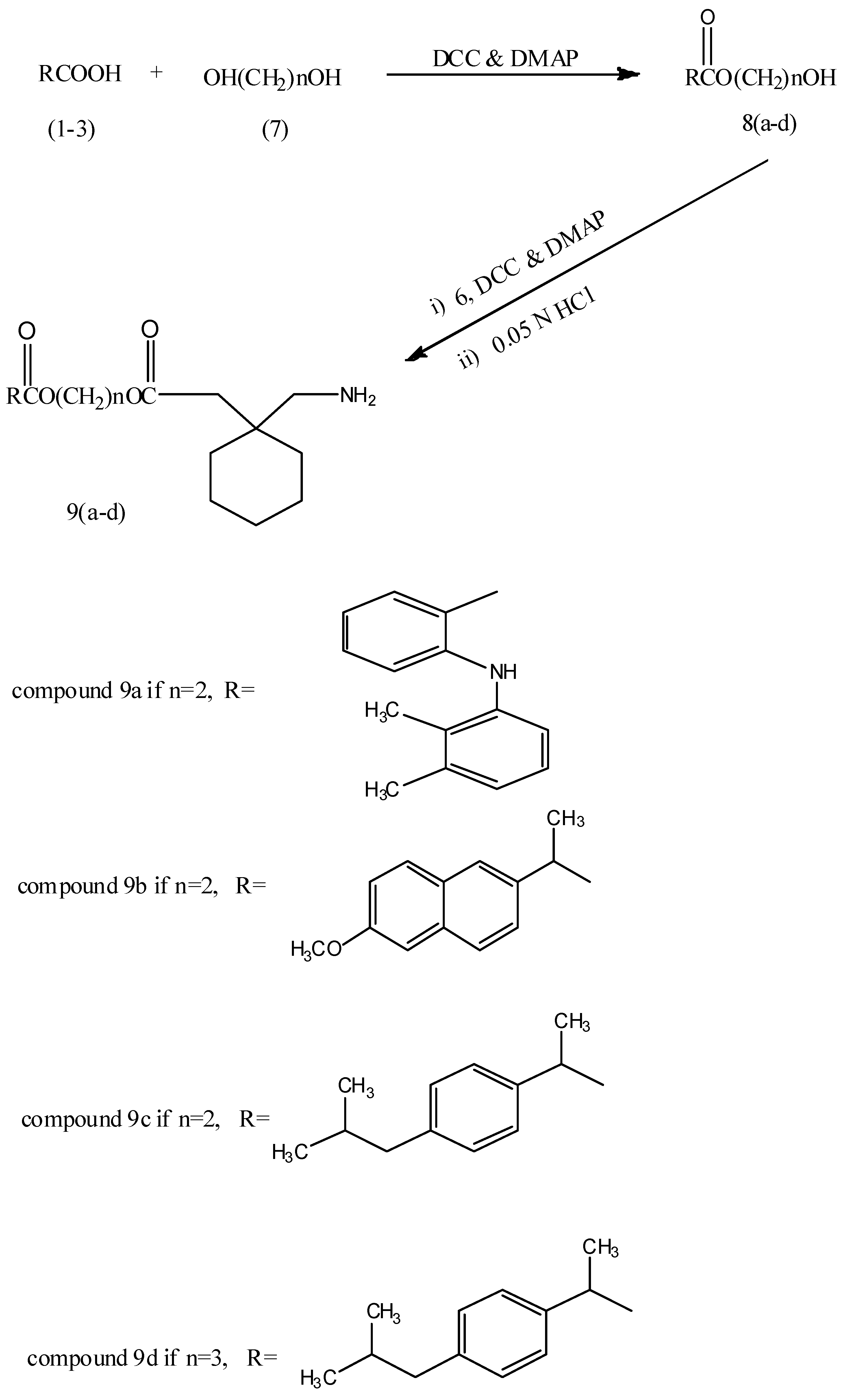
2.2. Hydrolysis Studies
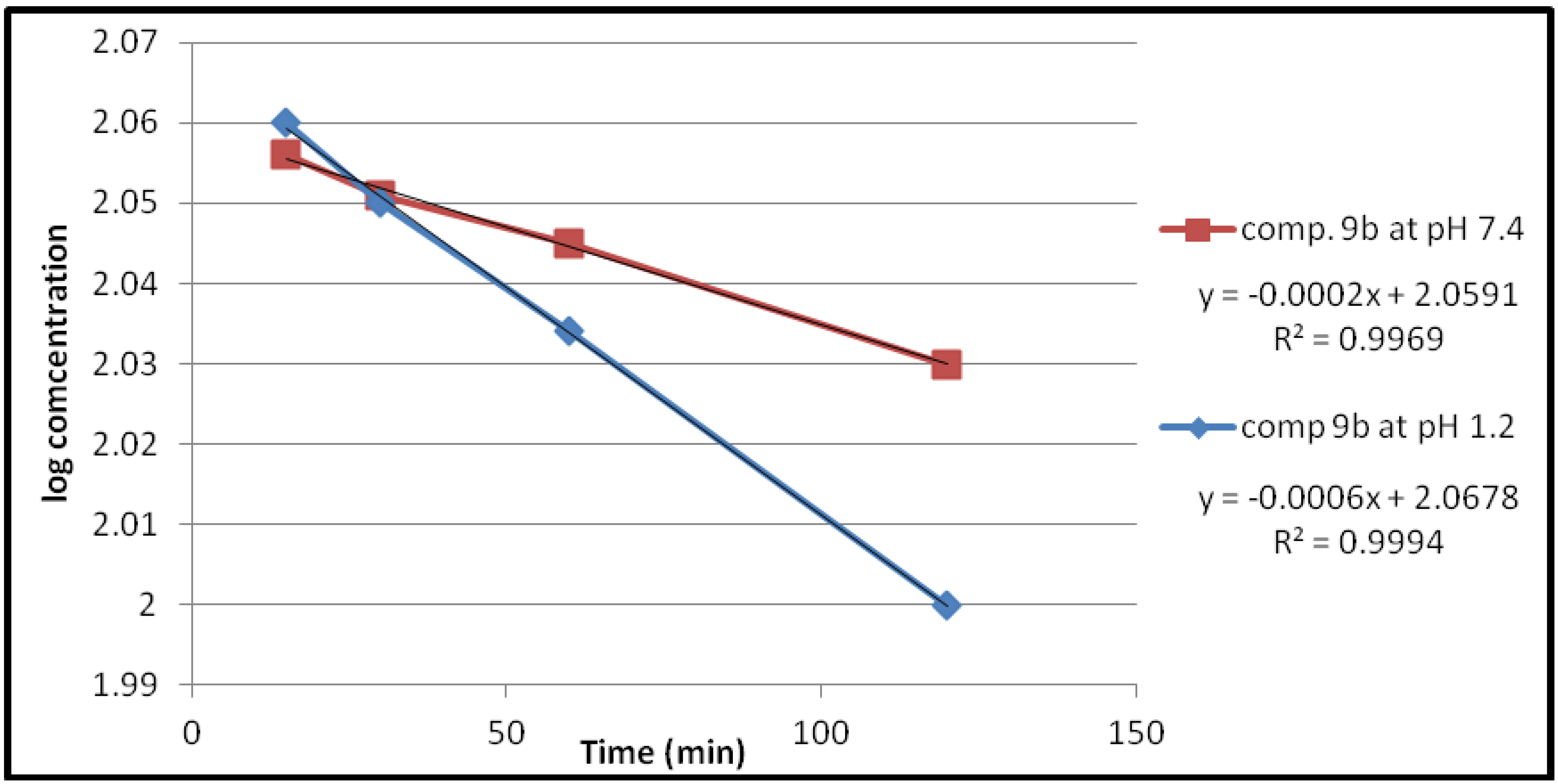
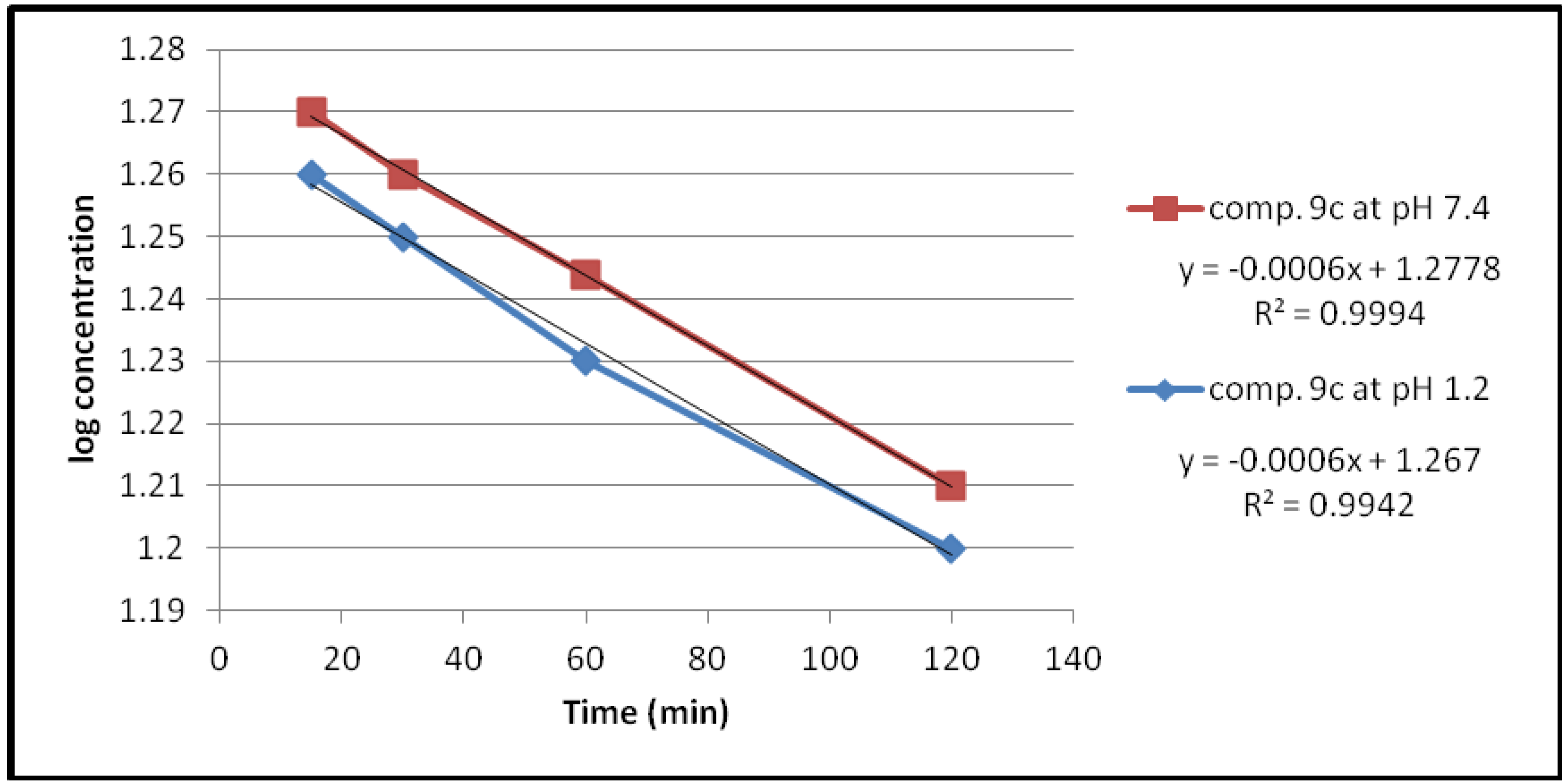
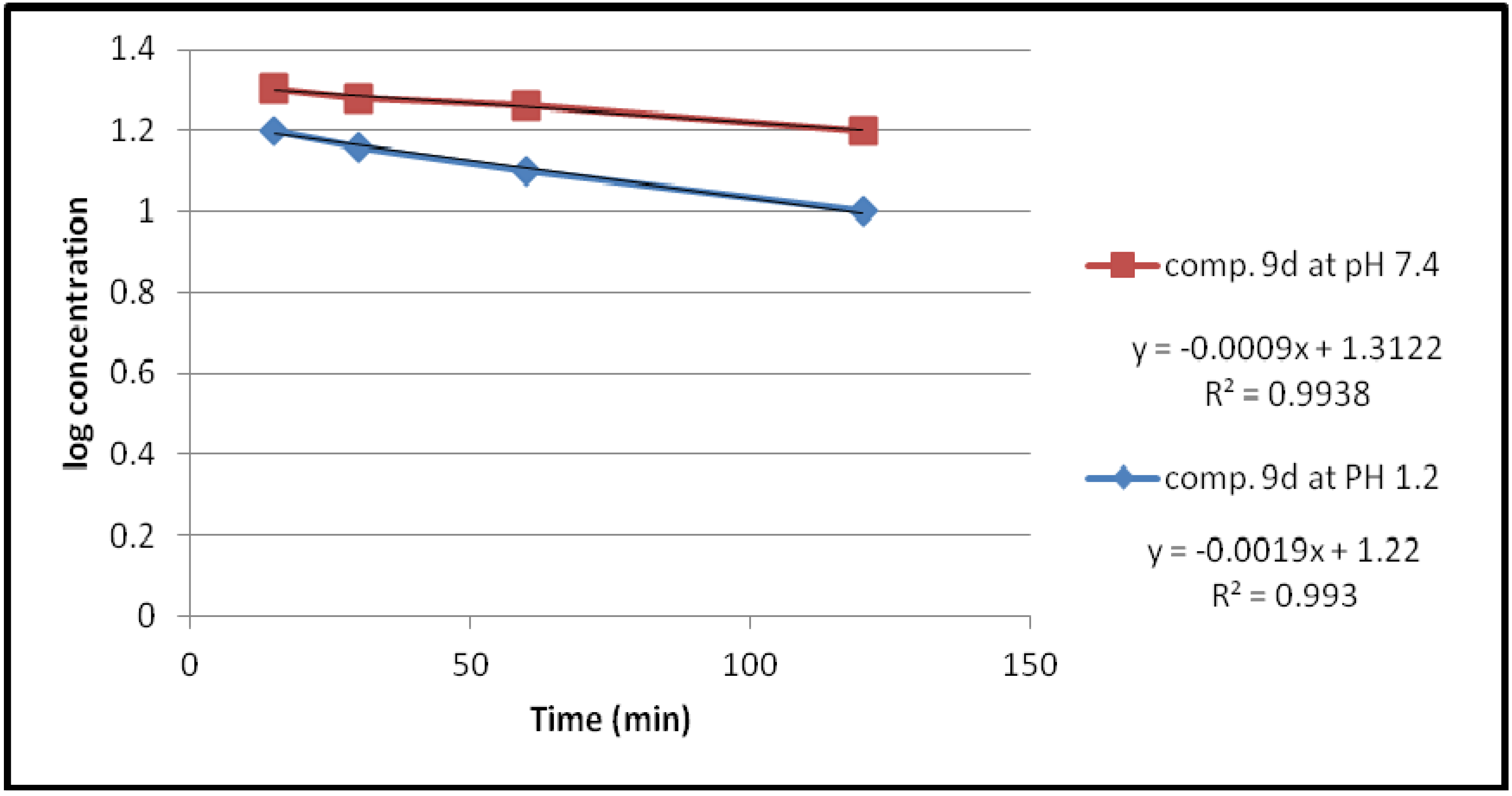
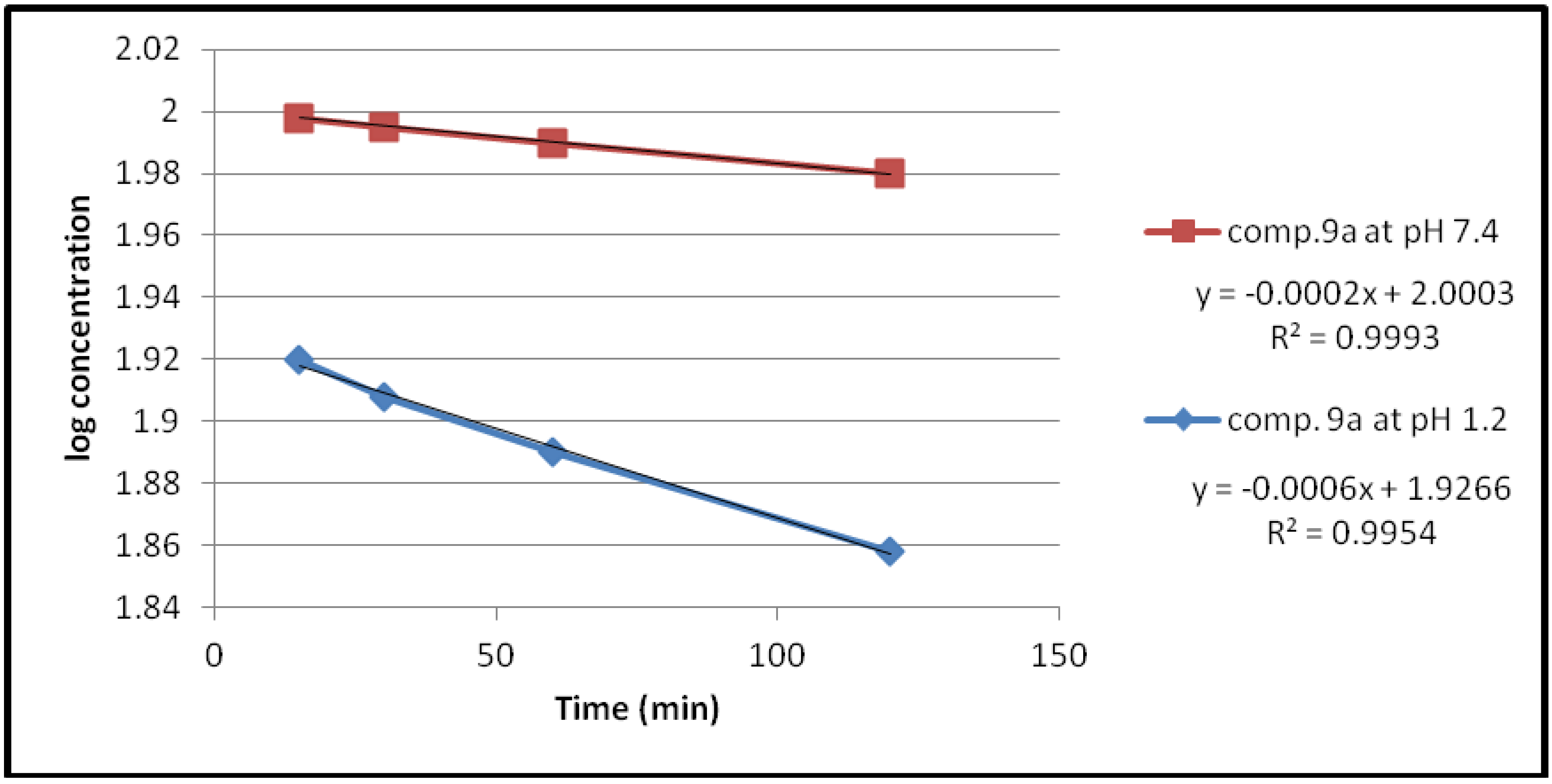
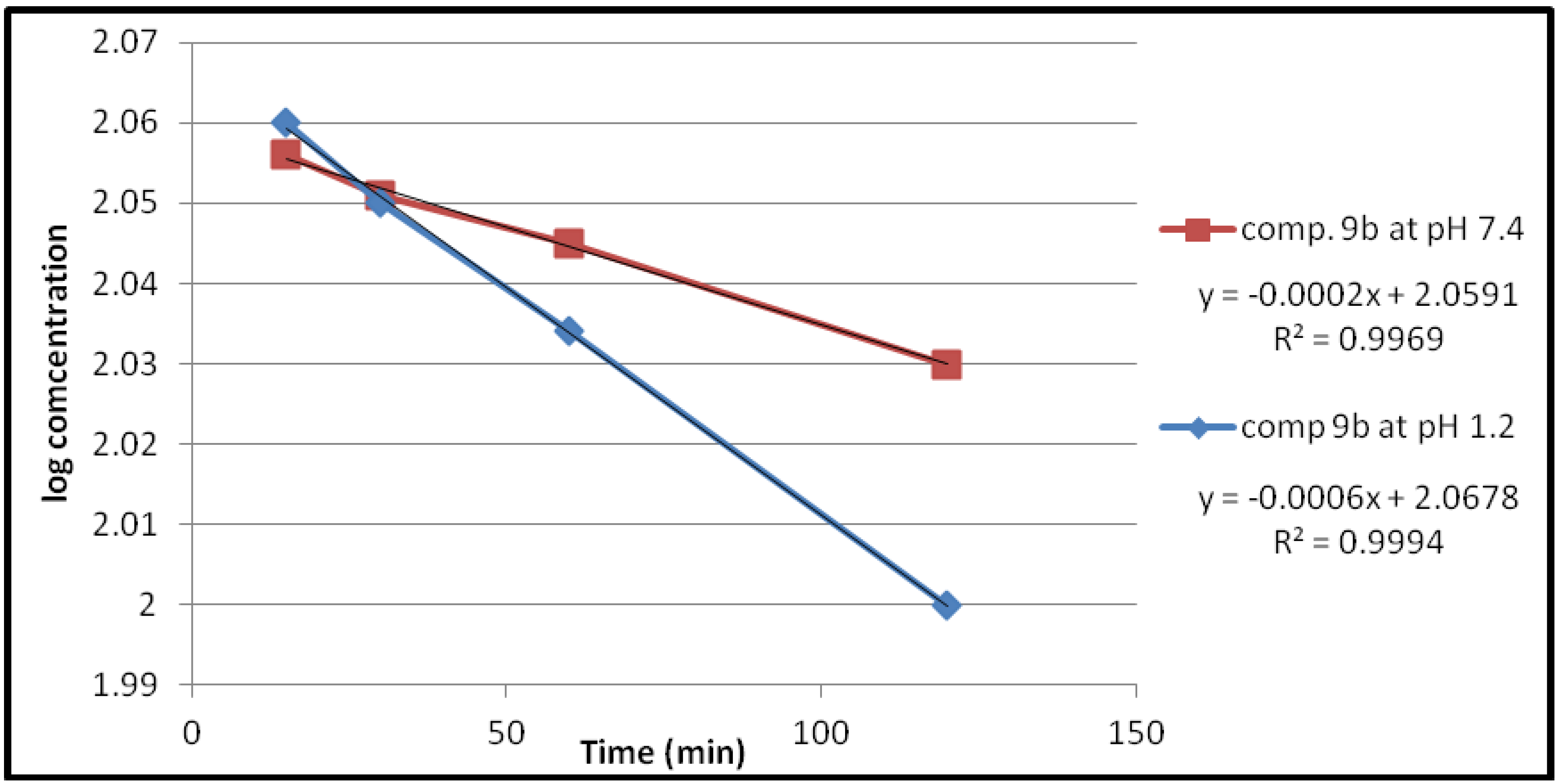
2.2.1. Chemical Hydrolysis
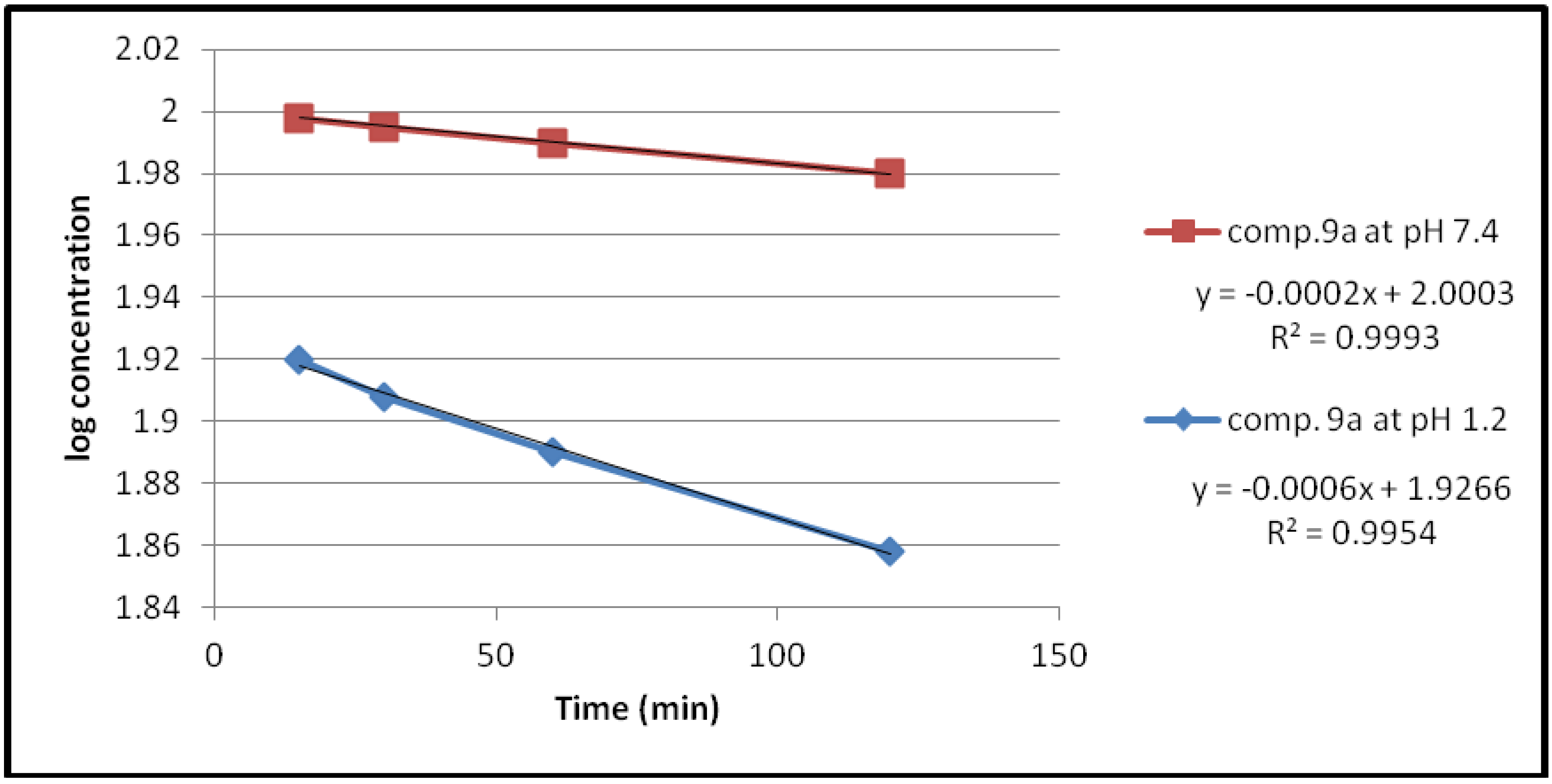

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | pH | Kobs (min−1) | t1/2(min) |
|---|---|---|---|
| 9a | 1.2 | 1.3818 × 10−3 | 501.51 |
| 7.4 | 0.4606 × 10−3 | 1504.55 | |
| 9b | 1.2 | 1.3818 × 10−3 | 501.51 |
| 7.4 | 0.4606 × 10−3 | 1504.55 | |
| 9c | 1.2 | 1.38 × 10−3 | 501.17 |
| 7.4 | 1.38 × 10−3 | 501.17 | |
| 9d | 1.2 | 4.3757 × 10−3 | 158.37 |
| 7.4 | 2.07 × 10−3 | 334.78 |
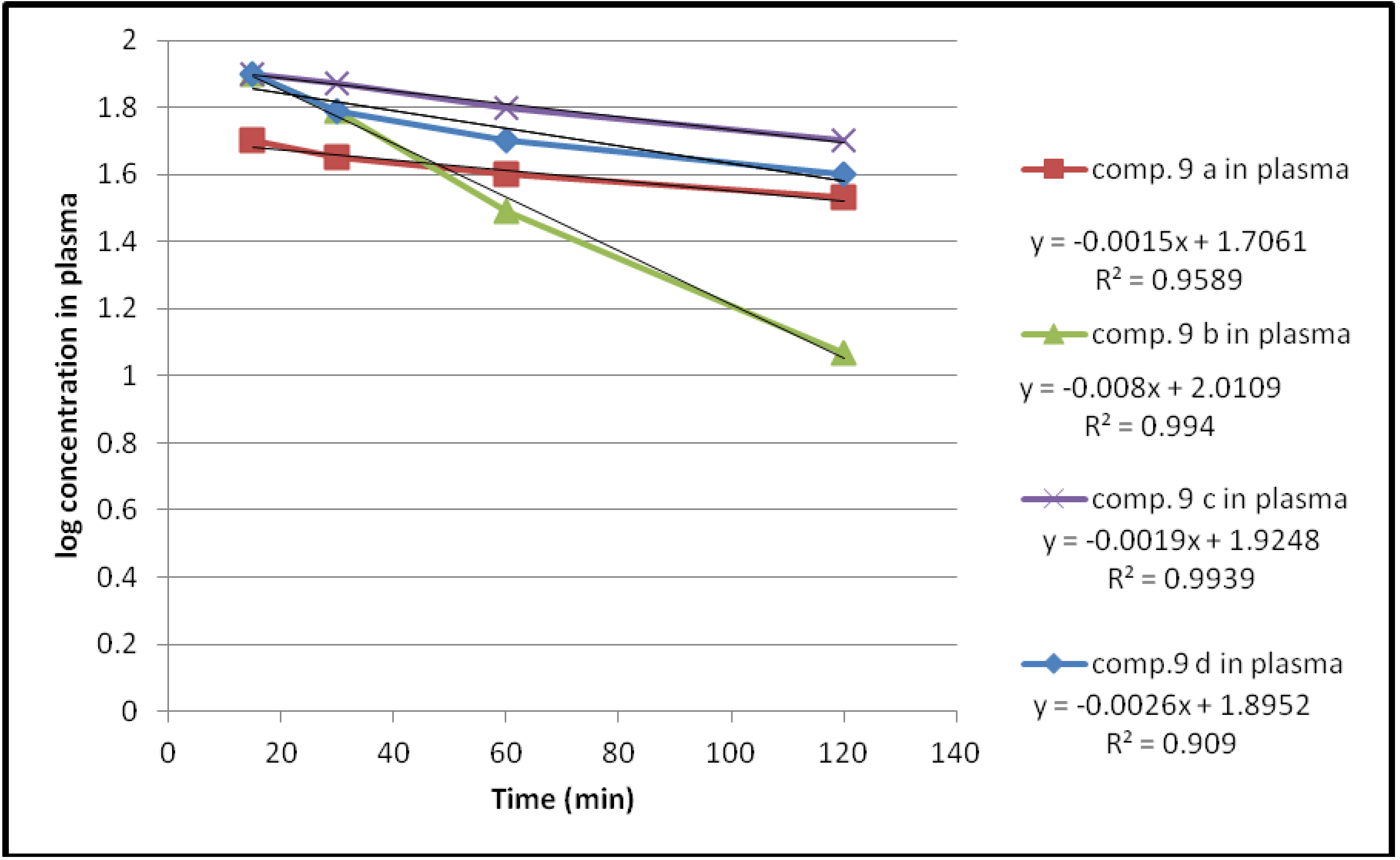
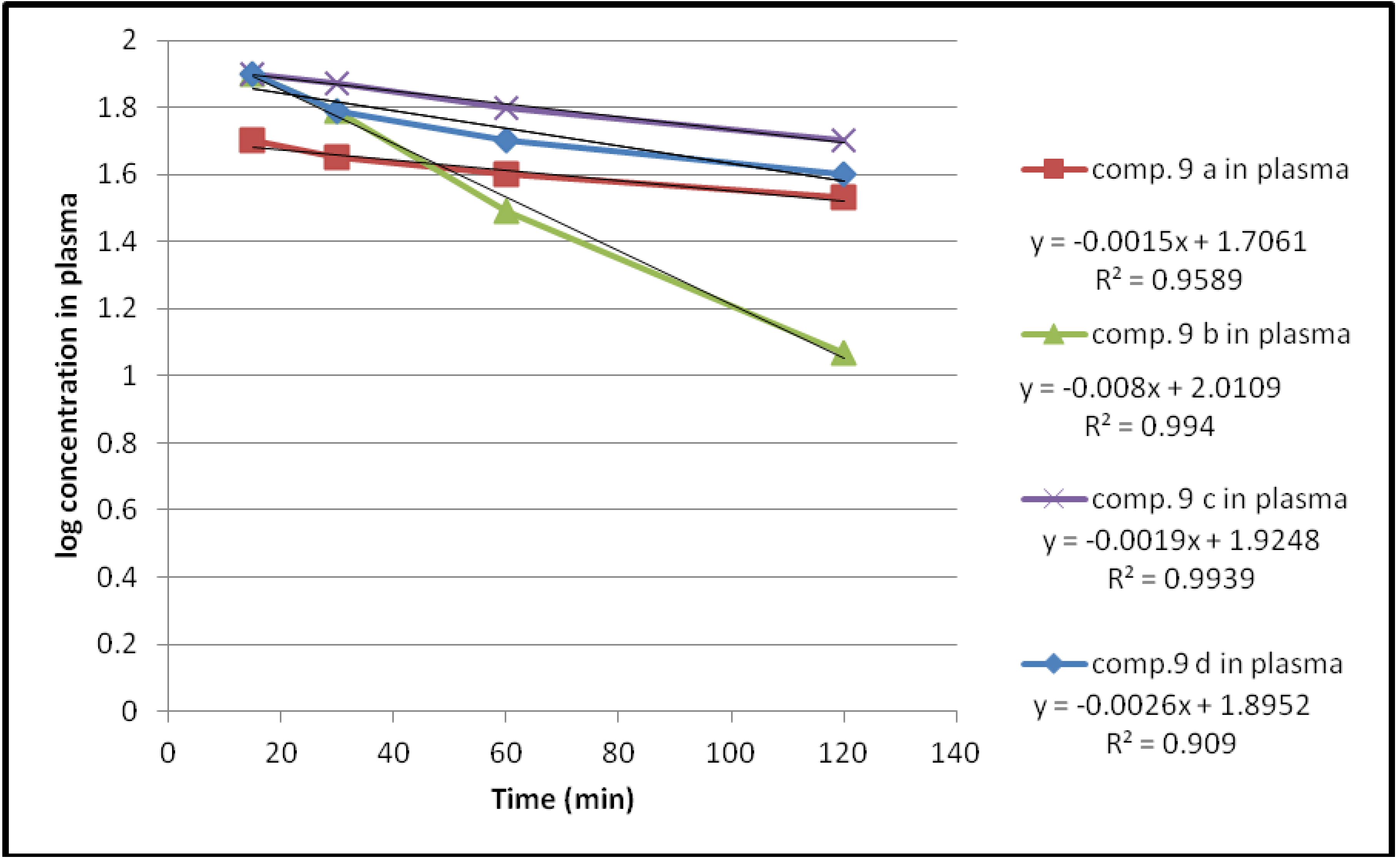
2.2.2. Enzymatic Hydrolysis
| Compound | Hydrolysis % | Kobs | T1/2(min) | |||
|---|---|---|---|---|---|---|
| 15 min | 30 min | 60 min | 120 min | |||
| 9a | 39 | 57.9 | 64 | 66 | 3.4545 × 10−3 | 200 |
| 9b | 1 | 37 | 68.5 | 88 | 18.424 × 10−3 | 37.6 |
| 9c | 19 | 21.4 | 37.8 | 49 | 4.3757 × 10−3 | 158.3 |
| 9d | 13 | 37 | 47 | 59 | 5.9878 × 10−3 | 115.7 |
3. Experimental
3.1. General
3.2. Synthesis of N-Protected Gabapentin 6
3.3. General Procedure for the Synthesis of Glycol Ester Derivatives of NSAIDs 8a–d
3.4. General Procedure for the Synthesis of the Final Compounds 9a–d
3.5. Chemical Hydrolysis
3.6. Enzymatic Hydrolysis
4. Conclusions
Acknowledgments
References
- Ajmone-Cat, M.A.; Bernardo, A.; Greco, A.; Minghetti, L. Non-steroidal anti-inflammatory drugs and brain inflammation: Effects on microglial functions. Pharmaceuticals 2010, 3, 1949–1964. [Google Scholar] [CrossRef]
- Uludag, M.O.; Ergün, B.C.; Alkan, D.A.; Ercan, N.; Özkan, G.Y.; Banoglu, E. Stable ester and amide conjugates of some NSAIDs as analgesic and anti-inflammatory compounds with improved biological activity. Turk. J. Chem. 2011, 35, 427–439. [Google Scholar]
- James, M.S.; Clemence, E.H. Strategies to optimize treatment with NSAIDs in patients at risk for gastrointestinal and cardiovascular adverse events. Clin. Ther. 2010, 32, 667–677. [Google Scholar] [CrossRef]
- Parto, S.K.; Robert, F.H. Evidence for neuroprotection by the fenamate NSAID, mefenamic acid. Neurochem. Int. 2009, 55, 683–688. [Google Scholar] [CrossRef]
- Mizushima, T. Molecular mechanism for various pharmacological activities of NSAIDS. Pharmaceuticals 2010, 3, 1614–1636. [Google Scholar] [CrossRef]
- Wittine, K.; Benci, K.; Rajic, Z.; Zorc, B.; Kralj, M.; Marjanovic, M.; Pavelic, K.; De Clercq, E.; Andrei, G.; Snoeck, R.; et al. The novel phosphoramidate derivatives of NSAIDs 3-hydroxypropylamides: Synthesis, cytostatic and antiviral activity evaluations. Eur. J. Med. Chem. 2009, 44, 143–151. [Google Scholar] [CrossRef]
- Tomisato, W.; Tsutsumi, S.; Hoshino, T.; Hwang, H.; Mio, M.; Tsuchiya, T.; Mizushima, T. Role of direct cytotoxic effects of NSAIDs in the induction of gastric lesions. Biochem. Pharmacol. 2004, 67, 575–585. [Google Scholar] [CrossRef]
- Burian, M.; Geisslinger, G. COX-dependent mechanisms involved in the antinociceptive action of NSAIDs at central and peripheral sites. Pharmacol. Ther. 2005, 107, 139–154. [Google Scholar] [CrossRef]
- Jensen, T.S.; Finnerup, N.B. Neuropathic pain: Peripheral and central mechanisms. Eur. J. of Pain Supplements 2009, 3, 33–36. [Google Scholar] [CrossRef]
- Jensen, T.S. Pathophysiology of pain: From theory to clinical evidence. Eur. J. Pain Suppl. 2008, 2, 13–17. [Google Scholar] [CrossRef]
- Sarah, M.S.; Cristina, E.M.; Steven, P.W.; Srinivasa, N.R. Peripheral opioid analgesia for the treatment of neuropathic pain: Gene mutation to virus mediated gene transfer. Eur. J. Pain Suppl. 2010, 4, 251–256. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Sindrup, S.H.; Jensen, T.S. The evidence for pharmacological treatment of neuropathic pain. Pain 2010, 150, 573–581. [Google Scholar] [CrossRef]
- Bıyık, İ.; Gülcüler, M.; Karabiga, M.; Ergene, O.; Tayya, N. Efficacy of gabapentin versus diclofenac in the treatment of chest pain and paresthesia in patients with sternotomy. Anadolu Kardiyol. Derg. 2009, 9, 390–396. [Google Scholar]
- Gupta, S.K.; Mahajan, A.; Tandon, V. Gabapentin for the treatment of neuropathic pain. Palliat. Med. 2004. [Google Scholar] [CrossRef]
- Graeme, J.S. The mechanisms of action of gabapentin and pregabalin. Curr. Opin. Pharmacol. 2006, 6, 108–111. [Google Scholar] [CrossRef]
- Abdel-Azeem, A.Z.; Abdel-Hafez, A.A.; El-Karamany, G.S.; Farag, H.H. Chlorzoxazone esters of some non-steroidal anti-inflammatory (NSAI) carboxylic acids as mutual prodrugs: Design, synthesis, pharmacological Investigations and docking studies. Bioorg. Med. Chem. 2009, 17, 3665–3670. [Google Scholar] [CrossRef]
- Ohlan, S.; Nanda, S.; Pathak, D.P.; Jagia, M. Mutual prodrugs-A swot analysis. IJPSR 2011, 2, 719–729. [Google Scholar]
- Bhosle, D.; Bharambe, S.; Gairola, N.; Dhaneshwar, S.S. Mutual prodrug concept: Fundamentals and applications. Indian J. Pharm. Sci. 2006, 68, 3, 286–294. [Google Scholar]
- Amjad, M.Q.; Meriem, M.R. and Bassam, M.T. Synthesis, characterization and in vitro hydrolysis of a gemfibrozil-nicotinic acid codrug for improvement of lipid profile. Eur. J. Pharm. Sci. 2011, 43, 99–108. [Google Scholar]
- Hamad, M.O.; Kiptoo, P.K.; Stinchcomb, A.L.; Crooks, P.A. Synthesis and hydrolytic behavior of two novel tripartate codrugs of naltrexone and 6β-Naltrexol with hydroxybupropion as potential alcohol abuse and smoking cessation gents. Bioorg. Med. Chem. 2006, 14, 7051–7061. [Google Scholar] [CrossRef]
- Mechael, B.A. Kinetic of product stability. In Pharmaceutics the Design and Manufacture of Medicines, 3th ed; Elsevier Limited: New York, NY, USA, 2007; pp. 99–107. [Google Scholar]
- Kalgutkar, A.S.; Marnett, B.A.; Crews, B.C.; Remmel, R.P.; Marnett, L.J. Ester and amide derivatives of the nonsteroidal anti-inflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitor. J. Med. Chem. 2000, 43, 2860–2870. [Google Scholar] [CrossRef]
- Bonina, F.P.; Montenegro, L.; Caprariis, P.; Palagiano, F.; Capasso, A.; Sorrentino, L. Pharmacokinetic and pharmacodynemic profile of triethylene glycol indomethacin ester as a new oral prodrug. J. Controlled Release 1996, 41, 187–193. [Google Scholar] [CrossRef]
- Nawaz, H.; Akhter, Z.; Yameen, S.; Siddiqi, H.M.; Mirza, B.; Rifat, A. Synthesis and biological evaluation of some Schiff base ester of ferrocenyl aniline and simple aniline. J. Organomet. Chem. 2009, 694, 2198–2203. [Google Scholar] [CrossRef]
- Lee, B.S.; Yoon, C.W.; Osipov, A.; Moghavem, V.; Nwachokor, D.; Amatya, R.; Na, R.; Pantoja, J.L.; Pham, M.D.; Black, K.L.; et al. Nanoprodrugs of NSAIDs: Preparation and characterization of flufenamic acid nanoprodrugs. J. Drug Deliv. 2011. [Google Scholar] [CrossRef]
- Wadhwa, L.K.; Sharma, P.D. Glycolamide ester of 6-methoxy-2-naphthylacetic acid as potential prodrugs-physicochemical properties, chemical stability and enzymatic hydrolysis. Int. J. Pharm. 1995, 118, 31–39. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mahdi, M.F.; Alsaad, H.N. Design, Synthesis and Hydrolytic Behavior of Mutual Prodrugs of NSAIDs with Gabapentin Using Glycol Spacers. Pharmaceuticals 2012, 5, 1080-1091. https://doi.org/10.3390/ph5101080
Mahdi MF, Alsaad HN. Design, Synthesis and Hydrolytic Behavior of Mutual Prodrugs of NSAIDs with Gabapentin Using Glycol Spacers. Pharmaceuticals. 2012; 5(10):1080-1091. https://doi.org/10.3390/ph5101080
Chicago/Turabian StyleMahdi, Monther Faisal, and Hiba Najeh Alsaad. 2012. "Design, Synthesis and Hydrolytic Behavior of Mutual Prodrugs of NSAIDs with Gabapentin Using Glycol Spacers" Pharmaceuticals 5, no. 10: 1080-1091. https://doi.org/10.3390/ph5101080
APA StyleMahdi, M. F., & Alsaad, H. N. (2012). Design, Synthesis and Hydrolytic Behavior of Mutual Prodrugs of NSAIDs with Gabapentin Using Glycol Spacers. Pharmaceuticals, 5(10), 1080-1091. https://doi.org/10.3390/ph5101080